
Abstract
Background
Infusion-related reactions (IRRs) are the most common adverse event (AE) associated with infusion of rituximab, an anti-CD20 monoclonal antibody.
Objective
Our objective was to evaluate the impact of dosing/infusion patterns and certain baseline characteristics on IRR occurrence during the first rituximab infusion administered as the biosimilar PF-05280586 (RTX-PF) or reference rituximab sourced from the EU (RTX-EU, MabThera®) in patients with CD20+ low-tumor-burden follicular lymphoma.
Patients and methods
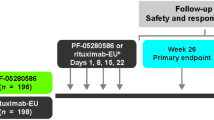
Rituximab (RTX-PF, n=196; RTX-EU, n=198) was administered (375 mg/m2) on days 1, 8, 15, and 22 (one cycle), with a follow-up period through 52 weeks. The relationships between infusion rate, drug exposure, and IRR incidence were assessed by logistic regression analysis and pharmacokinetic modeling and simulation. Baseline CD20 level, antidrug antibody (ADA) status, and tumor burden according to IRR occurrence (yes/no) were compared descriptively.
Results
Median rituximab infusion duration on day 1 was 3.50 h for each of the two groups. There was a positive correlation between infusion rate and all-grade IRRs occurring within 24 h after infusion (p < 0.0001). Patients who developed IRRs had a higher median baseline CD20+ level. IRR incidence was unaffected by baseline ADA status. Drug exposure did not predict IRR incidence. Baseline tumor burden was similar between patients with and without IRRs.
Conclusions
Results of this analysis provide a better understanding of IRRs after the first rituximab (RTX-PF or RTX-EU) infusion and demonstrate a potential correlation of infusion rate and other factors with IRR at the individual and population levels. Infusion-rate escalation steps continue to be needed to manage IRRs.
Trial registration (date of registration)
ClinicalTrials.gov Identifier: NCT02213263 (11 August 2014); and EudraCT: 2014-000132-41 (10 October 2014).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Infusion-related reactions (IRRs) are the most common adverse event associated with rituximab infusion. |
This retrospective analysis of data from a randomized, double-blind comparative trial in patients with CD20+ low-tumor-burden follicular lymphoma demonstrated a positive correlation between infusion rate and all-grade IRRs occurring within 24 h after infusion of the rituximab biosimilar PF-05280586 or reference rituximab sourced from the EU (p < 0.0001). |
Patients who developed IRRs had a higher median baseline CD20+ cell level. IRR incidence was unaffected by other baseline characteristics and by drug exposure. |
1 Introduction
Rituximab is a murine/human chimeric monoclonal immunoglobulin (Ig) G1 kappa antibody that is directed against the CD20 antigen expressed on the surface of pre-B and mature B-lymphocytes and on most malignant B cells [1, 2]. Rituximab has revolutionized the treatment of, and has become a standard component of care for patients with, B-cell malignancies [3]. Clinical studies in patients with relapsed or refractory low-grade non-Hodgkin’s lymphoma (NHL) have reported improved overall tumor response rates after rituximab monotherapy administered once weekly for 4 weeks [4,5,6,7,8]. Randomized controlled trials in patients with previously untreated follicular lymphoma demonstrated improved response rates and prolonged survival outcomes when rituximab was added to induction chemotherapy as compared with chemotherapy alone [9,10,11,12].
Biosimilars are biological drugs that are highly similar to licensed biologic reference products (RPs) such that there are no clinically meaningful differences between the two products in safety, purity, or potency [13, 14]. Biosimilar drug development follows a stepwise approach of rigorous comparative analytical and nonclinical testing, followed by comparative clinical studies (i.e., clinical pharmacokinetics/pharmacodynamics, efficacy, safety, and immunogenicity) [13, 14]. Biosimilar regulatory approval is granted when the totality of the evidence obtained from each stage of development establishes that it is as safe and effective as the biologic RP [13,14,15].
PF-05280586 (RTX-PF) (Ruxience™; rituximab-pvvr; Pfizer) is a biosimilar to Rituxan®/MabThera® that is approved for all eligible cancer indications in the USA, EU, and Japan and for additional indications that vary by country [16,17,18,19]. RTX-PF was approved based on a development program comprising analytical and nonclinical assessments that demonstrated highly similar structural and functional profiles of RTX-PF and rituximab RPs sourced from the USA (RTX-US; Rituxan®) and from the EU (RTX-EU; MabThera®) [20]; a pharmacokinetics trial that demonstrated pharmacokinetic bioequivalence among RTX-PF, RTX-US, and RTX-EU in patients with rheumatoid arthritis (RA) [21]; and a comparative clinical study that demonstrated therapeutic equivalence between RTX-PF and RTX-EU, and similar safety, immunogenicity, pharmacokinetics, and pharmacodynamics between treatments at week 26 in patients with previously untreated CD20-positive (CD20+) low-tumor-burden follicular lymphoma (LTB-FL) [22].
