
Abstract
Electrochemical reduction of CO2 is an effective method for storing intermittent renewable energy. This could result in fuel additives and chemical feedstocks such as alcohols. A challenge of electrochemical alcohol production is the transfer of electrons and protons, as well as the formation of C–C bonds. As of now, copper-based materials are the most commonly used and effective catalysts. Although CuOx is considered a promising catalyst for electrochemical CO2 reduction reactions (CO2RR), significant improvements in product selectivity are still needed. This paper presents some results obtained using copper oxide as a cathode, combined with 33% of ionomer, nickel iron as anode, and membrane Fumatech as electrolyte. As a result of physico-chemical experiments, morphological measurements of the cathode, electrochemical experiments carried out with a complete zero-gap cell operating under alkaline conditions, and gas-chromatographic (GC) analyses of the cathode outlet stream, we determined that methyl formate, ethanol, and propanol were mainly obtained at a rate of 116.3 μmol \({\text{g}}_{\text{cat}}^{-1} \, {\text{h}}^{-{1}}\) during operation at 2.2 V.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
To combat global warming caused by increasing carbon dioxide (CO2) emissions into the atmosphere, technological solutions for their removal are urgently needed [1, 2]. One of the greatest technological challenges of our time is the production of fuels and chemicals from carbon dioxide [3, 4]. It is possible to achieve this goal using electrochemical processes at room temperature and pressure [5, 6]. By using renewable energy to feed an electrochemical cell, carbon dioxide and water can be converted into “green fuels”, reducing CO2 emissions and storing renewable energy [3, 7]. Through a mechanism involving electron transfer, an electrochemical cell containing an active electrocatalyst at the cathode reduces CO2 into organic products. It is currently challenging to control which products could be achieved through this mechanism. Several reduction products can be formed in aqueous and non-aqueous based electrochemical devices, including methanol, formic acid, methane, and carbon monoxide [8, 9]. Due to the fact that one of the most promising electrochemical cells uses alkaline conditions, novel, hierarchical materials made of non-noble and noncritical raw materials (CRM-free) have recently gained attention [10, 11]. Among the most commonly used electrocatalysts for electrochemical CO2 reduction in alkaline conditions is copper, which electrochemically converts CO2 into more than 30 products, including hydrocarbons and alcohols [9, 12,13,14,15,16,17,18,19]. Specific studies evidenced that Cu-based electrocatalysts can enhance CO dimerization to promote the formation of C2 products [18, 20,21,22,23,24]. According to the literature, product distribution is influenced by morphology and the exposed face of Cu, although applied potential and electrolytes also play an important role [20, 25, 26]. While CO2 is being reduced, copper catalysts produce ethylene rather than ethanol or n-propanol [10, 27]. In a specific experiment conducted using Cu-based electrocatalysts, Roy et al. reported that n-propanol might be one of the most important C3 products [28]. Consequently, there is a great deal of motivation to develop Cu-derived oxide electrocatalysts so that C2 or higher products can be produced with high Faradaic efficiency.
Moreover, alkaline conditions improve oxygen evolution kinetics because less energy is required in comparison with acidic conditions. In addition, CO2 solubility in alkaline conditions could contribute to its easier reduction, since the reaction involves a lower order of reaction [29, 30]. In spite of this, alkaline electrolytes suffer from several disadvantages, including a lower ionic conductivity than H+ electrolytes and the need to recirculate caustic solutions [31,32,33,34]. The purpose of this manuscript is to examine the role of CuOx mixed with carbon in the cathodic reduction of CO2 in a single cell of 5 cm2 and under different cell potentials to evaluate the quality of products and the efficiency of their production [35,36,37,38]. Experiments were conducted at 1.8 V and 2.2 V cell voltages with 1 M KOH circulating on the anode as a refilling electrolyte, while the liquid phase in the outlet stream from the cathode was collected and analysed with gas chromatography (GC).
