
Abstract
Background
CT-P43 is a candidate ustekinumab biosimilar in clinical development.
Objectives
This paper aims to demonstrate equivalent efficacy of CT-P43 to originator ustekinumab in adults with moderate to severe plaque psoriasis.
Methods
This double-blind, phase III trial randomised patients (1:1) to receive subcutaneous CT-P43 or originator ustekinumab (45/90 mg for patients with baseline body weight ≤ 100 kg/> 100 kg) at week 0 and week 4 in Treatment Period I. Prior to week 16 dosing in Treatment Period II, patients receiving originator ustekinumab were re-randomised (1:1) to continue originator ustekinumab or switch to CT-P43; patients initially randomised to CT-P43 continued receiving CT-P43 (at weeks 16, 28 and 40). The primary endpoint of the trial was mean per cent improvement from baseline in Psoriasis Area Severity Index (PASI) score at week 12. Equivalence was concluded if confidence intervals (CIs) for the estimate of treatment difference were within pre-defined equivalence margins: ± 10% [90% CI; modified intent-to-treat set; Food and Drug Administration (FDA) approach] or ± 15% [95% CI; full analysis set for patients only receiving 45 mg doses in Treatment Period I; European Medicines Agency (EMA) approach]. Additional efficacy, pharmacokinetic, safety and immunogenicity endpoints were evaluated through week 52. Results to week 28 are reported here.
Results
In Treatment Period I, 509 patients were randomised (CT-P43: N = 256; originator ustekinumab: N = 253). The mean per cent improvement in PASI score at week12 was 77.93% and 75.89% for CT-P43 and originator ustekinumab, respectively (FDA approach); per the EMA approach, corresponding values were 78.26% and 77.33%. Estimated treatment differences were 2.05 (90% CI −0.23, 4.32) and 0.94 (95% CI −2.29, 4.16); equivalence was achieved for both sets of assumptions. Further efficacy parameters and pharmacokinetic, safety and immunogenicity outcomes were comparable between treatment groups, including after switching from originator ustekinumab to CT-P43.
Conclusions
CT-P43 demonstrated equivalent efficacy to originator ustekinumab in patients with moderate to severe plaque psoriasis, with comparable pharmacokinetic, safety and immunogenicity profiles.
Clinical Trial Registration
ClinicalTrials.gov Identifier: NCT04673786; date of registration: 17 December, 2020
Similar content being viewed by others

Avoid common mistakes on your manuscript.
This phase III trial was designed to compare a candidate ustekinumab biosimilar, CT-P43, to originator ustekinumab in adults with moderate to severe plaque psoriasis. |
CT-P43 showed equivalent efficacy to originator ustekinumab, with comparable pharmacokinetics, safety and immunogenicity. |
1 Introduction
Plaque psoriasis is a chronic, immune-mediated inflammatory disease, with an estimated prevalence of ≥ 1.5% in regions such as Australasia and Western and Central Europe, and in areas of North America [1]. Ustekinumab is a monoclonal antibody approved for the treatment of adults with moderate to severe plaque psoriasis, Crohn’s disease, ulcerative colitis or active psoriatic arthritis (PsA) [2, 3]. It specifically targets the p40 subunit of the cytokines interleukin (IL)-12 and IL-23 and attenuates downstream cytokine signalling [3, 4], reducing expression of inflammatory transcripts [3, 5]. Previous trials evaluating ustekinumab have consistently demonstrated a favourable safety profile [6,7,8,9,10].
The high cost of biologic therapies such as ustekinumab can be a barrier to patient access. Biosimilars, which have no clinically meaningful differences from the approved reference biologic, can help to address this challenge as lower-cost alternatives, encouraging more competitive pricing [11, 12]. Regulatory approval of biosimilars by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) is subject to demonstration of the absence of clinically meaningful differences in terms of safety, quality, biological activity and efficacy with respect to the originator biologic [11, 12]. There are currently no FDA- or EMA-approved ustekinumab biosimilars [13, 14] .
CT-P43 is a candidate ustekinumab biosimilar currently in development. The safety and pharmacokinetics (PK) of CT-P43 have been investigated in a phase I study. We report week 28 findings from a phase III study comparing CT-P43 with originator ustekinumab in patients with moderate to severe plaque psoriasis. The primary objective was to determine equivalence of subcutaneously administered CT‑P43 and originator ustekinumab in terms of efficacy at week 12, and to evaluate using two sets of equivalence margins (± 10% and ± 15% to align with FDA and EMA approaches, respectively). Quality of life, PK, safety and immunogenicity were evaluated in addition to efficacy endpoints.