Rituximab has a well-defined safety profile when administered as monotherapy or in combination with chemotherapy or targeted therapy (e.g., lenalidomide, ibrutinib, idelalisib) for the treatment of patients with NHL, such as follicular lymphoma and chronic lymphocytic leukemia (CLL) [3, 23,24,25]. Mild-to-moderate infusion-related reactions (IRRs) are the most common adverse event (AE) associated with rituximab administration [1, 2, 26]. Most IRRs (77%) occur during or after the first rituximab infusion [1, 2, 26]. Premedication with acetaminophen, along with antihistamines and corticosteroid-containing prophylaxis, can reduce the incidence and severity of IRRs [1, 2, 26].
The predictive factors of rituximab IRR AEs are not well understood. Previously, Paul and Cartron [26] reported lymphocyte count, bulky disease, low-grade B-cell NHL, the level of CD20 expression on CLL cells, and Fc/Fc gamma receptor (FcγR)-mediated interactions between natural killer (NK) cells and CD20+ cells as being associated with the occurrence of IRRs. In the observational setting in patients with hematologic malignancies, predictive indicators of rituximab IRRs have included bone marrow involvement, body weight, low-grade lymphomas, and bulky disease [27,28,29].
Additional data that reinforce the similarity between biosimilars and their RPs, including in terms of their safety profiles, may help to remove perceived barriers to the use/adoption of biosimilars by clinicians. Here, we investigate the impact of dosing/infusion patterns and patients’ baseline characteristics on the occurrence of IRR AEs during the first infusion of RTX-PF and RTX-EU in a comparative randomized clinical trial of monotherapy in patients with CD20+ LTB-FL (NCT02213263) [22].
2 Methods
2.1 Study Design
This analysis used a retrospective approach that incorporated data from a randomized, double-blind comparative trial of RTX-PF and RTX-EU monotherapy in patients with CD20+ LTB-FL [22]. The study conduct and results have been described in detail previously [22] and are summarized briefly here.
Patients were randomized (1:1) to RTX-PF or RTX-EU, which were administered at a dose of 375 mg/m2 intravenously once weekly for 4 weeks (1 cycle) on days 1, 8, 15, and 22, with a follow-up period through 52 weeks [22]. All patients received premedication with intravenous methylprednisolone 100 mg or its equivalent before rituximab (RTX-PF or RTX-EU) infusion. Premedication consisting of an antipyretic and an antihistamine (e.g., paracetamol [acetaminophen] and diphenhydramine) was administered before rituximab infusion.
On day 1, rituximab infusion was initiated at a rate of 50 mg/h. After 30 min, and in the absence of infusion toxicity, the infusion rate was allowed to increase by 50 mg/h increments at 30-min intervals, to a maximum of 400 mg/h. On days 8, 15, and 22, infusion was initiated at a rate of 100 mg/h. After 30 min, and in the absence of infusion toxicity, the infusion rate was allowed to increase by 100 mg/h increments at 30-min intervals, to a maximum of 400 mg/h.
Safety was characterized by the type, incidence, severity, timing, seriousness, and relatedness of AEs to study treatment, including infections, IRRs, and other clinical outcomes associated with immunogenicity, and laboratory abnormalities. The severity of AEs was graded based on the National Cancer Institute Common Terminology Criteria for AEs (NCI CTCAE, v4.03) and the seriousness of AEs was determined based on study protocol criteria. Immunogenicity-associated AEs were referred to as immune-based AEs and included events relating to Standardized Medical Dictionary for Regulatory Activities queries of anaphylaxis and hypersensitivity reactions and events meeting Sampson’s criteria. IRRs were defined following the summary of product characteristics for RTX-EU and included cytokine release syndrome, tumor lysis syndrome, and anaphylactic and hypersensitivity reactions [1]. Individual signs and symptoms of IRRs were documented in addition to the preferred term of “infusion-related reaction” for the purpose of comparing the overall percentage of patients by treatment arm. The reporting of IRR AEs and timing of these events in relation to the infusion were determined by the investigator.
Patients were closely monitored for the onset of cytokine release syndrome. Infusions were immediately interrupted in patients who developed evidence of severe reactions, especially severe dyspnea, bronchospasm, or hypoxia. Mild or moderate IRRs usually responded to a reduction in the rate of infusion. The infusion rate could be increased upon improvement of symptoms. The patient was observed post-infusion, if necessary, based on the investigator’s medical judgment.
The objectives of this retrospective analysis were to evaluate infusion-rate patterns; determine total infusion duration, and the incidence of IRRs during the first infusion (within 24 h after the start) of rituximab, administered as RTX-PF or RTX-EU; and assess whether IRR incidence was associated with infusion rates of rituximab, pharmacokinetic exposure, and patients’ baseline characteristics, including CD20+ B-cell level, tumor burden, and immunogenicity status.