Materials and methods
Copper-based cathode electrocatalyst was prepared by the oxalate method (Patent WO2004049491), described in detail in our previous paper [19]. In brief, Cu nitrate was dissolved in distilled water and mixed with a solution of oxalic acid neutralized with NaOH at pH 6.5. The metal and chelating agent were mixed at a molar ratio of 10. The metal complex was formed and then decomposed at 80 °C with hydrogen peroxide to obtain a precipitate. This precipitate was then filtered, washed, and dried at 100 °C for 24 h. Afterward, the raw powders were calcined at 350 degrees Celsius for 120 min, then milled for 6 h with Ketjenblack (KB 600) carbon in the ratio 70:30 (metal oxide:carbon).
As part of the physicochemical characterization of the cathode electrocatalyst, transmission electron microscopy was used to study the morphology of the material at nanoscale magnification (TEM-FEI CM12 fitted with a LaB6 filament). The structure and crystallinity of the cathode electrocatalyst were determined using a D8 Advance diffractometer (Bruker AXS, Germany) equipped with Ni β-filtered Cu–Kα radiation (λ = 1.5406 Å), based on the following experimental parameters: 2θ range 20°–100°, 40 kV and 40 mA, scan step of 0.03° s−1.
A complete cell architecture was used to conduct electrochemical experiments. First, anode and cathode inks were prepared. The anode for all electrochemical experiments was NiFeOx, which has already been thoroughly characterized and described in our previous paper [11]. The inks contained 33 wt% anionic ionomer (i.e. Fumatech) and 67 wt% electrocatalyst. These materials were dispersed and sonicated in ethanol for 30 min before being used to manufacture electrodes by spray coating technique. Gas diffusion layer (GDL) purchased from Sigracet was used as support for the cathode and nickel felt purchased from Bekaert for the anode. This procedure resulted in a total catalyst loading of 2.5 mg cm−2 for the anode and 6 mg cm−2 for the cathode. A Fumatech (FAA3-50) anion exchange membrane was used as a polymer electrolyte [39]. After anode and cathode electrodes were cold assembled with electrolyte in the middle, the membrane electrode assembly (MEA) was completed.
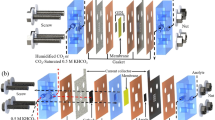
A setup cell with an active area of 5 cm2 is shown in Fig. 1. This apparatus was designed for electrolysis of CO2 in an alkaline environment with 1 M KOH recirculated at the anode, as described in this paper. Figure 1a illustrates the inlet and outlet streams of each electrode, in addition to the wire connections and materials placed within the housing. The cryogenic system shown in Fig. 1c was used to collect liquid from the cathode side.

Setup for electrochemical cell testing. a Cell housing, b connection between test bench and cell housing, c cryogenic system for capturing the cathode outlet stream
In this setup, a nickel housing maintained at 50 °C was used to conduct electrochemical experiments on MEAs. A peristaltic pump was used to circulate a KOH solution (1 M) at a rate of 5 ml min−1 in the anode compartment. The cathode was fed with humidified CO2 at a rate of 50 ml min−1 after being preheated to cell temperature. Keithley power supply equipment (Tektronic, Rome, Italy) was used to conduct electrochemical investigations (polarization curves versus current density, I–V curves) and chronoamperometry. A gas chromatographic analysis of the cathodic outlet was performed during endurance testing to identify electrochemically produced organic soluble species. For this purpose, the liquid was entrapped in a vial maintained in a cryogenic apparatus at a temperature below 1 °C. These analyses were performed using a GC model Agilent 7890 A. The instrument included a capillary column (30 m by 0.53 mm by 0.2 m film thickness), model Supelcowax 10 (Merck KGaA, Darmstadt, Germany) connected directly to a flame ionization detector. Helium was used as a carrier gas for eluting the analytes. To determine the “sensitivity factors” of each compound, the GC was calibrated with standards before use.