2 Methods
2.1 Study Design
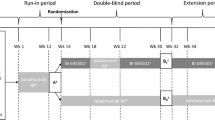
This was a randomised, active-controlled, double-blind, phase III trial (ClinicalTrials.gov: NCT04673786) conducted at 34 centres in Estonia, Poland, Republic of Korea and Ukraine (Supplementary Table 1). Following initial screening from day 42 to day 1, patients were randomised (1:1) on day 1 to receive either CT-P43 or European Union-sourced originator ustekinumab in Treatment Period I (weeks 0–16; Fig. 1). Randomisation was conducted using an interactive web response system, which linked sequential patient randomisation numbers to treatment codes. At the first randomisation, patients were stratified by country, body weight (≤ 100 kg versus > 100 kg) and prior use of biologic treatment for psoriasis (yes versus no).

Study design. aAt week 12, it was recommended that patients (in either group) who achieved at least PASI 50 continued study drug administration in Treatment Period II. bPrior to dosing at week 16, patients who were initially randomised to originator ustekinumab were re-randomised 1:1 to either continue receiving originator ustekinumab or to switch to CT-P43. cAt week 28, it was recommended that only patients who achieved at least PASI 75 continued further study drug administration (for all groups). dInvestigator-reported outcome assessments were performed by a qualified efficacy assessor per site. It was recommended that the same assessor perform the investigator-reported outcome assessments throughout the entire study period, if possible. EOS end of study, F/U follow-up, PASI 50/75 Psoriasis Area and Severity Index 50/75% improvement from baseline
Following Treatment Period I, patients who had received originator ustekinumab were re-randomised (1:1) to either continue originator ustekinumab or switch to CT-P43 in Treatment Period II (weeks 16–40). The second randomisation was stratified by week 16 dose (45 mg versus 90 mg). Patients previously assigned to CT-P43 continued to receive CT-P43. The study drug was administered at weeks 0 and 4 (Treatment Period I) and weeks 16, 28 and 40 (Treatment Period II) by subcutaneous injection via pre-filled syringe. During Treatment Period I, doses were 45 mg or 90 mg (two 45 mg/0.5 mL doses) for patients with baseline body weight ≤ 100 kg or > 100 kg, respectively. Dosing for Treatment Period II was adjusted if a significant change in body weight had occurred pre-dose at week 16. There was a 12-week follow-up period after Treatment Period II and an end-of-study visit at week 52. This manuscript reports findings up to week 28.
Pre-defined personnel were unblinded for reporting purposes, following database lock for week 28 data. Other pre-defined personnel, investigators and patients remained blinded until end of study. Study drug administration was conducted by pre-defined unblinded staff at the site (as drug solutions had slightly different appearances).
The study was conducted according to Good Clinical Practice and the principles of the Declaration of Helsinki, as well as national, state and local laws or regulations. Study materials were approved by independent ethics committee/institutional review boards at each site (Supplementary Table 1). All patients in this manuscript have given written informed consent for participation in the study and the use of their de-identified, anonymized, aggregated data and their case details (including photographs) for publication. The original protocol was revised three times during the study period, including generation of two country-specific protocols (Supplementary Methods).
2.2 Patients
Full inclusion/exclusion criteria are included in the Supplementary Methods. Eligible patients were aged 18–80 years (inclusive), with plaque-type psoriasis diagnosed for ≥ 24 weeks prior to first study drug administration. Eligible patients had moderate to severe plaque-type psoriasis (defined as Psoriasis Area and Severity Index [PASI] score ≥ 12, involved body surface area ≥ 10%, and static Physician’s Global Assessment [sPGA] score ≥ 3), with or without PsA, at screening and first study drug administration, and were candidates for phototherapy or systemic therapy. Key exclusion criteria were diagnosis of other forms of psoriasis or skin conditions that could interfere with efficacy assessments, previous treatment with ustekinumab or candidate ustekinumab biosimilars and other drugs directly targeting IL-12 or IL‑23, or ≥ 2 biologics approved for psoriasis treatment. Previous treatment with one biologic was allowed (with a sufficient washout period).
2.3 Study Endpoints and Assessments
The primary efficacy endpoint was mean per cent improvement from baseline in PASI score at week 12. Secondary efficacy endpoints included: PASI scores, improvement from baseline PASI scores, proportions of patients achieving 50/75/90/100% improvement from baseline PASI scores (PASI 50/75/90/100), proportions of patients with sPGA scores of 0 (clear) or 1 (almost clear) and change from baseline in Dermatology Life Quality Index (DLQI) scores. PK, safety and immunogenicity were also assessed as secondary endpoints. Testing for anti-drug antibodies (ADAs) and neutralising antibodies (for patients with positive ADA results) against CT-P43 and originator ustekinumab was performed using an electrochemiluminescence assay (Meso Scale Discovery, Meso Scale Diagnostics, Rockville, MD, USA). PK assessment was based on serum drug concentrations at weeks 0, 4, 12, 16, 28, 40 and 52. Safety evaluations included treatment-emergent adverse events (TEAEs), coded according to the Medical Dictionary for Regulatory Activities version 24.1 and graded according to the Common Terminology Criteria for Adverse Events version 5.0. TEAEs of special interest were infections/serious infections, injection-site reactions (ISRs), hypersensitivity reactions and malignancies. Study assessments are summarised in Supplementary Table 2.
