
Abstract
Background
Palmoplantar pustulosis (PPP) is a pruritic, painful, recurrent, and chronic dermatitis with limited therapeutic options.
Objective
To evaluate the efficacy and safety of apremilast for the treatment of Japanese patients with PPP and inadequate response to topical treatment.
Methods
This phase 2, randomized, double-blind, placebo-controlled study enrolled patients with Palmoplantar Pustulosis Area and Severity Index (PPPASI) total score ≥ 12 and moderate or severe pustules/vesicles on the palm or sole (PPPASI pustule/vesicle severity score ≥ 2) at screening and baseline with an inadequate response to topical treatment. Patients were randomized (1:1) to apremilast 30 mg twice daily or placebo for 16 weeks, followed by a 16-week extension phase during which all patients received apremilast. The primary endpoint was achievement of PPPASI-50 response (≥ 50% improvement from baseline in PPPASI). Key secondary endpoints included change from baseline in PPPASI total score, Palmoplantar Pustulosis Severity Index (PPSI), and patient’s visual analog scale (VAS) for PPP symptoms (pruritus and discomfort/pain).
Results
A total of 90 patients were randomized (apremilast: 46; placebo: 44). A significantly greater proportion of patients achieved PPPASI-50 at week 16 with apremilast versus placebo (P = 0.0003). Patients receiving apremilast showed greater improvement in PPPASI at week 16 versus placebo (nominal P = 0.0013), as well as PPSI and patient-reported pruritus and discomfort/pain (nominal P ≤ 0.001 for all). Improvements were sustained through week 32 with apremilast treatment. The most common treatment-emergent adverse events included diarrhea, abdominal discomfort, headache, and nausea.
Conclusions
Apremilast treatment demonstrated greater improvements in disease severity and patient-reported symptoms versus placebo at week 16 in Japanese patients with PPP with sustained improvements through week 32. No new safety signals were observed.
ClinicalTrials.gov
NCT04057937.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Significant improvements were seen in PPPASI, PGA, PPSI, pruritus, and pain with apremilast versus placebo at week 16, which were sustained through week 32. |
Adverse events were consistent with the known safety profile of apremilast. |
1 Introduction
Palmoplantar pustulosis (PPP) is a pustular eruption characterized by a combination of intra-epidermal vesicles, pustules, erythema, and scales/desquamation located on the palms and soles [1, 2]. PPP is characterized by intra-epidermal infiltration of polymorphonuclear neutrophils resulting in sterile pustules [3]. Patients with PPP have elevated levels of tumor necrosis factor (TNF)-α, interleukin (IL)-17, IL-22, IL-8, IL-1, and IL-36 in skin lesions [4, 5]. PPP is rare, with a prevalence of 0.12% in Japan [6]. PPP lesions are often pruritic and/or painful [7]. Other symptoms of PPP include painful fissures [1]. These symptoms can be chronic, repeating relapsing, or remitting, and they may be disabling and severely impair quality of life [2, 7,8,9,10]. Although PPP is very similar to palmoplantar pustular psoriasis (PPPP), some evidence has indicated that the two are genetically distinct [11, 12]. For example, psoriasis is correlated with the HLA-Cw6 allele, but PPP is not, and the incidence of mutations in the IL-36 receptor antagonist gene (IL36RN) is much greater in generalized pustular psoriasis than in PPP [12, 13]. Thus, it is still unclear whether PPP and PPPP are distinct or the same.
Treatment for PPP includes topical treatments (e.g., corticosteroids and active vitamin D3), oral systemic treatments (e.g., acitretin or etretinate, cyclosporine, methotrexate), phototherapy, biologic injectable treatments, and granulocyte and monocyte adsorption apheresis [7, 14,15,16,17,18,19]. However, many of these treatments are contraindicated in certain patient populations and are associated with side effects ranging from skin atrophy with corticosteroids to increased risk of malignancy with phototherapy and biologics [7]. In addition, PPP can be very recalcitrant, and many patients with PPP do not fully respond to their treatment, highlighting an unmet medical need in this patient population [7].
Apremilast is an oral phosphodiesterase 4 (PDE4) inhibitor that has demonstrated efficacy and tolerability for the treatment of psoriatic arthritis and plaque psoriasis, including palmoplantar psoriasis [20,21,22,23,24,25,26,27,28]. PDE4 degrades cyclic 3′,5′-adenosine monophosphate (cAMP), a key regulator of inflammatory signaling. Inhibition of PDE4 with apremilast causes an increase in cAMP, which inhibits production of several inflammatory cytokines that are related to the pathogenesis of PPP, such as TNF-α, IL-17, IL-22, and IL-8 [1, 4, 29]. Due to its mechanism of action and efficacy in other cytokine-mediated diseases [24,25,26,27,28, 30], apremilast theoretically may also be an effective treatment for PPP. However, until this study, the efficacy and safety of apremilast for the treatment of PPP had not been studied in randomized, placebo-controlled clinical trials. This randomized, phase 2 study (NCT04057937) evaluated the efficacy and safety of apremilast versus placebo for the treatment of patients with PPP after inadequate response to topical treatment in Japan.
2 Methods
2.1 Study Design
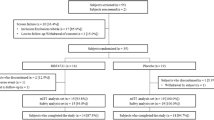
This was a multicenter, randomized, double-blind, placebo-controlled, parallel-group, phase 2 study of apremilast in Japanese patients with PPP and inadequate response to treatment with topical steroid and/or topical vitamin D3 derivative preparations (Online Resource 1). After a 4 week screening phase, patients were randomized in a 1:1 ratio to receive apremilast 30 mg twice daily (BID) or placebo for 16 weeks. Randomization was stratified according to rounded Palmoplantar Pustulosis Area and Severity Index (PPPASI) score (≤ 20, 21–30, ≥ 31) and focal infection status (yes, no). At week 16, all patients continued treatment with apremilast 30 mg BID or were switched from placebo to apremilast 30 mg BID until week 32. An observational follow-up evaluation was performed 4 weeks after the last dose of study medication or early discontinuation.