Results and discussion
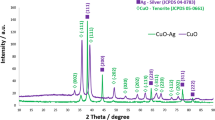
Figure 2 shows the XRD spectrum of fresh CuOx/KB used as a cathode in electrochemical experiments. There are typical crystal reflections of cuprite (Cu2O—JCPDS card no. 05-0667), tenorite (CuO—JCPDS card no. 05-0661), and metallic copper (Cu—JCPDS 04-0836). A semi-quantitative determination of the weight fraction of each phase was performed using native software on XRD equipment. As a result, 30.4 wt% of cuprite, 51.1 wt% of tenorite, and 18.5 wt% of metallic copper were found. Results from an XRD spectrum showed no apparent impurities.

XRD pattern of the cathode electrocatalyst prepared by the oxalate method
Fresh cathode electrocatalyst morphology was investigated using a TEM. As can be seen in Fig. 3, the nanoparticles were well dispersed on the Ketjenblack support. In spite of this, regions with a high particle density exhibit significant agglomeration, which could lead to larger particles. It is very likely that the high copper oxide content of the electrocatalyst (70 wt%) caused the particle distribution observed by microscopy.

TEM image of the fresh cathode
Using an I–V curve for a preliminary diagnosis, the fresh cell showed a linear dependence of V from current density at potentials higher than 1.8 V, mainly due to ohmic constraints (Fig. 5). Under these conditions, the fresh cell appeared to be stable as well as barely affected by activation constraints. Consequently, the cathodic outlet stream was measured at 1.8 V, where I–V profiles significantly changed, and at 2.2 V, where best performance was obtained. The first chronoamperometry test was conducted at 1.8 V (Fig. 4a). Cell performance was quite poor at this voltage, allowing a current density of approximately 90 mA cm−2, which rapidly dropped to approximately 30 mA cm−2. In the liquid collected from the cathode stream after 6 h, methyl formate, ethanol, and propanol were detected (Fig. 4c). The current density was approximately 200 mA cm−2 when the cell was placed at 2.2 V. It took approximately 1 h for the cell to achieve steady state behaviour around a current density of 530 mA cm−2 Chromatographic analysis of the liquid collected from the cathode outlet stream showed a significant increase in methyl formate and propanol, whereas the signal of ethanol remained substantially unchanged (Fig. 4c). It was in agreement with findings from two previous studies published by Kim et al. [10] and Ting and collaborators [27].

Chronoamperometric tests at 1.8 V (a) and 2.2 V (b) and corresponding liquid-phase analyses of cathode outlet streams (c)
Table 1 summarizes the gas-chromatography results for the cathode’s outlet stream. These data demonstrated a significant improvement in the products at 2.2 V, partly due to an additional hour of liquid collection. However, most of this improvement can be attributed to the significant increase in current density by cell operation at 2.2 V. On the other hand, despite a one order of magnitude increase in current density, liquid product formation and relative composition did not increase as expected. According to the literature, electrochemical reduction of CO2 in an alkaline environment results in intermediates such as carbon monoxide (Eq. 1), formic acid (Eq. 2), and ethene (Eq. 3). However, upon combination, or through other means of di- or trimerization, other C2+ final products can be observed. This aspect was highlighted in a study by Ai et al. [40], where these results were theoretically predicted and experimentally confirmed. Based on these preliminary results, a complex mechanism involving intermediates and secondary reactions was observed in the cell, which depended on the voltage and partial pressure of species.
Figure 5 illustrates a comparison of characteristic current–voltage curves collected at various cell life moments.

A comparison of polarization curves measured at various experimental moments
At the beginning of durability tests, there was a very low open circuit potential (OCP) and significant activation overpotential (blue curve). A moderate increase in current density was observed between approximately 1 V and 1.77 V (i.e. 4 mA cm−2 and 50 mA cm−2, respectively). This feature of the curve and slightly improved performance with increased cell potential could be attributed to the multiple Cu oxidation states. Above 50 mA cm−2, the curve feature was quite smooth and controlled by the ohmic mechanism. The electrolysis products were investigated at 1.8 and 2.2 V based on this specific cell behaviour (Fig. 4 and Table 1).