2.4 Statistical Analysis
Using FDA assumptions (described below), a minimum sample size of 400 patients was determined to provide ≥ 90% statistical power for demonstrating equivalence of the primary efficacy endpoint using a 90% confidence interval (CI) approach, corresponding to two one-sided tests at the 5% significance level. Calculations assumed an expected between-group difference of 0 and a standard deviation (SD) of 29.0 for the primary endpoint. Using EMA assumptions (described below), including only patients who received 45 mg doses of study drug during Treatment Period I, a minimum sample size of 166 patients was determined to provide ≥ 80% statistical power for demonstrating equivalence of the primary efficacy endpoint using a 95% CI approach, corresponding to two one-sided tests at the 2.5% significance level. Calculations assumed an expected between-group difference of 0 and an SD of 29.6 for the primary endpoint. A minimum sample size of 400 patients was estimated for the safety follow-up. With a hypothesised drop-out rate of 10%, a total sample size of 446 patients was planned.
The primary endpoint was analysed using analysis of covariance, as detailed in the Supplementary Methods. Covariates comprised country, body weight, use of prior biologic approved for psoriasis treatment and baseline PASI score. Using FDA assumptions, equivalence was concluded if the 90% CIs for the treatment difference were entirely contained within the pre-defined −10% to 10% equivalence margin [using the modified intent-to-treat (mITT) set]. Using EMA assumptions, equivalence was concluded if the 95% CIs for treatment difference were entirely contained within the pre-defined −15% to 15% equivalence margin (in the full analysis set of patients who received only 45 mg doses of study drug in Treatment Period I, using complete case analysis). Analysis populations are detailed in the Supplementary Methods. Statistical analyses were conducted using Statistical Analysis System (SAS®) software (SAS Institute Inc., Cary, NC, USA) version 9.4.
3 Results
3.1 Patient Disposition
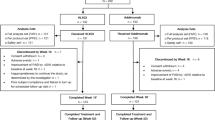
The first patient was randomised on 11 January 2021; the last patient’s visit for week 28 occurred on 25 November 2021. Patient disposition is summarised in Fig. 2. In Treatment Period I, 509 patients were randomised (CT-P43: N = 256; originator ustekinumab: N = 253). There were three (1.2%) and four (1.6%) patients in the CT-P43 and originator ustekinumab groups, respectively, who discontinued the study. In Treatment Period II, 502 participants were re-randomised (253 continued CT-P43, 125 continued originator ustekinumab and 124 switched to CT-P43). Five patients discontinued the study or study drug during Treatment Period II up to week 28 (three in the continued CT-P43 group and two in the continued originator ustekinumab group).

Patient disposition (ITT set). aThe second randomisation process was carried out at week 16. Patients randomised to the CT-P43 group for Treatment Period I continued to receive CT-P43 in Treatment Period II. Patients assigned to originator ustekinumab for Treatment Period I were randomised (1:1) to either continue originator ustekinumab or switch to CT-P43 in Treatment Period II. ITT, intent-to-treat
Patient demographics were similar across groups (Table 1 for intent-to-treat [ITT] set; Supplementary Table 3 for ITT-Treatment Period II subset). Overall, 461 (90.6%) patients were White and 377 (74.1%) from Poland. Mean (SD) body mass index was 28.41 (5.60) kg/m2, 397 (78.0%) patients had baseline body weight ≤ 100 kg and mean (SD) involved body surface area was 25.2% (13.7). Mean (SD) time since plaque-type psoriasis diagnosis was 16.69 (11.899) years, 163 (32.0%) patients had concomitant PsA and 82 (16.1%) had received prior biologics approved for psoriasis treatment.
3.2 Efficacy
Equivalence was demonstrated between CT-P43 and originator ustekinumab for the primary efficacy endpoint of mean per cent improvement from baseline in PASI score at week 12 in both FDA and EMA analyses, as CIs for treatment differences were within pre-defined equivalence margins (Table 2). Results in the per-protocol set supported the primary analyses. ‘Best-worst case’ and ‘worst-best case’ sensitivity analyses for the primary endpoint were also supportive (Supplementary Table 4). Absolute values and mean per cent improvement from baseline in PASI scores were generally similar between groups at all timepoints through week 28 (Fig. 3; Supplementary Tables 5, 6), as was the proportion of patients achieving PASI 50/75/90/100 (Supplementary Fig. 1; Supplementary Tables 7, 8).