2.2 Key Inclusion Criteria
Adults (age ≥ 20 years) with a diagnosis of PPP with or without pustulotic arthro-osteitis (not requiring treatment by an immunosuppressant) for at least 24 weeks before screening, regardless of the presence or absence of concurrent extra-palmoplantar lesions, were eligible for enrollment. Diagnostic criteria were sterile pustules located on palms and/or soles, change from vesicles to pustules via pustulo-vesicles with progression of disease, and repeat recurrence at the same skin lesion.
Patients were required to have a PPPASI total score ≥ 12 at screening and baseline, moderate or severe pustules/vesicles on the palm or sole (PPPASI pustule/vesicle severity score ≥ 2) at screening and baseline, and inadequate response to treatment with topical steroid and/or topical vitamin D3 derivative preparations before or at screening. Inadequate response was defined as repeated relapsing–remitting in the same lesion observed during a 6 month treatment period. Eligible patients met the following laboratory criteria at screening: white blood cell count ≥ 3000/mm3 (≥ 3.0 × 109/L) and ≤ 14,000/mm3 (≤ 14 × 109/L); platelet count ≥ 100,000/μL (≥ 100 × 109/L); serum creatinine ≤ 1.5 mg/dL (≤ 132.6 μmol/L); aspartate aminotransferase (AST)/serum glutamic-oxaloacetic transaminase (SGOT), and alanine aminotransferase (ALT)/serum glutamic pyruvic transaminase (SGPT) ≤ 2.0× upper limit of normal (ULN); total bilirubin ≤ 2.0 mg/dL; and hemoglobin > 9 g/dL.
2.3 Key Exclusion Criteria
Patients were excluded from the study if they had any significant medical condition, laboratory abnormality, or psychiatric illness that would prevent study participation or that would place the patient at unacceptable risk upon study participation or any condition that confounds the ability to interpret data from the study. Other key exclusion criteria were diagnosis of plaque-type psoriasis, presence of pustular psoriasis in any part of the body other than the palms and soles (excluding those derived from PPP, which have characteristics distinct from psoriasis [31]); obvious improvement (≥ 5 PPPASI total score improvement) during screening, having any procedures for focal infection (e.g., tonsillectomy or dental therapy) within 24 weeks of baseline, periodontitis obviously requiring treatment at screening, or chronic or recurrent tonsillitis or sinusitis requiring any continuous treatment for a month or more at screening.
Patients with planned or concurrent use of the following therapies were not eligible to participate in the study. This included topical therapy within 2 weeks prior to randomization (except an unmedicated skin moisturizer as needed but not within 24 h prior to clinic visit), conventional systemic therapy within 4 weeks prior to randomization, adsorptive granulocyte and monocyte apheresis (GMA) within 2 weeks prior to randomization (which has been shown to have efficacy in PPP [19, 32]), phototherapy treatment within 4 weeks prior to randomization (i.e., UVB, psoralen and UVA), and biologic therapy within 12 weeks or 5 times of half-lives, whichever is longer, prior to randomization.
2.4 Study Endpoints
The primary efficacy endpoint was the proportion of patients achieving PPPASI-50 response at week 16. Secondary endpoints assessed every 2 weeks during the placebo-controlled phase included proportion of patients achieving PPPASI-50 response, proportion of patients achieving PPPASI-75 response, percent change from baseline in PPPASI total score, percent change from baseline in Palmoplantar Severity Index (PPSI) total score, and proportion of patients achieving Physician Global Assessment (PGA) score of 0 (cleared) or 1 (minimal) and ≥ 2 point reduction/improvement. Change from baseline in patient’s VAS for PPP symptoms (pruritus and skin discomfort/pain) was evaluated at baseline and weeks 2, 4, 6, 8, 12, and 16 during the placebo-controlled phase. The PPSI (range: 0–12) evaluates change of disease status on the most severe skin lesion at screening. The PGA (range: 0–5) for palms and soles represents a global assessment of skin lesions in PPP. The patient’s VAS for PPP symptoms (pruritus and skin discomfort/pain; range 0–100) is a patient-rated assessment of pruritus and discomfort/pain related to PPP on the hands and feet.
Exploratory endpoints evaluated through week 32 included change from baseline in Dermatology Life Quality Index (DLQI) total score, achievement of PPPASI-50, achievement of PPPASI-75, change from baseline in PPPASI, change from baseline in PPSI, and proportion of patients achieving PGA score of 0 or 1 and ≥ 2 point reduction. The patient’s VAS for PPP symptoms was also evaluated at weeks 24 and 32. DLQI (range 0–30) evaluates patient-rated quality of life and was assessed at baseline and Weeks 8, 16, 24, and 32. PPPASI-90 response was evaluated as a post hoc analysis. Safety was evaluated throughout the study, including frequency, severity, and incidence rate of treatment-emergent adverse events (TEAEs), and treatment discontinuations due to TEAEs.
2.5 Statistical Analysis
For this study to have 80% power (based on a chi-square test at a two-sided significance level of 0.10) for the primary endpoint, assuming a treatment difference of 25% in PPPASI-50 rates between apremilast 30 mg BID and placebo at week 16, the sample size of 86 patients was estimated using SAS 9.4 POWER procedure (SAS, Cary NC, USA). Patients were randomized (1:1) to receive apremilast 30 mg BID or placebo using a centralized interactive response technology. Efficacy endpoints were analyzed in the intent-to-treat (ITT) population, defined as all randomized patients who received at least one dose of study drug. Patients were included in the treatment group for the treatment to which they were randomized. Safety was analyzed in the safety population, which included all randomized patients who received at least one dose of study drug.