2.2 Data Analysis Method and Statistical Analysis
2.2.1 Rituximab Infusion Duration and Infusion-Rate Distribution
The percentages of patients who had total infusion durations of ≤ 2 or > 3.5 h were calculated. If a patient experienced multiple short infusions during one dose/the same day, the total duration is the sum of all records related to that dose. Infusion-rate distributions describing the protocol-recommended infusion rate (range 50–400 mg/h) and the theoretical highest infusion rate (using a cut-off of ≤ 760 mg/h, based on the highest dose in the study population) were summarized.
2.2.2 Population Pharmacokinetics Model and Simulation
A population pharmacokinetics model that describes the pharmacokinetic characteristics of RTX-PF and RTX-EU was previously established with drug concentration versus time data from the comparative clinical study of RTX-PF and RTX-EU [22] using a nonlinear mixed-effect modeling approach (internal report). A total of 3217 pharmacokinetic concentrations following four weekly doses of RTX-EU or RTX-PF, administered to 386 patients, were well described by a two-compartment pharmacokinetic structural model following intravenous infusion in the central compartment and first-order linear elimination from the central compartment. The following fixed-effect pharmacokinetic parameters were estimated: intrinsic clearance (CL; L/day); central volume of distribution (V1; L); peripheral volume of distribution (V2; L); and intercompartmental clearance (Q; L/day) between the central and peripheral compartments following intravenous infusion into the central compartment.
For a typical 60-year-old patient weighing 73 kg, the CL (L/day), V1 (L), V2 (L), and Q (L/day) point estimates (95% bootstrapped confidence intervals) were 0.277 (0.251–0.298) L/day, 2.80 (2.19–2.94) L, 5.75 (5.46–6.78) L, and 6.94 (5.23–17.4) L, respectively. Significant covariate effects were observed in age on CL and in body weight on CL, V1, and V2. The formulation, antidrug antibody (ADA) status, and race (Japanese and Asian) variables were not significant predictors of CL. The typical individual (73 kg, age 60 years) exhibited an elimination half-life (t½) of 21.4 (20.0–23.7) days; CL of 0.277 (0.251–0.298) L/day; V1 of 2.80 (2.19–2.94) L; and V2 of 5.75 (5.46–6.78) L.
To further understand the pharmacokinetic profile and maximum drug concentration level under different infusion doses and/or different infusion rates, two simulations were conducted. Simulation 1 was based on a normal infusion speed scenario and tested a single dose (679 mg) of rituximab under three different infusion-rate escalation steps. Simulation 2 was based on a potential rapid-infusion protocol with an overall infusion duration of 90 min and tested two doses (500 and 679 mg) of rituximab given the same infusion duration.
2.2.3 Logistic Regression Analysis of the Relationship Between IRRs, Infusion Rate, and Drug Exposure
Logistic regression analyses were performed to evaluate the potential correlation between the incidence of IRRs during the first dose and infusion rates of the respective dose, with infusion rate as the independent variable and the probability of IRR as the dependent variable. Significance levels and odds ratios were produced. In addition, similar methods were used to evaluate the potential correlation between the incidence of IRRs and the rituximab pharmacokinetic parameters Cmax (the peak concentration data collected at the end of infusion of the first dose, i.e., day 1 at 3 h 30 min after the start of the infusion) and Ctrough (the trough serum concentration collected before the second dose, i.e., day 8, predose). Data from both treatment arms (RTX-PF and RTX-EU) were combined in this model. Treatment (as a covariate of the combined model) was dropped from the final model if it was not found to be significant. The logistic regression equation for the final model was:
where b0 and b1 are the intercept and slope, respectively, in the model for the log of the odds ratio, and x is the independent variable (e.g., infusion rate, Cmax, Ctrough).
2.2.4 Other Analyses of Baseline Characteristics and IRR Occurrence
Patient baseline characteristics, including CD20+ B-lymphocyte count (cells/µL), ADA status (positive or negative), tumor burden (Ann Arbor stage II, III, or IV), and bone marrow biopsy lymphoma results (positive, negative, or indeterminate), were summarized descriptively by IRR occurrence (yes or no) and treatment group (RTX-PF or RTX-EU).
Analyses were conducted using the modified intent-to-treat population, previously defined as all patients who were randomized and received one or more dose of any study drug [22].
3 Results
A total of 394 patients were randomized to RTX-PF (n = 196) or RTX-EU (n = 198); 196 and 197 patients, respectively, were included in the current analysis. The median infusion duration on day 1 was 3.50 h for each of the RTX-PF or RTX-EU groups. A total of 42 (21.4%) patients in the RTX-PF group and 53 (26.9%) patients in the RTX-EU group had dose interruption after the first dose of rituximab. Overall, a small percentage (4.5%) of patients had relatively rapid overall infusion, reflected by a total infusion duration of ≤ 2 h. Around one-third (33.1%) of patients had a total infusion duration of > 3.5 h, and the majority (62.4%) of patients completed their infusion in > 2 but ≤ 3.5 h.