Percentage improvement from baseline in PASI score (mITT set). mITT modified intent-to-treat, PASI Psoriasis Area and Severity Index, SD standard deviation
The proportion of patients with an sPGA score of 0 or 1 was similar between treatment groups (Supplementary Fig. 2; Supplementary Tables 9, 10). The reduction in DLQI score from baseline was similar across treatment groups at weeks 12 and 28 (Table 3; Supplementary Tables 11, 12).
3.3 Pharmacokinetics
At all timepoints up to week 28, serum ustekinumab concentrations were generally similar between treatment groups and by dose (adjusted for baseline body weight), including after a single transition from originator ustekinumab to CT-P43 (Supplementary Tables 13, 14).
3.4 Safety
In Treatment Period I, 95 (37.1%) and 75 (29.6%) patients in the CT-P43 and originator ustekinumab groups, respectively, experienced at least one TEAE (Table 4). TEAEs related to study drug occurred in 18 (7.0%) and 15 (5.9%) patients in the CT-P43 and originator ustekinumab groups, respectively. Four (1.6%) patients in each group experienced treatment-emergent serious adverse events (TESAEs). One patient (in the CT-P43 group) experienced a TESAE of coronavirus disease 2019 (COVID-19) pneumonia that was considered possibly related to the study drug by the investigator. The TESAE was considered resolved 11 days after onset following appropriate treatment. Study drug was interrupted but later resumed. No TEAEs resulted in discontinuation of study drug.
In Treatment Period II (up to week 28), 40 (15.8%), 28 (22.4%) and 26 (21.0%) patients in the continued CT-P43 group, continued originator ustekinumab group and switched to CT-P43 group, respectively, experienced at least one TEAE (Table 5); correspondingly, four (1.6%), five (4.0%) and six (4.8%) patients experienced TEAEs related to study drug. There were no TESAEs during Treatment Period II. Two patients in the continued CT-P43 group and one patient in the continued originator ustekinumab group discontinued study drug owing to TEAEs related to hepatitis B [hepatitis B and hepatitis B surface antigen positive (0.8%), and hepatitis B DNA assay positive (0.8%), respectively]. No patients discontinued in the switched to CT-P43 group. There were no TEAEs resulting in death.
With regard to TEAEs of special interest, no TEAEs were classified as hypersensitivity reactions or malignancies (Tables 4 and 5). TEAEs of infection occurred with similar frequencies across groups in both periods, with no serious infections during Treatment Period II. ISRs occurred at comparable frequencies across groups (Tables 4 and 5).
Overall, the majority of TEAEs were grade 1–2 in intensity; grade ≥ 3 TEAEs are detailed in Supplementary Table 15. The most frequent TEAEs are presented in Supplementary Table 16. There were no notable between-group differences in vital sign measurements or local injection-site pain. No active tuberculosis was reported.
3.5 Immunogenicity
In Treatment Period I, the proportion of patients with positive ADA results was generally lower in the CT-P43 group compared with the originator ustekinumab group (Supplementary Table 17). In Treatment Period II, there was no overall change in ADA frequency following the single transition from originator ustekinumab to CT-P43 at week 16, compared with continuing originator ustekinumab. Pre-dose patients at week 28, 26 (10.3%), 21 (16.8%) and 22 (17.7%) in continued CT-P43, continued originator ustekinumab and switched to CT-P43 groups, respectively, were ADA positive. Overall, few patients (< 5%) without positive ADA results during Treatment Period I had positive ADA/neutralising antibody results in Treatment Period II.
Primary efficacy endpoint data were similar regardless of ADA positivity at week 12 and were also comparable between treatment groups within ADA-positive or ADA-negative subgroups (Supplementary Table 18). In Treatment Period I, serum ustekinumab concentrations were slightly lower in ADA-positive versus ADA-negative subgroups in both treatment groups (regardless of dose received); within subgroups, serum ustekinumab concentrations were generally similar between treatment groups (Supplementary Table 19). In addition, there was no apparent correlation between ADA positivity and the occurrence of TEAEs, TESAEs, ISRs or hypersensitivity reactions during Treatment Period I (Supplementary Table 20).
4 Discussion
This study demonstrated equivalence of the candidate ustekinumab biosimilar CT-P43 to originator ustekinumab with respect to efficacy, as determined by mean per cent improvement from baseline in PASI score at week 12. Equivalence was demonstrated using both FDA and EMA assumptions (in the mITT set and full analysis set of patients with a baseline body weight of ≤ 100 kg and therefore only receiving 45 mg doses of study drug in Treatment Period I, respectively). Secondary efficacy endpoints also demonstrated comparability between treatment groups, and findings remained comparable after patients receiving originator ustekinumab switched to CT-P43. PK and safety profiles were also comparable between CT-P43 and originator ustekinumab.