The primary endpoint was analyzed using a chi-square test at the two-sided 0.10 level. Missing data were handled using nonresponder imputation (NRI). Categorical secondary endpoints were analyzed using the Cochran–Mantel–Haenszel test adjusting for baseline PPPASI score range (≤ 20, 21–30, ≥ 31) and focal infection status (yes, no) at baseline. Missing data were handled using NRI. Change from baseline in PPPASI, PPSI, pruritus, and skin discomfort/pain VAS were analyzed by mixed-effects model for repeated measures (MMRM) for the change from baseline, with treatment group, visit, treatment-by-visit interaction, PPPASI total score range, and focal infection status at baseline as fixed effects and the baseline value as a covariate. An unstructured covariance matrix that is homogeneous across treatment groups was used. Secondary efficacy endpoints were tested without multiplicity adjustment, and P-values for those endpoints were regarded as nominal. Exploratory efficacy endpoints were summarized descriptively.
3 Results
3.1 Patients
From 16 October 2019 to 7 June 2021, a total of 90 patients from 22 institutions in Japan were randomized (apremilast: 46; placebo: 44); 87 (96.7%) completed the placebo-controlled phase (apremilast: 46; placebo: 41) and entered the active treatment phase (weeks 16 to 32) (Online Resource 2). Of the 41 patients who were switched from placebo to apremilast (placebo/apremilast group) at week 16, 40 (97.6%) completed the active treatment phase. Forty-six patients were initially randomized to apremilast and continued with apremilast (apremilast/apremilast group) at week 16, and 44 (95.7%) of them completed the active treatment phase. The three patients who discontinued active treatment early discontinued due to pregnancy, physician decision, and withdrawal by patient.
Demographics and baseline characteristics were generally similar between groups (Table 1). Mean age was 54.9 years in the apremilast group and 54.7 years in the placebo group, at least three-quarters of patients were female, and more than 80% of patients used tobacco. Mean duration of PPP was 7.3 years in the apremilast group and 7.5 years in the placebo group. Most patients [65.6% (59/90)] had a PPPASI score ≥ 21 at randomization, and mean PPSI score was 8.3 and 8.2 in the apremilast and placebo groups, respectively.
3.2 Efficacy in Disease Severity Outcomes
More than three-quarters of patients treated with apremilast achieved PPPASI-50 response at week 16 (primary endpoint), which is nearly twice the proportion of patients in the placebo group [78.3% and 40.9%, respectively; treatment difference (95% CI) 37.4% (18.6%, 56.1%), P = 0.0003; Fig. 1; Online Resource 3].Greater PPPASI-50 response rates with apremilast versus placebo were observed as early as week 2 and at every time point up to week 16 (Online Resource 4). The proportion of patients who achieved PPPASI-50 response increased from week 16 [40.9% (18/44)] to week 32 [75.0% (33/44)] in the placebo/apremilast group. In the apremilast/apremilast group, the proportion of patients achieving PPPASI-50 response was generally maintained over 32 weeks of treatment [week 16: 78.3% (36/46); week 32: 71.7% (33/46)].

Proportion of patients with PPPASI-50, PPPASI-75, and PPPASI-90 responses at Week 16. Intent-to-treat population. *Two-sided P-value is based on chi-square test. Missing data were imputed by nonresponder imputation. †Two-sided P-value (nominal) based on Cochran–Mantel–Haenszel test adjusting for baseline PPPASI score range and baseline focal infection status at baseline. BID twice daily, CI confidence interval, PPPASI Palmoplantar Pustulosis Area and Severity Index; PPPASI-50/75/90, ≥50%/75%/90% improvement from baseline in PPPASI total score
Achievement of PPPASI-75 [adjusted treatment difference: 26.3% (8.3%, 44.2%), nominal P = 0.0074] response was nearly three-fold greater and PPPASI-90 [treatment difference: 6.2% (−6.0%, 18.5%); nominal P = 0.3278] response was almost double with apremilast versus placebo at week 16 (Fig. 1). PPPASI-75 response rates were greater with apremilast versus placebo over time, with nominal P values < 0.05 versus placebo at weeks 10, 14, and 16 (Online Resource 4). In the apremilast/apremilast group, the proportion of patients achieving PPPASI-75 response was maintained over 32 weeks of treatment [week 16: 43.5% (20/46); week 32: 52.2% (24/46)] (Online Resource 4).
Greater improvement from baseline in PPPASI total score was observed with apremilast versus placebo at week 16. For least-squares (LS) mean change from baseline in PPPASI total score, the treatment difference (95% CI) at week 16 was −5.5 [(−8.7, −2.2); nominal P = 0.0013; Fig. 2]. Improvements in PPPASI total score were greater with apremilast versus placebo as early as week 2 and at every time point up to week 16 (Online Resource 5). Improvements in PPPASI total score were maintained up to week 32; the percent change in mean PPPASI score from baseline with apremilast was −64.3% at week 16 and −68.3% at week 32. In addition, PPPASI total score improved in the placebo group from −42.4% at week 16 to −72.1% at week 32 in the placebo/apremilast group.

Change from Baseline in PPPASI total score and PPSI score at Week 16 after treatment with apremilast or placebo (ITT population). Error bars represent 95% CI. Intent-to-treat population. Differences are based on MMRM model for the change from baseline, with treatment group, visit, treatment-by-visit interaction, PPPASI total score range, and focal infection status at baseline as fixed effects and the baseline value as a covariate. An unstructured covariance matrix that is homogeneous across treatment groups was used. BID twice daily, CI confidence interval, MMRM mixed-effects model for repeated measures, PPPASI Palmoplantar Pustulosis Area and Severity Index, PPSI Palmoplantar Severity Index.