The distribution of maximum infusion rate by treatment arm is shown in Fig. 1. Few patients had a maximum infusion rate of 400 mg/h; rather, the majority of patients had a maximum infusion rate of 250–300 mg/h. The median maximum infusion rate was 277.8 mg/h for RTX-PF and 273.2 mg/h for RTX-EU.

Distribution of maximum infusion ratea by treatment for a RTX-PF and b RTX-EU. aInfusion rate is a derived variable, calculated by dividing dose amount by infusion time, where infusion time is the difference between infusion start and stop times. bRate data is the amount (mg) of drug dose infused in time (h); hence, units of mg/h. The maximum rate for each individual patient is plotted and represents the maximum/highest rates used within a patient per visit. The vertical dotted lines represent the median value and the 95% percentile-based confidence intervals. RTX rituximab, RTX-EU reference RTX sourced from the European Union, RTX-PF PF-05280586
The incidence of IRR overall for the duration of the study was 25.0% (49/196) and 29.9% (59/197) in the RTX-PF and RTX-EU groups, respectively. The majority of IRRs were grade 1 or 2 in severity. No treatment-emergent IRRs of grade 4 or higher were reported in either treatment group. One patient in the RTX-EU group experienced serious treatment-emergent AEs of IRR (grade 3) and dyspnea (grade 2) on day 15; no serious events of IRR were reported for patients in the RTX-PF group. One patient in the RTX-PF group permanently discontinued treatment and withdrew from the study because of treatment-emergent AEs of IRR and angioedema, both of which were grade 3 in severity (based on NCI CTCAE v4.03 criteria) and not considered serious AEs (based on the study protocol criteria).
In total, 24.0% (47/196) and 28.9% (57/197) of patients in the RTX-PF and RTX-EU groups, respectively, reported IRRs within 24 h after the start of the first infusion on day 1. The logistic regression analysis demonstrated a significant correlation (p < 0.0001) between infusion rate of rituximab (RTX-PF and RTX-EU) and the probability of IRR AEs on day 1 (Fig. 2). For all patients, the estimated odds ratio for probability of developing an IRR with an increase in infusion rate by 100 mg/h was 7.7. There was no apparent difference between RTX-PF and RTX-EU groups.

Probability of developing an infusion-related reaction by infusion rate in patients treated with RTX-PF or RTX-EU. RTX rituximab, RTX-EU reference RTX sourced from the European Union, RTX-PF PF-05280586
There was a nonsignificant positive trend between Cmax of rituximab (RTX-PF and RTX-EU) and developing an IRR (Fig. 3a); the estimated probability of developing an IRR was 0.26 at the median (196.5 μg/mL) Cmax of rituximab and increased to 0.37 at the highest (481 μg/mL) Cmax of rituximab. The Ctrough of rituximab (RTX-PF and RTX-EU), collected before the second dose, did not correlate with increased IRR incidence (Fig. 3b). Rather, the probability of developing an IRR was approximately 25%, regardless of Ctrough level.

Probability of developing an infusion-related reaction by a Cmax and b Ctrough collected before the second dose (day 8) of rituximab (RTX-PF and RTX-EU). Cmax maximum serum concentration, Ctrough trough serum concentration, RTX rituximab, RTX-EU reference RTX sourced from the European Union, RTX-PF PF-05280586
Patients who developed IRRs had a median baseline CD20+ B-lymphocyte level of 149.87 (RTX-PF) and 136.91 (RTX-EU) cells/µL, compared with patients who did not develop IRRs and who had a median baseline CD20+ B-lymphocyte level of 108.21 (RTX-PF) and 105.04 (RTX-EU) cells/µL (Fig. 4). Baseline CD20+ B-lymphocyte level was similar between treatment arms but slightly higher in patients who experienced an IRR. The percentages of patients with IRRs were unaffected by the level of tumor burden at baseline, based on Ann Arbor stage (Table 1).

Baseline CD20+ B-lymphocyte level by infusion-related reaction status and treatment. Diamonds represent the mean value. IRR infusion-related reaction, Q1 first quartile, Q3 third quartile, RTX rituximab, RTX-EU reference rituximab sourced from the EU, RTX-PF PF-05280586
The percentages of patients with IRRs were unaffected by ADA status at baseline (Table 1). In the RTX-PF group, 14.3% (2/14) and 24.9% (45/181) of patients who tested ADA positive and ADA negative at baseline, respectively, reported IRR AEs. In the RTX-EU group, 23.5% (4/17) and 29.2% (52/178) of patients who tested ADA positive and ADA negative at baseline, respectively, reported IRR AEs.
No clinically meaningful differences were observed in terms of potential immunogenicity-associated AEs between patients who were ADA positive and those who were ADA negative at any time during the study. In the RTX-PF group, 25.6% (11/43) and 23.7% (36/152) of patients who tested ADA positive and ADA negative at any time during the study, respectively, reported IRR AEs. In the RTX-EU group, 25.6% (10/39) and 29.1% (46/158) of patients who tested ADA positive and ADA negative at any time during the study, respectively, reported IRR AEs.