The mean per cent improvement from baseline in PASI score at week 12 (primary endpoint) in both treatment groups was comparable with findings from the PHOENIX 1 and 2 studies [15, 16] and other phase II/III studies of originator ustekinumab for the treatment of psoriasis [17,18,19]. Similarly, the proportions of patients achieving PASI 50/75/90/100 at weeks 12 and 28 were broadly comparable with previous originator ustekinumab studies [15,16,17,18,19].
The safety profiles of CT-P43 and originator ustekinumab were similar in this study, including after switching from originator ustekinumab to CT-P43. Safety findings were consistent with the reported safety profile of originator ustekinumab [2]. Most TEAEs were grade 1–2 in intensity, and there were no hypersensitivity reactions, malignancies or deaths reported through 28 weeks. Unlike previous studies of ustekinumab, COVID-19 infection was one of the most frequently reported TEAEs, reflecting the period during which the study was conducted; however, the overall infection rate in this study was broadly comparable to previous reports [15,16,17,18,19]. Except for COVID-19 pneumonia, reported in two patients in the CT-P43 group and one in the originator ustekinumab group, the rate of TESAEs was also similar to previous reports for originator ustekinumab [15,16,17,18,19].
The proportion of patients with positive ADA results was lower in the CT-P43 versus originator ustekinumab group in Treatment Period I, and similar trends were observed across groups in Treatment Period II. There was no change in ADA frequency after switching from originator ustekinumab to CT-P43. The impact of immunogenicity on clinical outcomes was similar between CT-P43 and originator ustekinumab during Treatment Period I; while positive ADA status was associated with reduced serum drug concentrations (in line with originator ustekinumab) [2, 3], there was no clinically significant impact of ADA status on efficacy or safety in terms of the occurrence of TEAEs, TESAEs, ISRs or hypersensitivity reactions. Although the incidence of TEAEs was slightly higher in the CT-P43 versus originator ustekinumab treatment group (regardless of ADA positivity), this was not clinically significant.
The main strength of this study was the controlled study design and adequate statistical power to demonstrate equivalence of efficacy between CT-P43 and originator ustekinumab in patients with moderate to severe plaque psoriasis. One limitation was the absence of multiple switches in the study design, with only a single switch from originator ustekinumab to CT-P43 evaluated. Staff administering study drug could not be blinded to treatment assignment (due to marginal visual differences between the originator and biosimilar study drugs) but did not conduct other study assessments; patients and other investigators remained blinded. In addition, the study was conducted during the COVID-19 pandemic; while this did not result in any patients missing primary endpoint assessments, one and two patients in CT-P43 and originator ustekinumab groups, respectively, were excluded from the per-protocol set owing to protocol deviations arising from COVID-19. Data were collected on serum ustekinumab concentrations, although PK data had also been collected previously [20]. This manuscript reports data up to week 28; final study results (up to week 52) will be published once available.
5 Conclusions
This study demonstrated the equivalence in terms of efficacy of CT-P43 and originator ustekinumab in patients with moderate to severe plaque psoriasis. The PK, safety and immunogenicity profiles of CT-P43 and originator ustekinumab were comparable. No notable safety issues or increase in immunogenicity were observed following a single switch from originator ustekinumab to CT-P43 group, compared with continued therapy with CT-P43 or originator ustekinumab.
References
Parisi R, Iskandar IYK, Kontopantelis E, Augustin M, Griffiths CEM, Ashcroft DM, et al. National, regional, and worldwide epidemiology of psoriasis: Systematic analysis and modelling study. BMJ. 2020;369:m1590. https://doi.org/10.1136/bmj.m1590.
European Medicines Agency. Stelara: Summary of product characteristics. 2022. https://www.ema.europa.eu/en/documents/product-information/stelara-epar-product-information_en.pdf. Accessed 30 May 2023.
US Food & Drug Administration. Stelara: prescribing information. 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761044s010lbl.pdf. Accessed 30 May 2023.
Benson JM, Peritt D, Scallon BJ, Heavner GA, Shealy DJ, Giles-Komar JM, et al. Discovery and mechanism of ustekinumab: a human monoclonal antibody targeting interleukin-12 and interleukin-23 for treatment of immune-mediated disorders. MAbs. 2011;3(6):535–45. https://doi.org/10.4161/mabs.3.6.17815.