Greater improvement from baseline in PPSI total score was observed with apremilast versus placebo at week 16. For LS mean change from baseline in PPSI total score at week 16, the treatment difference (95% CI) was −1.6 [(–2.4, –0.9); nominal P < 0.0001; Fig. 2]. Improvements in PPSI total score were greater with apremilast versus placebo as early as week 2 and at every time point up to week 16 (Online Resource 5). Improvements in PPSI total score were maintained up to week 32 with apremilast treatment; the percent change in mean from baseline was −49.0% at week 16 and −51.3% at week 32. In addition, PPSI total score improved in the placebo group at week 16 from −30.9 to −56.7% at week 32 in the placebo/apremilast group. Apremilast treatment also demonstrated greater achievement of PGA response [treatment difference: 12.0% (–0.6%, 24.6%); nominal P = 0.0773] compared with placebo at week 16. PGA response rates improved in the placebo/apremilast group and were maintained in the apremilast/apremilast group at week 32 (Fig. 3).

PGA response rate (score of 0 or 1 and ≥ 2 grade improvement) and change from Baseline in patient’s VAS for PPP symptoms at Week 16 after treatment with apremilast or placebo (ITT population). Data shown for week 16 are LS mean changes and for week 32 are mean changes from baseline. Error bars represent 95% CI. Intent-to-treat population. For PGA response, missing data were imputed by nonresponder imputation. Differences in patient’s VAS assessments are based on MMRM model for the change from baseline, with treatment group, visit, treatment-by-visit interaction, PPPASI total score range, and focal infection status at baseline as fixed effects and the baseline value as a covariate. An unstructured covariance matrix that is homogeneous across treatment groups was used. For patient’s VAS assessments, LS mean values are shown for week 16, and mean values are shown for week 32. APR apremilast 30 mg twice daily, BID twice daily, CI confidence interval, MMRM mixed-effects model for repeated measures, NRI nonresponder imputation, PBO placebo, PGA Physician Global Assessment, PPPASI Palmoplantar Pustulosis Area and Severity Index, VAS visual analog scale.
3.3 Efficacy in Patient-Reported Outcomes
Greater improvements in pruritus VAS (treatment difference: −20.3; nominal P = 0.0002) and skin discomfort/pain VAS (treatment difference: −16.1; nominal P = 0.0010) assessments were observed with apremilast compared with placebo at week 16 (Fig. 3). Greater improvements were observed as early as week 2 compared with placebo. Improvements were maintained in the apremilast/apremilast group and improved in the placebo/apremilast group at week 32 (Online Resource 6). Changes from baseline in DLQI total score were −4.7 with apremilast versus −2.7 with placebo at week 16 and −4.9 in the placebo/apremilast group and −4.3 in the apremilast/apremilast group at week 32 (Fig. 4). These changes in DLQI in the apremilast group met the minimal clinically important difference of a four point change in inflammatory skin diseases [33].

Change from Baseline in DLQI at Week 16 and Week 32 (exploratory endpoint). Error bars represent 95% CIs. Intent-to-treat population. Data are presented as observed. BID twice daily, CI confidence interval, DLQI Dermatology Life Quality Index.
3.4 Safety
In the placebo-controlled period, 82.6% (38/46) of patients in the apremilast group and 70.5% (31/44) of patients in the placebo group experienced ≥ 1 TEAE at week 16, and 83.9% (73/87) of all apremilast-treated patients experienced ≥ 1 TEAE at week 32 (Table 2). The most commonly reported TEAEs were consistent with previous clinical trials for apremilast and included diarrhea, abdominal discomfort, headache, and nausea (Table 2). Most TEAEs were mild to moderate in severity (Table 2). There were no serious TEAEs during the placebo-controlled period. During the apremilast exposure period, three patients experienced serious TEAEs, including peritonitis in one patient in the placebo/apremilast group and COVID-19 pneumonia, constipation, and volvulus in the apremilast/apremilast group. Few patients reported TEAEs leading to treatment discontinuation (apremilast: 0% (0/46); placebo: 6.8% (3/44); Table 2). The three TEAEs that led to discontinuation in the placebo group were pustulotic arthro-osteitis, dysuria, and peripheral edema. There were no deaths reported (Table 2).
4 Discussion
The patient population in this study had significant disease burden, as indicated by baseline scores on PPPASI, PPSI, and PGA assessments, and a high proportion of patients had focal infections at baseline. Patients also exhibited PPP symptoms such as pruritus, pain, and substantial quality-of-life impairment.
In this randomized, phase 2 study of the efficacy and safety of apremilast in patients with PPP, significantly more patients achieved a PPPASI-50 response at week 16 with apremilast versus placebo [treatment difference (95% CI) 37.4% (18.6%, 56.1%), P = 0.0003]. PPPASI-50 response rates were maintained with continued apremilast treatment over 32 weeks. In patients who switched from placebo to apremilast, the proportion of patients who achieved PPPASI-50 response increased nearly twofold from week 16 to week 32. The treatment effects of apremilast were also consistently observed in the secondary endpoints. Tolerability and AEs were consistent with the known safety profile of apremilast. There was no evidence of increased incidence of TEAEs with longer apremilast exposure.
Apremilast treatment resulted in significant improvements in patient-reported PPP symptoms and quality-of-life outcomes. Improvements in pruritus, skin discomfort/pain, and DLQI assessments at week 16 were significantly greater with apremilast versus placebo, with continued mean score improvements from week 16 to week 32. Treatment efficacy in VAS measures was noted as early as week 2. Improvements were generally maintained over the 32 week treatment period in the apremilast/apremilast group.
The efficacy and safety profiles of apremilast in this study are similar to those in studies of apremilast in psoriasis [24,25,26,27,28]. In the phase 3 ESTEEM 1 and 2 trials in patients with moderate-to-severe psoriasis, approximately 56–60% of patients experienced a ≥ 50% reduction in Psoriasis Area and Severity Index (PASI-50) at week 16 [24, 25]. This response was maintained over 32 weeks of treatment in ESTEEM 2 [24, 25]. Significant decreases in pruritus VAS and improvements in DLQI have also been seen in studies of apremilast in patients with psoriasis over an extended period of time [24,25,26,27].