The median total dose of the study by individual patient was 679 mg (based on 375 mg/m2 dosing). A faster infusion rate (with the same total dose) led to a higher and earlier peak concentration, with no effect on area under the curve (AUC) or Ctrough (Fig. 1 in the electronic supplementary material [ESM]). Higher doses (with the same infusion rate and rate-escalation steps) produced a higher peak concentration and overall AUC. Cmax was reached at the end of the infusion at 1.5 h and was 171.1 μg/mL for a total dose of 500 mg and 232.4 μg/mL for a total dose of 679 mg (Fig. 2 in the ESM). Therefore, the infusion rate tended to be higher for a larger dose. The Cmax depends on both the total dose and the infusion rate.
4 Discussion
Biosimilars are expected to have no clinically meaningful differences from the reference biologic product in terms of drug safety, purity, and potency [13, 14]. A systematic literature review and meta-analysis of rituximab biosimilar clinical trials (11 studies in patients with RA or NHL) demonstrated that there were no clinically meaningful differences between the rituximab biosimilars and reference rituximab in terms of the safety profile, including IRR AEs [30].
Theoretically, rituximab IRRs could result from different mechanisms, such as IgE-mediated hypersensitivity or IgE-independent anaphylactoid reaction, immunogenicity of rituximab, and complement activation [26, 31, 32]. However, it is suggested that the occurrence of IRRs with rituximab is mainly linked to cytokine release by immune (B and NK) cells, mediated through binding of the Fc portion of rituximab to FcγRIIIA/CD16 [26, 33]. This is supported by the following evidence. First, rituximab-induced cytokine release has been shown to depend on the binding of rituximab to FcγR on immune cells rather than to CD20+ cells [34]. Second, IgE-mediated hypersensitivity reactions to rituximab are rare and require prior exposure; therefore, such reactions are usually not observed at first infusion [26, 32]. Third, the immunogenicity of rituximab may not contribute to the occurrence of rituximab IRRs in some patient populations. For example, while the presence of anti-rituximab antibodies predicted the occurrence of IRRs in patients with systemic lupus erythematosus [35], there was no apparent association between the presence of human anti-chimeric antibodies and the occurrence of IRRs in patients with multiple sclerosis [36]. Finally, while complement activation occurs rapidly after rituximab infusion and the level of complement activation is correlated with IRR severity, it does not appear to be a main contributor to IRRs. This is based on the observation that higher occurrence and severity of IRRs with obinutuzumab, a third-generation humanized anti-CD20 monoclonal antibody, is associated with reduced complement activation as compared with rituximab [26].
Here, we explored the effect of patient baseline characteristics (CD20+ B-lymphocyte level; tumor burden, as measured by Ann Arbor stage and bone marrow biopsy lymphoma results; and immunogenicity status) and infusion rate on the occurrence of IRRs and whether IRR AEs for RTX-PF and RTX-EU were subject to the same influences based on data from a randomized controlled trial of RTX-PF and RTX-EU monotherapy in patients with CD20+ LTB-FL [22]. Descriptive analyses of patient baseline characteristics suggested that the occurrence of IRRs may be influenced by CD20+ B-lymphocyte level. This finding is consistent with results from a previous study that indicated that patients with higher expression of CD20 on the surface of CLL cells at baseline were at a greater risk of developing severe-grade, early IRRs from obinutuzumab or rituximab, especially patients treated with obinutuzumab (p = 0.02) [37].
Results from our analyses of patient baseline characteristics also demonstrated that IRR incidence was unaffected by tumor burden level, based on Ann Arbor stage. Although other evidence has suggested a possible relationship between the severity of certain IRRs and disease burden at the time of exposure in patients with leukemia [38], our study population comprised patients with LTB-FL; therefore, the range of tumor burden in our dataset may not be large enough to detect a relationship with IRRs. Other characteristics, such as metabolic tumor volume, may provide an appropriate measure of tumor burden level. While such data were not available for this study, it could be considered for correlative analyses for future studies.
Using a rich dataset collected for the evaluation of rituximab infusion duration enabled us to conduct a detailed analysis of rituximab infusion rate and calculate the distribution of infusion rate data. As described in the results section, the majority of IRRs occurred during the first infusion. Therefore, this analysis focused only on the IRRs recorded on day 1 or day 2 (i.e., within 24 h after the start of the infusion on day 1). The results of this analysis provide a better understanding of IRRs after the first rituximab (RTX-PF or RTX-EU) infusion and demonstrate a potential correlation of infusion rate and other factors with IRRs at the individual and population levels. Furthermore, the incidence of IRRs was similar between the RTX-PF and RTX-EU groups.