Brodmerkel C, Li K, Garcet S, Hayden K, Chiricozzi A, Novitskaya I, et al. Modulation of inflammatory gene transcripts in psoriasis vulgaris: differences between ustekinumab and etanercept. J Allergy Clin Immunol. 2019;143(5):1965–9. https://doi.org/10.1016/j.jaci.2019.01.017.
Ghosh S, Gensler LS, Yang Z, Gasink C, Chakravarty SD, Farahi K, et al. Ustekinumab safety in psoriasis, psoriatic arthritis, and Crohn’s disease: an integrated analysis of phase II/III clinical development programs. Drug Saf. 2019;42(6):751–68. https://doi.org/10.1007/s40264-019-00797-3.
McInnes IB, Kavanaugh A, Gottlieb AB, Puig L, Rahman P, Ritchlin C, et al. Efficacy and safety of ustekinumab in patients with active psoriatic arthritis: 1 year results of the phase 3, multicentre, double-blind, placebo-controlled PSUMMIT 1 trial. Lancet. 2013;382(9894):780–9. https://doi.org/10.1016/s0140-6736(13)60594-2.
Papp KA, Griffiths CE, Gordon K, Lebwohl M, Szapary PO, Wasfi Y, et al. Long-term safety of ustekinumab in patients with moderate-to-severe psoriasis: final results from 5 years of follow-up. Br J Dermatol. 2013;168(4):844–54. https://doi.org/10.1111/bjd.12214.
Ritchlin C, Rahman P, Kavanaugh A, McInnes IB, Puig L, Li S, et al. Efficacy and safety of the anti-IL-12/23 p40 monoclonal antibody, ustekinumab, in patients with active psoriatic arthritis despite conventional non-biological and biological anti-tumour necrosis factor therapy: 6-month and 1-year results of the phase 3, multicentre, double-blind, placebo-controlled, randomised PSUMMIT 2 trial. Ann Rheum Dis. 2014;73(6):990–9. https://doi.org/10.1136/annrheumdis-2013-204655.
Sands BE, Sandborn WJ, Panaccione R, O’Brien CD, Zhang H, Johanns J, et al. Ustekinumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2019;381(13):1201–14. https://doi.org/10.1056/NEJMoa1900750.
US Food & Drug Administration. Scientific considerations in demonstrating biosimilarity to a reference product: guidance for industry. 2015. https://www.fda.gov/media/82647/download. Accessed 30 May 2023.
European Medicines Agency. Biosimilars in the EU: Information guide for healthcare professionals. 2019. https://www.ema.europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_en.pdf. Accessed 30 May 2023.
US Food & Drug Administration. Biosimilar product information. 2023. https://www.fda.gov/drugs/biosimilars/biosimilar-product-information. Accessed 30 May 2023.
European Medicines Agency. Medicines. 2023. https://www.ema.europa.eu/en/medicines/search_api_aggregation_ema_medicine_types/field_ema_med_biosimilar?search_api_views_fulltext=omalizumab. Accessed 30 May 2023.
Leonardi CL, Kimball AB, Papp KA, Yeilding N, Guzzo C, Wang Y, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet. 2008;371(9625):1665–74. https://doi.org/10.1016/S0140-6736(08)60725-4.
Papp KA, Langley RG, Lebwohl M, Krueger GG, Szapary P, Yeilding N, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet. 2008;371(9625):1675–84. https://doi.org/10.1016/S0140-6736(08)60726-6.
Igarashi A, Kato T, Kato M, Song M, Nakagawa H, Group TJUS. Efficacy and safety of ustekinumab in Japanese patients with moderate-to-severe plaque-type psoriasis: long-term results from a phase 2/3 clinical trial. J Dermatol. 2012;39:242–52. https://doi.org/10.1111/j.1346-8138.2011.01347.x.
Tsai TF, Ho JC, Song M, Szapary P, Guzzo C, Shen YK, et al. Efficacy and safety of ustekinumab for the treatment of moderate-to-severe psoriasis: a phase III, randomized, placebo-controlled trial in Taiwanese and Korean patients (PEARL). J Dermatol Sci. 2011;63(3):154–63. https://doi.org/10.1016/j.jdermsci.2011.05.005.
Zhu H, Zheng M, Song M, Shen Y-K, Chan D, Szapary PO, et al. Efficacy and safety of ustekinumab in Chinese patients with moderate to severe plaque-type psoriasis: results from a phase 3 clinical trial (LOTUS). J Drugs Dermatol. 2013;12:166–74.
ClinicalTrials.gov. Study to compare PK and safety of subcutaneous injection of ustekinumab and CT-P43 in healthy subjects (NCT04428814). 2021. https://clinicaltrials.gov/ct2/show/NCT04428814. Accessed 30 May 2023.