The expression of PDE4 in a broad range of cell types and its role in immunomodulation have made it an attractive therapeutic target for a range of diseases. Four PDE4 inhibitors have been successfully developed: roflumilast for the treatment of chronic obstructive pulmonary disease and psoriasis; crisaborole for atopic dermatitis; apremilast for psoriasis, psoriatic arthritis, and Behçet’s disease; and ibudilast for Krabbe disease [34]. Apremilast is the only PDE 4 inhibitor to be evaluated in a clinical trial for PPP. Apremilast has been shown to decrease plasma levels of various cytokines including TNF-α, IL-17, IL-22, and IL-8 in patients with psoriasis and psoriatic arthritis [29, 35, 36]. Increases in levels of these cytokines have been detected in the serum or lesions of patients with PPP [4, 37,38,39]. Therefore, it is possible that the inhibition of these cytokines may contribute to the efficacy of apremilast in improving symptoms of PPP. Apremilast has also been shown to reduce neutrophil infiltration, inhibit cell death, and promote production of the anti-inflammatory cytokine IL-10 [40]. Fibroblast migration and keratinocyte proliferation, which is a key cause of epidermal thickening in psoriasis, are also attenuated by apremilast treatment [40]. These may represent potential mechanisms by which apremilast improves symptoms of PPP, although this remains to be tested.
Other therapies being investigated for use in PPP/PPPP include the IL-17 inhibitor secukinumab and the IL-23 inhibitor guselkumab. Secukinumab 300 mg was evaluated in a phase 3b study in patients with moderate-to-severe PPPP, and guselkumab 100 mg was evaluated in a phase 3 study of Japanese patients with PPP [41, 42]. PPPASI-50 was achieved by 52.2% of patients receiving secukinumab (versus 32.9% receiving placebo) and 57.4% of patients receiving guselkumab (versus 34.0% receiving placebo) at week 16 in these studies. In the current study, 78.3% of patients receiving apremilast achieved PPPASI-50 at week 16 compared with 40.9% receiving placebo. PPPASI-75 response at week 16 was achieved by 43.5% of patients with apremilast in the current study. In the respective studies, PPPASI-75 response rates were 26.6% with secukinumab and 20.4% with guselkumab. At week 16 in each study, LS mean change from baseline in PPPASI total score was −16.5 with apremilast and −15.3 with guselkumab.
There is a need for more studies of oral systemic treatments for PPP. There have been only a few randomized controlled trials in a small number of patients that assessed the efficacy of oral systemic treatments in patients with PPP [7]. Thus, evidence is lacking for use of these therapies in PPP. There are also a number of limitations associated with current oral systemic therapies. Although retinoids such as acitretin and etretinate have been shown to be effective in the treatment of pustular psoriasis, especially when in combination with ultraviolet light, oral retinoids are associated with side effects that affect the liver, mucocutaneous, musculoskeletal, and neurological systems and are contraindicated in women of childbearing potential due to teratogenicity [43]. Cyclosporine has shown efficacy in PPP in clinical trials [44, 45]; however, treatment is recommended only in short periods for 1–2 years because of side effects such as increased blood pressure and renal impairment [46, 47]. Side effects of methotrexate use include myelosuppression, hepatic dysfunction, alopecia, and interstitial pneumonia [48]. Furthermore, although cyclosporine and methotrexate are commonly used to treat PPP, they may not be covered by insurance for PPP in Japan. Smaller, exploratory studies and case reports have suggested the potential benefits of apremilast for the treatment of PPP [49,50,51]. This randomized, placebo-controlled, phase 2 study extends understanding of the potential benefits of apremilast in a larger population of patients with PPP. It represents one of few randomized controlled trials to demonstrate the efficacy of an oral systemic treatment in patients with PPP.
This study is limited by its short duration and small population size. A phase 3 study in a larger population with longer follow-up is needed to confirm these findings. As the patient population here was limited to patients with a rare disease in Japan, generalizability of findings is limited.
5 Conclusion
Apremilast may represent an effective and safe treatment option for patients with PPP with inadequate response to topical treatment. The data from this phase 2 study have assisted in the development of a larger, long-term phase 3 study in a similar PPP patient population.
Change history
07 December 2023
A Correction to this paper has been published: https://doi.org/10.1007/s40257-023-00825-0
References
Murakami M, Terui T. Palmoplantar pustulosis: current understanding of disease definition and pathomechanism. J Dermatol Sci. 2020;98(1):13–9.
Misiak-Galazka M, Zozula J, Rudnicka L. Palmoplantar pustulosis: recent advances in etiopathogenesis and emerging treatments. Am J Clin Dermatol. 2020;21(3):355–70.
Masuda-Kuroki K, Murakami M, Kishibe M, Kobayashi S, Okubo Y, Yamamoto T, et al. Diagnostic histopathological features distinguishing palmoplantar pustulosis from pompholyx. J Dermatol. 2019;46(5):399–408.
Murakami M, Hagforsen E, Morhenn V, Ishida-Yamamoto A, Iizuka H. Patients with palmoplantar pustulosis have increased IL-17 and IL-22 levels both in the lesion and serum. Exp Dermatol. 2011;20(10):845–7.
Liang Y, Xing X, Beamer MA, Swindell WR, Sarkar MK, Roberts LW, et al. Six-transmembrane epithelial antigens of the prostate comprise a novel inflammatory nexus in patients with pustular skin disorders. J Allergy Clin Immunol. 2017;139(4):1217–27.
Kubota K, Kamijima Y, Sato T, Ooba N, Koide D, Iizuka H, et al. Epidemiology of psoriasis and palmoplantar pustulosis: a nationwide study using the Japanese national claims database. BMJ Open. 2015;5(1): e006450.
Obeid G, Do G, Kirby L, Hughes C, Sbidian E, Le Cleach L. Interventions for chronic palmoplantar pustulosis. Cochrane Database Sys Rev. 2020. https://doi.org/10.1002/14651858.CD011628.pub2.