With regard to the investigation of the relationship between pharmacokinetic exposure and IRR incidence, Cmax and Ctrough are considered indirect measures of the total dose and the infusion rate, respectively. For example, a patient with a low body surface area (BSA) would be prescribed a lower total dose (based on 375 mg/m2) as compared with a patient with a high BSA. In addition, a patient with a low BSA would not require a fast infusion rate and would not exhibit a high Cmax. Therefore, the likelihood of developing an IRR might be lower for patients receiving smaller doses of rituximab administered using a normal infusion rate than for patients who require a larger dose of rituximab administered using a rapid infusion rate.
In this study, 4.5% of patients had a fairly fast infusion duration of ≤ 2 h. This was observed in both the RTX-EU and the RTX-PF groups. The majority of patients completed their infusion in > 2 but ≤ 3.5 h. The potential relationship between the occurrence of IRRs and rituximab efficacy was beyond the scope of the current analysis. However, in the comparative clinical study reported by Sharman et al. [22], there was no difference in efficacy between RTX-PF and RTX-EU.
Finally, the small number of baseline ADA-positive patients may have limited the power of our analysis to investigate the relationship between ADA status and IRR outcome. However, the immunogenicity of rituximab may not influence the occurrence of IRRs, as a correlation between the presence of human anti-chimeric antibody and the occurrence of IRRs has not been demonstrated [26, 36]. In spite of this limitation of the current study, the availability of baseline ADA status data provides the opportunity for an exploratory analysis of the relationship between rituximab immunogenicity and the incidence of IRRs.
5 Conclusion
The results of this analysis demonstrated that the incidence of IRR AEs was similar between RTX-PF and RTX-EU. Furthermore, the results demonstrated a potential correlation of infusion rate and other factors with IRRs at the individual and population levels. We mathematically modeled the data with a predication estimate between an increase in the infusion rate and the likelihood of a corresponding increase in IRR incidence. Rituximab infusion is managed with rate escalation, which is in line with the expectation that a higher infusion rate increases the risk of IRR occurrence. Inclusion of immunogenicity assessment as part of the biosimilarity assessment provided a valuable source of potential factors that may influence IRRs, including baseline ADA status. Nevertheless, IRR incidence was unaffected by baseline ADA status or baseline tumor burden level. Baseline CD20+ B-lymphocyte level was similar between treatment arms but slightly higher in patients who experienced an IRR, which could suggest a role for baseline CD20+ B-lymphocyte level in the occurrence of IRR.
The same pattern of results, in terms of the distribution of infusion rate and the incidence of IRRs, was observed for RTX-PF and RTX-EU and was consistent with expectations for rituximab based on clinical experience. Finally, although the majority of patients completed their infusion in > 2 but ≤ 3.5 h, a small percentage (4.5%) of patients completed their infusion in ≤ 2 h, demonstrating the safety and tolerability of the treatment in the short infusion duration setting.
References
European Medicines Agency. MabThera (rituximab): Summary of product characteristics. 2019. https://www.ema.europa.eu/en/documents/product-information/mabthera-epar-product-information_en.pdf. Accessed 25 Feb 2020.
Genentech Inc. Rituxan (rituximab): US prescribing information. 2019. https://www.gene.com/download/pdf/rituxan_prescribing.pdf. Accessed 25 Feb 2020.
Salles G, Barrett M, Foa R, Maurer J, O’Brien S, Valente N, et al. Rituximab in B-Cell hematologic malignancies: a review of 20 years of clinical experience. Adv Ther. 2017;34:2232–73.
Davis TA, White CA, Grillo-Lopez AJ, Velasquez WS, Link B, Maloney DG, et al. Single-agent monoclonal antibody efficacy in bulky non-Hodgkin’s lymphoma: results of a phase II trial of rituximab. J Clin Oncol. 1999;17:1851–7.
Feuring-Buske M, Kneba M, Unterhalt M, Engert A, Gramatzki M, Hiller E, et al. IDEC-C2B8 (rituximab) anti-CD20 antibody treatment in relapsed advanced-stage follicular lymphomas: results of a phase-II study of the German Low-Grade Lymphoma Study Group. Ann Hematol. 2000;79:493–500.
Foran JM, Gupta RK, Cunningham D, Popescu RA, Goldstone AH, Sweetenham JW, et al. A UK multicentre phase II study of rituximab (chimaeric anti-CD20 monoclonal antibody) in patients with follicular lymphoma, with PCR monitoring of molecular response. Br J Haematol. 2000;109:81–8.
McLaughlin P, Grillo-Lopez AJ, Link BK, Levy R, Czuczman MS, Williams ME, et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol. 1998;16:2825–33.
Piro LD, White CA, Grillo-Lopez AJ, Janakiraman N, Saven A, Beck TM, et al. Extended rituximab (anti-CD20 monoclonal antibody) therapy for relapsed or refractory low-grade or follicular non-Hodgkin’s lymphoma. Ann Oncol. 1999;10:655–61.