Acknowledgements
We thank all patients and investigators involved in the study. Medical writing support, including development of a draft outline and subsequent drafts in consultation with the authors, collating author comments, copyediting, fact checking and referencing, was provided by Beatrice Tyrrell, DPhil, CMPP, at Aspire Scientific Limited (Bollington, UK). Funding for medical writing support for this article was provided by Celltrion, Inc. (Incheon, Republic of Korea).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Celltrion, Inc. (Incheon, Republic of Korea).
Conflicts of Interest
K.A. Papp has received grants for clinical studies from AbbVie, Akros, Amgen, Anacor, Arcutis, Avillion, Bausch Health/Valeant, Boehringer Ingelheim, Bristol Myers Squibb, Can-Fite Biopharma, Celgene, Coherus, Dermavant, Dermira, Dow Pharma, Eli Lilly, Evelo, Galderma, Gilead, GSK, Incyte, Janssen, Kyowa Hakko Kirin, LEO Pharma, Merck (MSD), Novartis, Pfizer, Regeneron, Roche, Sanofi-Aventis/Genzyme, Sun Pharma, Takeda, and UCB; consulting fees from AbbVie, Acelyrin, Akros, Amgen, Aralez Pharmaceuticals, Arcutis, Avillion, Bausch Health/Valeant, Boehringer Ingelheim, Bristol Myers Squibb, Can-Fite Biopharma, Celgene, Celltrion, Coherus, Dermavant, Dermira, Dice Pharmaceuticals, Dow Pharma, Eli Lilly, Evelo, Forbion, Galderma, Incyte, Janssen, Kyowa Hakko Kirin, LEO Pharma, Meiji Seika Pharma, Merck (MSD), Mitsubishi Pharma, Novartis, Pfizer, Regeneron, Reistone, Roche, Sanofi-Aventis/Genzyme, Sandoz, Sun Pharma, Takeda, UCB, vTv Therapeutics, and Xencor; has participated in speakers bureaus for AbbVie, Amgen, Bausch Health/Valeant, Celgene, Eli Lilly, Galderma, Incyte, Janssen, Kyowa Hakko Kirin, LEO Pharma, Merck (MSD), Novartis, Pfizer, and Sanofi-Aventis/Genzyme; has received honoraria for attending meetings and/or travel from AbbVie, Acelyrin, Akros, Amgen, Aralez Pharmaceuticals, Bausch Health/Valeant, Boehringer Ingelheim, Celgene, Celltrion, Coherus, Dermavant, Dice Pharmaceuticals, Eli Lilly, Forbion, Galderma, Janssen, Kyowa Hakko Kirin, LEO Pharma, Meiji Seika Pharma, Mitsubishi Pharma, Merck (MSD), Novartis, Pfizer, Reistone, Sanofi-Aventis/Genzyme, Sandoz, Sun Pharma, Takeda, UCB, vTv Therapeutics, and Xencor; has served on advisory boards for AbbVie, Amgen, Bausch Health/Valeant, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Dice Pharmaceuticals, Dow Pharma, Eli Lilly, Galderma, Janssen, Merck (MSD), Novartis, Pfizer, Regeneron, Sanofi-Aventis/Genzyme, Sun Pharma, and UCB; has served as a scientific officer for Akros, Anacor, Arcutis, Dice Pharmaceuticals, Kyowa Hakko Kirin; and has served on steering committees for AbbVie, Amgen, Bausch Health/Valeant, Boehringer Ingelheim, Celgene, Eli Lilly, Janssen, Kyowa Hakko Kirin, Merck (MSD), Novartis, Pfizer, Regeneron, Reistone, and Sanofi-Aventis/Genzyme. M.G. Lebwohl has received grants/contracts from AbbVie, Amgen, Arcutis, Avotres, Boehringer Ingelheim, Cara Therapeutics, Dermavant Sciences, Eli Lilly, Incyte, Janssen Research & Development, LLC, Ortho Dermatologics, Regeneron, and UCB, Inc.; and has received consulting fees from Aditum Bio, Almirall, AltruBio Inc., AnaptysBio, Apogee Therapeutics, Arcutis, Inc., Aristea Therapeutics, Atomwise, Avotres Therapeutics, Brickell Biotech, Boehringer Ingelheim, Bristol Myers Squibb, Cara Therapeutics, Castle Biosciences, Celltrion, Corevitas, Dermavant Sciences, Dr. Reddy, EPI, Evommune, Inc., Facilitation of International Dermatology Education, Forte Biosciences, Foundation for Research and Education in Dermatology, Galderma, Genentech, Helsinn, Incyte, LEO Pharma, Meiji Seika Pharma, Mindera, National Society for Cutaneous Medicine, Pfizer, Seanergy, Strata, Trevi, and Verrica, and Vial Health Technologies. D. Thaçi has received grants/contracts from AbbVie, LEO Pharma and Novartis; received consulting fees from AbbVie, Almirall, Boehringer Ingelheim, Bristol Myers Squibb, Eli Lilly, Galderma, Janssen-Cilag, LEO