Benzian-Olsson N, Dand N, Chaloner C, Bata-Csorgo Z, Borroni R, Burden AD, et al. Association of clinical and demographic factors with the severity of palmoplantar pustulosis. JAMA Dermatol. 2020;156(11):1216–22.
Masuda-Kuroki K, Kawakami H, Abe N, Mori M, Tobita R, Fukushi R, et al. Nail lesions in palmoplantar pustulosis and pustulotic arthro-osteitis impairs patients’ quality of life: suggesting new assessment tool of PPP nail lesions. J Dermatol Sci. 2022;106(1):29–36.
Trattner H, Blüml S, Steiner I, Plut U, Radakovic S, Tanew A. Quality of life and comorbidities in palmoplantar pustulosis—a cross-sectional study on 102 patients. J Eur Acad Dermatol Venereol. 2017;31(10):1681–5.
Bissonnette R, Suárez-Fariñas M, Li X, Bonifacio KM, Brodmerkel C, Fuentes-Duculan J, et al. Based on molecular profiling of gene expression, palmoplantar pustulosis and palmoplantar pustular psoriasis are highly related diseases that appear to be distinct from psoriasis vulgaris. PLoS ONE. 2016;11(5): e0155215.
Yamamoto T. Similarity and difference between palmoplantar pustulosis and pustular psoriasis. J Dermatol. 2021;48(6):750–60.
Twelves S, Mostafa A, Dand N, Burri E, Farkas K, Wilson R, et al. Clinical and genetic differences between pustular psoriasis subtypes. J Allergy Clin Immunol. 2019;143(3):1021–6.
Umezawa Y, Nakagawa H, Tamaki K. Phase III clinical study of maxacalcitol ointment in patients with palmoplantar pustulosis: a randomized, double-blind, placebo-controlled trial. J Dermatol. 2016;43(3):288–93.
Muro M, Kawakami H, Matsumoto Y, Abe N, Tsuboi R, Okubo Y. Topical combination therapy with vitamin D3 and corticosteroid ointment for palmoplantar pustulosis: a prospective, randomized, left-right comparison study. J Dermatol Treat. 2016;27(1):51–3.
Hayama K, Inadomi T, Fujisawa D, Terui T. A pilot study of medium-dose cyclosporine for the treatment of palmoplantar pustulosis complicated with pustulotic arthro-osteitis. Eur J Dermatol. 2010;20(6):758–62.
Terui T, Kobayashi S, Okubo Y, Murakami M, Hirose K, Kubo H. Efficacy and safety of guselkumab, an anti-interleukin 23 monoclonal antibody, for palmoplantar pustulosis: a randomized clinical trial. JAMA Dermatol. 2018;154(3):309–16.
Okubo Y, Morishima H, Zheng R, Terui T. Sustained efficacy and safety of guselkumab in patients with palmoplantar pustulosis through 1.5 years in a randomized phase 3 study. J Dermatol. 2021;48(12):1838–53.
Kawakami H, Nagaoka Y, Hirano H, Matsumoto Y, Abe N, Tsuboi R, et al. Evaluation of the efficacy of granulocyte and monocyte adsorption apheresis on skin manifestation and joint symptoms of patients with pustulotic arthro-osteitis. J Dermatol. 2019;46(2):144–8.
Kavanaugh A, Mease PJ, Gomez-Reino JJ, Adebajo AO, Wollenhaupt J, Gladman DD, et al. Treatment of psoriatic arthritis in a phase 3 randomized, placebo-controlled trial with apremilast, an oral phosphodiesterase 4 inhibitor. Ann Rheum Dis. 2014;73(6):1020–6.
Cutolo M, Myerson GE, Fleischmann R, Liote F, Diaz-Gonzalez F, Van den Bosch F, et al. A phase III, randomized, controlled trial of apremilast in patients with psoriatic arthritis: results of the PALACE 2 trial. J Rheumatol. 2016;43(9):1724–34.
Edwards CJ, Blanco FJ, Crowley J, Birbara CA, Jaworski J, Aelion J, et al. Apremilast, an oral phosphodiesterase 4 inhibitor, in patients with psoriatic arthritis and current skin involvement: a phase III, randomised, controlled trial (PALACE 3). Ann Rheum Dis. 2016;75(6):1065–73.
Wells AF, Edwards CJ, Kivitz AJ, Bird P, Nguyen D, Paris M, et al. Apremilast monotherapy in DMARD-naive psoriatic arthritis patients: results of the randomized, placebo-controlled PALACE 4 trial. Rheumatology. 2018;57(7):1253–63.
Papp K, Reich K, Leonardi CL, Kircik L, Chimenti S, Langley RG, et al. Apremilast, an oral phosphodiesterase 4 (PDE4) inhibitor, in patients with moderate to severe plaque psoriasis: results of a phase III, randomized, controlled trial (Efficacy and Safety Trial Evaluating the Effects of Apremilast in Psoriasis [ESTEEM 1]). J Am Acad Dermatol. 2015;73(1):37–49.
Paul C, Cather J, Gooderham M, Poulin Y, Mrowietz U, Ferrandiz C, et al. Efficacy and safety of apremilast, an oral phosphodiesterase 4 inhibitor, in patients with moderate to severe plaque psoriasis over 52 weeks: a phase III, randomized, controlled trial (ESTEEM 2). Br J Dermatol. 2015;173(6):1387–99.
Stein Gold L, Bagel J, Lebwohl M, Jackson JM, Chen R, Goncalves J, et al. Efficacy and safety of apremilast in systemic- and biologic-naive patients with moderate plaque psoriasis: 52-week results of UNVEIL. J Drugs Dermatol. 2018;17(2):221–8.
Strober B, Bagel J, Lebwohl M, Stein Gold L, Jackson JM, Chen R, et al. Efficacy and safety of apremilast in patients with moderate plaque psoriasis (UNVEIL phase IV study) [poster 4892]. Annual Meeting of the American Academy of Dermatology; March 3–7, 2017; Orlando, FL.