Herold M, Haas A, Srock S, Neser S, Al-Ali KH, Neubauer A, et al. Rituximab added to first-line mitoxantrone, chlorambucil, and prednisolone chemotherapy followed by interferon maintenance prolongs survival in patients with advanced follicular lymphoma: an East German Study Group Hematology and Oncology Study. J Clin Oncol. 2007;25:1986–92.
Hiddemann W, Kneba M, Dreyling M, Schmitz N, Lengfelder E, Schmits R, et al. Frontline therapy with rituximab added to the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) significantly improves the outcome for patients with advanced-stage follicular lymphoma compared with therapy with CHOP alone: results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood. 2005;106:3725–32.
Marcus R, Imrie K, Belch A, Cunningham D, Flores E, Catalano J, et al. CVP chemotherapy plus rituximab compared with CVP as first-line treatment for advanced follicular lymphoma. Blood. 2005;105:1417–23.
Marcus R, Imrie K, Solal-Celigny P, Catalano JV, Dmoszynska A, Raposo JC, et al. Phase III study of R-CVP compared with cyclophosphamide, vincristine, and prednisone alone in patients with previously untreated advanced follicular lymphoma. J Clin Oncol. 2008;26:4579–86.
European Medicines Agency. Guideline on similar biological medicinal products. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/10/WC500176768.pdf. Accessed 1 July 2020.
US Food and Drug Administration. Scientific considerations in demonstrating biosimilarity to a reference product. Guidance for industry. 2015. https://www.fda.gov/media/82647/download. Accessed 16 July 2020.
US Food and Drug Administration. Biosimilar basics for patients. 2019. https://www.fda.gov/drugs/biosimilars/patient-materials. Accessed 29 June 2020.
Pfizer Inc. Ruxience (rituximab-pvvr) US prescribing information. 2019. http://labeling.pfizer.com/ShowLabeling.aspx?id=12090. Accessed 26 February 2020.
European Medicines Agency. Ruxience (rituximab): Summary of product characteristics. 2020. https://www.ema.europa.eu/en/documents/product-information/ruxience-epar-product-information_en.pdf. Accessed 07 April 2020.
Pfizer Inc. Pfizer receives European approval for oncology biosimilar, Ruxience™ (rituximab). 2020. https://investors.pfizer.com/investor-news/press-release-details/2020/Pfizer-Receives-European-Approval-for-Oncology-Biosimilar-RUXIENCE-rituximab/default.aspx. Accessed 20 Aug 2020.
Japan Pharmaceuticals and Medical Devices Agency. New drugs approved in FY 2019. 2019. https://www.pmda.go.jp/files/000235289.pdf. Accessed 6 Nov 2020.
Ryan AM, Sokolowski SA, Ng CK, Shirai N, Collinge M, Shen AC, et al. Comparative nonclinical assessments of the proposed biosimilar PF-05280586 and rituximab (MabThera(R)). Toxicol Pathol. 2014;42:1069–81.
Cohen S, Emery P, Greenwald M, Yin D, Becker JC, Melia LA, et al. A phase I pharmacokinetics trial comparing PF-05280586 (a potential biosimilar) and rituximab in patients with active rheumatoid arthritis. Br J Clin Pharmacol. 2016;82:129–38.
Sharman JP, Liberati AM, Ishizawa K, Khan T, Robbins J, Alcasid A, et al. A randomized, double-blind, efficacy and safety study of PF-05280586 (a rituximab biosimilar) compared with rituximab reference product (MabThera(R)) in subjects with previously untreated CD20-positive, low-tumor-burden follicular lymphoma (LTB-FL). BioDrugs. 2020;34:171–81.
Cheson BD, Morschhauser F, Martin P. Management of adverse events from the combination of rituximab and lenalidomide in the treatment of patients with follicular and low-grade non-Hodgkin lymphoma. Clin Lymphoma Myeloma Leuk. 2020;20:563–71.
Driscoll N. Idelalisib: practical tools for identifying and managing adverse events in clinical practice. J Adv Pract Oncol. 2016;7:604–13.
Fowler NH, Nastoupil L, De Vos S, Knapp M, Flinn IW, Chen R, et al. The combination of ibrutinib and rituximab demonstrates activity in first-line follicular lymphoma. Br J Haematol. 2020;189:650–60.
Paul F, Cartron G. Infusion-related reactions to rituximab: frequency, mechanisms and predictors. Expert Rev Clin Immunol. 2019;15:383–9.
Hayama T, Miura K, Uchiike A, Nakagawa M, Tsutsumi D, Sakagami M, et al. A clinical prediction model for infusion-related reactions to rituximab in patients with B cell lymphomas. Int J Clin Pharm. 2017;39:380–5.
Hong J, Kim JY, Ahn HK, Lee SM, Sym SJ, Park J, et al. Bone marrow involvement is predictive of infusion-related reaction during rituximab administration in patients with B cell lymphoma. Support Care Cancer. 2013;21:1145–52.
Kowalski KE, Adams CB, Voils SA, Wheeler SE. Use of patient and disease characteristics as predictive indicators of rituximab infusion-related reactions in adult malignant hematology patients at an academic medical center. Clin Lymphoma Myeloma Leuk. 2019;19:723–8.