Pharma, MorphoSys, Novartis, Pfizer, Regeneron Pharmaceuticals Inc., Samsung, Sanofi and UCB; received honoraria from AbbVie, Almirall, Amgen, Beiersdorf, Biogen, Boehringer Ingelheim, Bristol Myers Squibb, Eli Lilly, Galapagos, Galderma, Janssen-Cilag, LEO Pharma, MorphoSys, New-Bridge, Novartis, Pfizer, Regeneron Pharmaceuticals Inc., Samsung, Sandoz, Sanofi, Sun Pharma and UCB; and participated on data safety monitoring boards or advisory boards for AbbVie, Almirall, Boehringer Ingelheim, Bristol Myers Squibb, Eli Lilly, Galapagos, Galderma, Janssen-Cilag, LEO Pharma, Novartis, Pfizer, Regeneron Pharmaceuticals Inc., Sanofi and UCB. A. Dudek has received grants for clinical trials from AbbVie, Bristol Myers Squibb, Celltrion, Eli Lilly, GSK, Horizon, Janssen, UCB and VieLaBio. J.C. Szepietowski has received consulting fees from AbbVie, LEO Pharma, Novartis, Pfizer, Sanofi-Genzyme, Trevi, UCB and Vifor; and honoraria from AbbVie, Almirall, Janssen-Cilag, Eli Lilly, LEO Pharma, Novartis, Pfizer and Sanofi-Genzyme. N. Reznichenko has received grants for clinical trials from Alvotech, Argenx, Celltrion, Eli Lilly and Samsung. A. Reich has received grants for clinical trials from AnaptysBio, Argenx, Celltrion, Drug Delivery Solutions, Galderma, Genentech, Inflarx, Janssen, Kymab Ltd, LEO Pharma, Menlo Therapeutics, MetrioPharm, MSD, Novartis, Pfizer, Trevi, Eli Lilly, UCB and VielaBio; and speaker fees from AbbVie, Bausch Health, Bioderma, Celgene, Chema Elektromet, Eli Lilly, Galderma, Janssen, LEO Pharma, Medac, Novartis, Pierre Fabre, Pfizer, Sandoz and Trevi. Y. Andrashko has received grants for clinical trials from Alfa Sigma, Alvotech, Amgen, Celltrion, Dong-A Pharmaceutical, Eli Lilly, Galderma, Mayne Pharma and Samsung Bioepis; and speaker fees from Blausch Health, Delta Medical, GSK, Novartis and Pfizer. S. Kim, Y. Bae, D. Jeon, J. Jung, H. Lee and T. Pyo are employees of Celltrion and own stocks in Celltrion. W. Ko is an employee of Celltrion. J. Jaworski, J. Trefler, J. Narbutt, W. Baran, J. Kolinek, S. Daniluk, K. Bartnicka-Maslowska and B. Kwiek report no conflicts of interest.
Data Availability
The data that support the findings of this study are available in the supplementary material of this article.
Code Availability
Not applicable (no software or custom code).
Ethics Approval
The study was conducted according to Good Clinical Practice and the principles of the Declaration of Helsinki, as well as national, state and local laws or regulations. Study materials were approved by independent ethics committee/institutional review boards at each site.
Consent to Participate
All patients in this manuscript have given written informed consent for participation in the study and the use of their de-identified, anonymized, aggregated data and their case details (including photographs) for publication.
Consent for Publication
Not applicable (no identifiable data).
Author Contributions
KAP, MGL, DT, SK, YB, DJ, JJ, HL, TP, and WK contributed to the study design. JJ, BK, JT, AD, JCS, NR, JN, WB, JK, SD, KB-M, AR and YA collected study data. All authors contributed to data analysis or interpretation, reviewed and critically revised the manuscript, provided final approval of the version to be published, and agree to be accountable for accuracy and integrity.
Congress Presentations
Selected data from the CT-P43 3.1 study were shared in an oral presentation at the 31st European Academy of Dermatology and Venereology (EADV) congress (7–10 September 2022; Milan, Italy).
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Papp, K.A., Lebwohl, M.G., Thaçi, D. et al. Efficacy and Safety of Candidate Biosimilar CT-P43 Versus Originator Ustekinumab in Moderate to Severe Plaque Psoriasis: 28-Week Results of a Randomised, Active-Controlled, Double-Blind, Phase III Study. BioDrugs 38, 121–131 (2024). https://doi.org/10.1007/s40259-023-00630-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-023-00630-5