After 6 h of testing at 1.8 V, the characteristic curve (green curve) was determined. This led to an OCP value of 1.25 V in this case. Based on the results of this test, the cell performance was significantly depleted due to high ohmic constraints, probably as a result of partial reoxidation of metallic Cu, or Cu-based reactive sites were occupied by intermediates. In both cases, cathodic electronic percolation could have been adversely affected. After the cell was placed at 2.2 V, its performance gradually improved. As a result of the test, the characteristic I–V curve (red curve) showed the OCP dropping to around 700 mV. In the region where the current density was approximately 30 mA cm−2, the I–V curve was mainly influenced by the activation energies of the processes involved. Despite significant ohmic constraints up to 1.94 V, this cell showed very promising results above this value.
Conclusions
Our study investigated the feasibility of electrolysing CO2 in an alkaline environment to produce liquid added value synthetic fuels. To make this approach feasible, we developed a CRM-free cathode based on a CuOx electrocatalyst for its use in an alkaline zero-gap cell. Experimental results showed that using the oxalate method, we achieved a mixture of Cu-based structures having oxidizing states from 0 to 2. This specimen had a strong tendency to agglomerate. Electrochemical tests combined with liquid effluent analyses confirmed some results reported in the literature concerning the promotion of intermediates that can lead to secondary reactions producing liquid organic fuels. Our findings indicated that the recommended operating cell potential for this device should be above 1.9 V because of the high current density achievable and the quality of products.
Data Availability
This study included data to support its findings.
References
Küngas, R.: Review—electrochemical CO2 reduction for CO production: comparison of low- and high-temperature electrolysis technologies. J. Electrochem. Soc. 167, 044508 (2020)
Qiao, J., Liu, Y., Hong, F., Zhang, J.: A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 43, 631–675 (2014)
Rabinowitz, J.A., Kanan, M.W.: The future of low-temperature carbon dioxide electrolysis depends on solving one basic problem. Nat. Commun. 11, 5231 (2020)
Lee, S., Ocon, J.D., Son, Y.-I., Lee, J.: Alkaline CO2 electrolysis toward selective and continuous HCOO− production over SnO2 nanocatalysts. J. Phys. Chem. C 119, 4884–4890 (2015)
Zhang, W., Hu, Y., Ma, L., Zhu, G., Wang, Y., Xue, X., et al.: Progress and perspective of electrocatalytic CO(2) reduction for renewable carbonaceous fuels and chemicals. Adv Sci (Weinh). 5, 1700275 (2018)
Song, J.T., Song, H., Kim, B., Oh, J.: Towards higher rate electrochemical CO2 conversion: from liquid-phase to gas-phase systems. Catalysts 9, 224 (2019)
Kondratenko, E.V., Mul, G., Baltrusaitis, J., Larrazábal, G.O., Pérez-Ramírez, J.: Status and perspectives of CO2 conversion into fuels and chemicals by catalytic, photocatalytic and electrocatalytic processes. Energy Environ. Sci. 6, 3112–3135 (2013)
Marques da Silva, A.H., Raaijman, S.J., Santana, C.S., Assaf, J.M., Gomes, J.F., Koper, M.T.M.: Reprint of “Electrocatalytic CO2 reduction to C2+ products on Cu and CuxZny electrodes: effects of chemical composition and surface morphology.” J. Electroanal. Chem. 896, 115609 (2021)