Hatemi G, Mahr A, Ishigatsubo Y, Song YW, Takeno M, Kim D, et al. Trial of apremilast for oral ulcers of Behçet’s syndrome. N Engl J Med. 2019;381(20):1918–28.
Schafer PH, Chen P, Fang L, Wang A, Chopra R. The pharmacodynamic impact of apremilast, an oral phosphodiesterase 4 inhibitor, on circulating levels of inflammatory biomarkers in patients with psoriatic arthritis: substudy results from a phase III, randomized, placebo-controlled trial (PALACE 1). J Immunol Res. 2015;2015: 906349.
Hamzaoui K, Hamzaoui A, Guemira F, Bessioud M, Hamza M, Ayed K. Cytokine profile in Behçet’s disease patients: relationship with disease activity. Scand J Rheumatol. 2002;31(4):205–10.
Yamamoto T. Extra-palmoplantar lesions associated with palmoplantar pustulosis. J Eur Acad Dermatol Venereol. 2009;23(11):1227–32.
Fujisawa T, Tawada C, Mizutani Y, Doi T, Yoshida S, Ogura S, et al. Efficacy of granulocyte and monocyte adsorption apheresis for treatment of palmoplantar pustulosis. Ther Apher Dial. 2014;18(3):238–43.
Basra MK, Salek MS, Camilleri L, Sturkey R, Finlay AY. Determining the minimal clinically important difference and responsiveness of the Dermatology Life Quality Index (DLQI): further data. Dermatology. 2015;230(1):27–33.
Crocetti L, Floresta G, Cilibrizzi A, Giovannoni MP. An overview of PDE4 inhibitors in clinical trials: 2010 to early 2022. Molecules. 2022;27(15):4964.
Strober B, Alikhan A, Lockshin B, Shi R, Cirulli J, Schafer P. Apremilast mechanism of efficacy in systemic-naive patients with moderate plaque psoriasis: pharmacodynamic results from the UNVEIL study. J Dermatol Sci. 2019. https://doi.org/10.1016/j.jdermsci.2019.09.00.
Garcet S, Nograles K, Correa da Rosa J, Schafer PH, Krueger JG. Synergistic cytokine effects as apremilast response predictors in patients with psoriasis. J Allergy Clin Immunol. 2018;142(3):1010-3.e6.
Skov L, Beurskens FJ, Zachariae CO, Reitamo S, Teeling J, Satijn D, et al. IL-8 as antibody therapeutic target in inflammatory diseases: reduction of clinical activity in palmoplantar pustulosis. J Immunol. 2008;181(1):669–79.
Fukasawa T, Yoshizaki-Ogawa A, Enomoto A, Miyagawa K, Sato S, Yoshizaki A. Involvement of molecular mechanisms between T/B cells and IL-23: from palmoplantar pustulosis to autoimmune diseases. Int J Mol Sci. 2022;23(15):8261.
Murakami M, Kaneko T, Nakatsuji T, Kameda K, Okazaki H, Dai X, et al. Vesicular LL-37 contributes to inflammation of the lesional skin of palmoplantar pustulosis. PLoS ONE. 2014;9(10): e110677.
Pincelli C, Schafer PH, French LE, Augustin M, Krueger JG. Mechanisms underlying the clinical effects of apremilast for psoriasis. J Drugs Dermatol. 2018;17(8):835–40.
Mrowietz U, Bachelez H, Burden AD, Rissler M, Sieder C, Orsenigo R, et al. Secukinumab for moderate-to-severe palmoplantar pustular psoriasis: results of the 2PRECISE study. J Am Acad Dermatol. 2019;80(5):1344–52.
Terui T, Kobayashi S, Okubo Y, Murakami M, Zheng R, Morishima H, et al. Efficacy and safety of guselkumab in Japanese patients with palmoplantar pustulosis: a phase 3 randomized clinical trial. JAMA Dermatol. 2019;155(10):1153–61.
Van Zander J, Orlow SJ. Efficacy and safety of oral retinoids in psoriasis. Expert Opin Drug Saf. 2005;4(1):129–38.
Reitamo S, Erkko P, Remitz A, Lauerma AI, Montonen O, Harjula K. Cyclosporine in the treatment of palmoplantar pustulosis. A randomized, double-blind, placebo-controlled study. Arch Dermatol. 1993;129(10):1273–9.
Erkko P, Granlund H, Remitz A, Rosen K, Mobacken H, Lindelöf B, et al. Double-blind placebo-controlled study of long-term low-dose cyclosporin in the treatment of palmoplantar pustulosis. Br J Dermatol. 1998;139(6):997–1004.
Fujita H, Terui T, Hayama K, Akiyama M, Ikeda S, Mabuchi T, et al. Japanese guidelines for the management and treatment of generalized pustular psoriasis: the new pathogenesis and treatment of GPP. J Dermatol. 2018;45(11):1235–70.
Berth-Jones J, Exton LS, Ladoyanni E, Mohd Mustapa MF, Tebbs VM, Yesudian PD, et al. British Association of Dermatologists guidelines for the safe and effective prescribing of oral ciclosporin in dermatology 2018. Br J Dermatol. 2019;180(6):1312–38.
West J, Ogston S, Foerster J. Safety and efficacy of methotrexate in psoriasis: a meta-analysis of published trials. PLoS ONE. 2016;11(5): e0153740.
Kato N, Takama H, Ando Y, Yanagishita T, Ohshima Y, Ohashi W, et al. Immediate response to apremilast in patients with palmoplantar pustulosis: a retrospective pilot study. Int J Dermatol. 2021;60(5):570–8.
Freitas E, Rodrigues MA, Torres T. Diagnosis, screening and treatment of patients with palmoplantar pustulosis (PPP): a review of current practices and recommendations. Clin Cosmet Investig Dermatol. 2020;13:561–78.