Lee S, Lee H, Kim E. Comparative efficacy and safety of biosimilar rituximab and originator rituximab in rheumatoid arthritis and non-Hodgkin’s lymphoma: a systematic review and meta-analysis. BioDrugs. 2019;33:469–83.
van der Kolk LE, Grillo-Lopez AJ, Baars JW, Hack CE, van Oers MH. Complement activation plays a key role in the side-effects of rituximab treatment. Br J Haematol. 2001;115:807–11.
Vultaggio A, Matucci A, Nencini F, Pratesi S, Petroni G, Cammelli D, et al. Drug-specific Th2 cells and IgE antibodies in a patient with anaphylaxis to rituximab. Int Arch Allergy Immunol. 2012;159:321–6.
Winkler U, Jensen M, Manzke O, Schulz H, Diehl V, Engert A. Cytokine-release syndrome in patients with B-cell chronic lymphocytic leukemia and high lymphocyte counts after treatment with an anti-CD20 monoclonal antibody (rituximab, IDEC-C2B8). Blood. 1999;94:2217–24.
Wing M. Monoclonal antibody first dose cytokine release syndromes-mechanisms and prediction. J Immunotoxicol. 2008;5:11–5.
Wincup C, Menon M, Smith E, Schwartz A, Isenberg D, Jury EC, et al. Presence of anti-rituximab antibodies predicts infusion-related reactions in patients with systemic lupus erythematosus. Ann Rheum Dis. 2019;78:1140–2.
Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:676–88.
Freeman CL, Dixon M, Houghton R, Kreuzer KA, Fingerle-Rowson G, Herling M, et al. Role of CD20 expression and other pre-treatment risk factors in the development of infusion-related reactions in patients with CLL treated with obinutuzumab. Leukemia. 2016;30:1763–6.
Asselin B. Immunology of infusion reactions in the treatment of patients with acute lymphoblastic leukemia. Future Oncol. 2016;12:1609–21.
Acknowledgements
The authors thank Dr. Sandeep Parsad of the University of Chicago Medicine for contributions to the publication concept, and thank Peter Lee, BS, of Pfizer for contributions to the data analysis and guidance on statistical analysis and programming. Medical writing support was provided by Elyse Smith, PhD, of Engage Scientific Solutions and was funded by Pfizer.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Pfizer.
Conflicts of interest
NK is a full-time employee of and declares stock holdings and/or stock options from Pfizer. AA and JZ are full-time employees of and declare stock holdings and/or stock options from Pfizer; and had travel expenses paid or reimbursed by Pfizer. JK is a full-time employee of and declares stock holdings and/or stock options from Pfizer; and declares stock holdings and/or stock options from Abbot and AbbVie. PN-P is a full-time employee of and declares stock holdings and/or stock options from Pfizer; and declares stock holdings and/or stock options from Bristol-Myers Squibb and Celgene. JC was a full-time employee of and declares shareholdings, stock holdings, and/or stock options from Pfizer. LN has received honoraria from Bayer, Celgene, Gamida Cell, Gilead Sciences, Janssen Oncology, Juno Therapeutics, Novartis, Pfizer, Spectrum Pharmaceuticals, and TG Therapeutics; and research funding from Celgene, Genentech/Roche, Janssen Biotech, Karus Therapeutics, LAM Therapeutics, and TG Therapeutics.
Ethics approval
This study was conducted in compliance with the ethical principles originating in, or derived from, the Declaration of Helsinki and in compliance with all International Conference on Harmonization Good Clinical Practice Guidelines and was reviewed and approved by institutional review boards and/or independent ethics committees. All local regulatory requirements were followed, particularly those affording greater protection to the safety of trial participants. The study was sponsored by Pfizer and is registered on ClinicalTrials.gov (NCT02213263) and EudraCT (2014-000132-41).
Consent to participate
All patients provided informed consent before undergoing any screening procedures.
Consent for publication
Not applicable.
Code availability
Not applicable.
Availability of data and material
Upon request, and subject to certain criteria, conditions, and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de-identified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices (1) for indications that have been approved in the USA and/or EU or (2) in programs that have been terminated (i.e., development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de-identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
Author contributions
AA, JC, NK, JK, and PN-P contributed to the conception or design of the study; LN contributed to the acquisition of data; and JZ contributed to the data analysis. All authors participated in the interpretation of the data, contributed to the drafting or revision of the manuscript, read and gave final approval of the submitted manuscript, were involved in the decision to submit the manuscript for publication, and accept accountability for all aspects of the work.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Courville, J., Nastoupil, L., Kaila, N. et al. Factors Influencing Infusion-Related Reactions Following Dosing of Reference Rituximab and PF-05280586, a Rituximab Biosimilar. BioDrugs 35, 459–468 (2021). https://doi.org/10.1007/s40259-021-00487-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-021-00487-6