Ali, S., Razzaq, A., Kim, H., In, S.-I.: Activity, selectivity, and stability of earth-abundant CuO/Cu2O/Cu0-based photocatalysts toward CO2 reduction. Chem. Eng. J. 429, 131579 (2022)
Kim, D., Kley, C.S., Li, Y., Yang, P.: Copper nanoparticle ensembles for selective electroreduction of CO(2) to C(2)-C(3) products. Proc. Natl. Acad. Sci. USA 114, 10560–10565 (2017)
Campagna Zignani, S., Lo Faro, M., Trocino, S., Aricò, A.S.: Investigation of NiFe-based catalysts for oxygen evolution in anion-exchange membrane electrolysis. Energies 13(7), 1720 (2020)
Albo, J., Sáez, A., Solla-Gullón, J., Montiel, V., Irabien, A.: Production of methanol from CO2 electroreduction at Cu2O and Cu2O/ZnO-based electrodes in aqueous solution. Appl. Catal. B 176–177, 709–717 (2015)
Dong, F., Meng, Y., Han, W., Zhao, H., Tang, Z.: Morphology effects on surface chemical properties and lattice defects of Cu/CeO2 catalysts applied for low-temperature CO oxidation. Sci. Rep. 9, 12056 (2019)
Irfan Malik, M., Malaibari, Z.O., Atieh, M., Abussaud, B.: Electrochemical reduction of CO2 to methanol over MWCNTs impregnated with Cu2O. Chem. Eng. Sci. 152, 468–477 (2016)
Li, D., Huang, L., Liu, T., Liu, J., Zhen, L., Wu, J., et al.: Electrochemical reduction of carbon dioxide to formate via nano-prism assembled CuO microspheres. Chemosphere 237, 124527 (2019)
Liu, P., Liu, H., Zhang, S., Wang, J., Wang, C.: Significant role of reconstructed character on CuO-derived catalyst for enhanced electrocatalytic reduction of CO2 to multicarbon products. Electrochim. Acta 354, 136753 (2020)
Wang, X., Klingan, K., Klingenhof, M., Möller, T., Ferreira de Araújo, J., Martens, I., et al.: Morphology and mechanism of highly selective Cu(II) oxide nanosheet catalysts for carbon dioxide electroreduction. Nat. Commun. 12, 794 (2021)
Zahid, A., Shah, A., Shah, I.: Oxide derived copper for electrochemical reduction of CO(2) to C(2+) products. Nanomaterials (Basel) 12, 1380 (2022)
Zignani, S.C., Lo Faro, M., Palella, A., Spadaro, L., Trocino, S., Lo Vecchio, C., et al.: Bifunctional CuO-Ag/KB catalyst for the electrochemical reduction of CO2 in an alkaline solid-state electrolysis cell. Catalysts 12, 293 (2022)
Hongrutai, N., Watmanee, S., Pinthong, P., Panpranot, J.: Electrochemical reduction of carbon dioxide on the oxide-containing electrocatalysts. J. CO2 Util. 64, 102194 (2022)
Zhang, L., Merino-Garcia, I., Albo, J., Sánchez-Sánchez, C.M.: Electrochemical CO2 reduction reaction on cost-effective oxide-derived copper and transition metal–nitrogen–carbon catalysts. Curr. Opin. Electrochem. 23, 65–73 (2020)
Zhu, Q., Sun, X., Yang, D., Ma, J., Kang, X., Zheng, L., et al.: Carbon dioxide electroreduction to C2 products over copper-cuprous oxide derived from electrosynthesized copper complex. Nat. Commun. 10, 3851 (2019)
Giusi, D., Miceli, M., Genovese, C., Centi, G., Perathoner, S., Ampelli, C.: In situ electrochemical characterization of CuxO-based gas-diffusion electrodes (GDEs) for CO2 electrocatalytic reduction in presence and absence of liquid electrolyte and relationship with C2+ products formation. Appl. Catal. B 318, 121845 (2022)
Stalinraja, A., Gopalram, K., Venkatesan, S., Swamynathan, M.J., Ghosh, S., Selvaraj, T.: Electrochemical reduction of CO2 on Cu doped titanium nanotubes—an insight on ethylene selectivity. Electrochim. Acta. 431, 141078 (2022)
Rahaman, M., Dutta, A., Zanetti, A., Broekmann, P.: Electrochemical reduction of CO2 into multicarbon alcohols on activated Cu mesh catalysts: an identical location (IL) study. ACS Catal. 7, 7946–7956 (2017)