Wilsmann-Theis D, Kromer C, Gerdes S, Linker C, Magnolo N, Sabat R, et al. A multicentre open-label study of apremilast in palmoplantar pustulosis (APLANTUS). J Eur Acad Dermatol Venereol. 2021;35(10):2045–50.
Acknowledgements
Writing support was funded by Amgen Inc. and provided by Rebecca Lane, PhD, of Peloton Advantage, LLC, an OPEN Health company, and Dawn Nicewarner, PhD, employee of and stockholder in Amgen Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was sponsored by Amgen Inc.
Conflicts of interest
Tadashi Terui has received research grants, consulting fees, and/or speaker’s fees from AbbVie, Amgen, Boehringer Ingelheim, Eli Lilly, Eisai, Kyowa Hakko Kirin, LEO Pharma, Maruho, Mitsubishi Tanabe, Novartis, and Taiho Pharmaceutical. Yukari Okubo has received grants or contracts from AbbVie, Eisai, Jimro, Maruho, Shiseido, Sun Pharma, and Torii; consulting fees from AbbVie, Amgen, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Eisai, Eli Lilly, Janssen Pharma, Kyowa Kirin, LEO Pharma, Maruho, Pfizer, Sun Pharma, and UCB Pharma; and honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events from AbbVie, Amgen, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Eisai, Eli Lilly, Janssen Pharma, JIMRO, Kyowa Kirin, LEO Pharma, Maruho, Novartis Pharma, Pfizer, Sanofi, Sun Pharma, Taiho, Tanabe-Mitsubishi, Torii and UCB Pharma. Satomi Kobayashi has received research grants from Kyowa Kirin; received honoraria from AbbVie, Eli Lilly, Janssen Pharma, Maruho Pharmaceutical, Novartis, and Taiho Pharmaceutical; and participated in clinical trials for AbbVie, Amgen, Boehringer Ingelheim, Celgene, Eli Lilly, Janssen Pharma, Kyowa Kirin, Maruho Pharmaceutical, and Pfizer. Shigetoshi Sano has received research grants from Kaken, Maruho, Nihon, Nippon Zoki, Sanofi, Taiho, and Torii; and honoraria from AbbVie, Eisai, Eli Lilly, Janssen Pharma, Kyowa Kirin, Maruho, Sun Pharma, Taiho, and UCB. Akimichi Morita has received research grants, consulting fees, and/or speaker’s fees from AbbVie, Amgen, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Eli Lilly, Eisai, Janssen, Kyowa Hakko Kirin, LEO Pharma, Maruho, Mitsubishi Tanabe, Nichi-Iko, Nippon Kayaku, Novartis, Pfizer, Sun Pharma, Taiho Pharmaceutical, Torii Pharmaceutical, Ushio, and UCB Pharma. Shinichi Imafuku has received grants and/or personal fees from AbbVie, Amgen Inc., Boehringer Ingelheim, Bristol Myers Squibb, Daiichi-Sankyo, Eisai, Eli Lilly, GSK, Janssen, Kyowa Kirin, LEO Pharma, Maruho, Novartis, Sun Pharma, Taiho Pharmaceutical, Tanabe-Mitsubishi, Torii Pharmaceutical, and UCB Japan. Yayoi Tada has received honoraria and/or grants from AbbVie, Boehringer Ingelheim, Bristol Myers Squibb KK, Celgene, Eisai, Eli Lilly, Janssen, Kyowa Kirin, LEO Pharma, Maruho, Meiji Seika Pharma, Mitsubishi Tanabe Pharma, Novartis Pharma, Sun Pharma, Taiho Pharmaceutical, Torii Pharmaceutical, and UCB Pharma. Masatoshi Abe has received research grants, consulting fees, speaker fees, and/or participated in clinical trials for Celgene and Maruho. Masafumi Yaguchi, Natsuka Uehara, Takahiro Handa, and Masayuki Tanaka are employees of Amgen K.K. Wendy Zhang and Maria Paris are employees and stockholders of Amgen Inc. Masamoto Murakami has received research grants from AbbVie, ARISTEA Therapeutics, Eisai, Eli Lilly, Kyowa Kirin, and Novartis Pharma; honoraria from AbbVie, Amgen Inc., Boehringer Ingelheim, Celgene, Eisai, Eli Lilly, Janssen Pharma, Kyowa Kirin, Maruho, Novartis Pharma, Taiho Pharmaceutical, and Torii Pharmaceutical; and participated in clinical trials for AbbVie, Amgen Inc., Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Eli Lilly, Janssen Pharma, Maruho, and Novartis Pharma.
Ethics approval
This study was conducted in accordance with International Council for Harmonisation E6 and the ethical principles that are outlined in the Declaration of Helsinki. The study protocol and all amendments, the informed consent form, and any accompanying materials provided to the patients were reviewed and approved by an institutional review board or independent ethics committee at each study center.
Consent to participate
Patients provided written informed consent prior to study procedures.
Consent for publication
Not applicable.
Availability of data and material
Qualified researchers may request data from Amgen clinical studies. Complete details are available at http://www.amgen.com/datasharing.
Code availability
Not applicable.
Author contributions
Conceptualization: TT, YO, SK, MY, NU, WZ, MP, MM. Patient data collection: TT, YO, SK, SS, AM, SI, YT, MA, MY, NU, MM. Analysis and interpretation of data: TT, YO, SK, SS, AM, YT, MY, TH, MT, WZ, MP, MM.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Terui, T., Okubo, Y., Kobayashi, S. et al. Efficacy and Safety of Apremilast for the Treatment of Japanese Patients with Palmoplantar Pustulosis: Results from a Phase 2, Randomized, Placebo-Controlled Study. Am J Clin Dermatol 24, 837–847 (2023). https://doi.org/10.1007/s40257-023-00788-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40257-023-00788-2