Schizodimou, A., Kyriacou, G.: Acceleration of the reduction of carbon dioxide in the presence of multivalent cations. Electrochim. Acta 78, 171–176 (2012)
Ting, L.R.L., García-Muelas, R., Martín, A.J., Veenstra, F.L.P., Chen, S.T.-J., Peng, Y., et al.: Electrochemical reduction of carbon dioxide to 1-butanol on oxide-derived copper. Angew. Chem. Int. Ed. 59, 21072–21079 (2020)
Roy, A., Jadhav, H.S., Park, S.J., Seo, J.G.: Recent advances in the possible electrocatalysts for the electrochemical reduction of carbon dioxide into methanol. J. Alloy. Compd. 887, 161449 (2021)
Bandal, H.A., Jadhav, A.R., Kim, H.: Facile synthesis of bicontinuous Ni3Fe alloy for efficient electrocatalytic oxygen evolution reaction. J. Alloy Compd. 726, 875–884 (2017)
Pletcher, D., Li, X.: Prospects for alkaline zero gap water electrolysers for hydrogen production. Int. J. Hydrogen Energy 36, 15089–15104 (2011)
Aili, D., Kraglund, M.R., Tavacoli, J., Chatzichristodoulou, C., Jensen, J.O.: Polysulfone-polyvinylpyrrolidone blend membranes as electrolytes in alkaline water electrolysis. J. Membr. Sci. 598, 117674 (2020)
Davydova, E.S., Mukerjee, S., Jaouen, F., Dekel, D.R.: Electrocatalysts for hydrogen oxidation reaction in alkaline electrolytes. ACS Catal. 8, 6665–6690 (2018)
Dionigi, F., Strasser, P.: NiFe-based (oxy)hydroxide catalysts for oxygen evolution reaction in non-acidic electrolytes. Adv. Energy Mater. 6, 1600621 (2016)
Kevin, K.: Unstable cathode potential in alkaline flow cells for CO2 electroreduction driven by gas evolution. ACS Sustain. Chem. Eng. 9, 5570–5579 (2021)
Peterson, A.A., Abild-Pedersen, F., Studt, F., Rossmeisl, J., Nørskov, J.K.: How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 3, 1311–1315 (2010)
Benson, E.E., Kubiak, C.P., Sathrum, A.J., Smieja, J.M.: Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem. Soc. Rev. 38, 89–99 (2009)
Li, C.W., Kanan, M.W.: CO2 reduction at low overpotential on Cu electrodes resulting from the reduction of thick Cu2O films. J. Am. Chem. Soc. 134, 7231–7234 (2012)
Luo, H., Li, B., Ma, J.G., Cheng, P.: Surface modification of nano-Cu(2) O for controlling CO(2) electrochemical reduction to ethylene and syngas. Angew. Chem. Int. Ed. Engl. 61, e202116736 (2022)
Gatto I, Caprì A, Lo Vecchio C, Zignani S, Patti A, Baglio V. Optimal operating conditions evaluation of an anion-exchange-membrane electrolyzer based on FUMASEP® FAA3–50 membrane. International Journal of Hydrogen Energy. 2022.
Ai, L., Ng, S.-F., Ong, W.-J.: Carbon dioxide electroreduction into formic acid and ethylene: a review. Environ. Chem. Lett. 20, 3555–3612 (2022)
Acknowledgements
The authors acknowledge the financial support provided by the EU Horizon 2020 Framework Programme “LOTERCO2M” project “CRM-free Low Temperature Electrochemical Reduction of CO2 to Methanol” Grant Agreement number: 761093 and by the EU H2020 GREEN DEAL “ECO2FUEL” project “Large-scale low-temperature electrochemical CO2 conversion to sustainable liquid fuels” Grant Agreement number: 101037389.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zignani, S.C., Lo Faro, M., Carbone, A. et al. Alkaline electrolysis using CuOx cathode for the conversion of carbon dioxide into liquid fuels. Mater Renew Sustain Energy 12, 141–146 (2023). https://doi.org/10.1007/s40243-023-00235-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40243-023-00235-6