
Abstract
Direct carbon fuel cell (DCFC) is a promising technology with high energy efficiency and abundant fuel. To date, a variety of DCFC configurations have been investigated, with molten hydroxide, molten carbonate or oxides being used as the electrolyte. Recently, there has been particular interest in DCFC with molten carbonate involved. The molten carbonate is either an electrolyte or a catalyst in different cell structures. In this review, we consider carbonate as the clue to discuss the function of carbonate in DCFCs, and start the paper by outlining the developments in terms of molten carbonate (MC)-based DCFC and its electrochemical oxidation processes. Thereafter, the composite electrolyte merging solid carbonate and mixed ionic–electronic conductors (MIEC) are discussed. Hybrid DCFC (HDCFCs ) combining molten carbonate and solid oxide fuel cell (SOFC) are also touched on. The primary function of carbonate (i.e., facilitating ion transfer and expanding the triple-phase boundaries) in these systems, is then discussed in detail. Finally, some issues are identified and a future outlook outlined, including a corrosion attack of cell components, reactions using inorganic salt from fuel ash, and wetting with carbon fuels.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Fuel cells (FCs) offer an environmentally friendly and highly efficient approach to energy-conversion technology for power generation. This approach has been intensively investigated for over a hundred years, since Grove et al. [1] developed the first FC in 1839. This technology is steadily evolving and improving, as the demand for electricity and new electricity generation technologies continues to grow. The FC device generates electric power from gaseous fuels (hydrogen or syngas) by reforming liquefied petroleum gas or liquid fuels. However, the complex synthesis process and the storage and transportation requirements increase the cost. At present, the utilization of carbon fuels is still a preferred option for the development of FCs, due to the abundant resources, easy access and relatively high-energy density. Direct carbon fuel cells (DCFCs), as an energy-conversion technology, generate electricity from solid carbon fuels through electrochemical reactions [2, 3]. The device has a higher energy-conversion efficiency rate than traditional power generation devices of heat engines, which minimize system complexity and thus ensure a lower cost [4].
The first DCFC was reported by Becquerel [5] in the middle of the nineteenth century; the configuration was a carbon rod in a KNO3 solution inside a platinum container. In 1896, William Jacques [6] developed a large-scale DCFC using 100 single cells, and baked coal as the anode. The electrolyte was an unspecified mixture of KOH and/or NaOH and the cathode was iron pots. Initially, the actual reaction in this system was only deemed to be the chemical reaction of carbon with nitrate [7]. Later research reported that gases from solid carbon pyrolysis are involved in the electrochemical reaction [8, 9]. Other researchers from Stanford Research Institute (SRI, International) investigated this technology thoroughly and invented the actual devices for electricity generation by the electrochemical oxidation of carbon [10, 11].
DCFC technology is again receiving interest, especially in terms of the direct carbon oxidation mechanism. Gür [12, 13] proposed that it is appropriate to name the carbon fuel cell a “direct” carbon fuel cell if the reaction is achieved in a single procedure or a single chamber. This proposal is based on the simplified reactions in the anode. However, it is much more complicated in practice. Generally, more than one reaction happens in the anode simultaneously. Among those reactions, carbon is often oxidized into carbon monoxide as an intermediate product, and then CO is oxidized into CO2 as the final product. We use the term “direct carbon fuel cells” for most carbon fuel cells to be consistent with most publications.
One advantage of DCFC is the emission of pure CO2, unlike other coal-fired power plants, which reduce the technical difficulties of CO2 capture and separation [14]. Notably, the DCFC power station has a modular structure design, which can be adjusted regarding the cost [15]. The direct utilization of various solid carbonaceous materials as fuels is the most distinctive feature of DCFC. Some solid carbon fuels, like graphite, carbon black [16,17,18,19,20,21,22,23] and different types of coal fuel [24,25,26,27,28,29] have been used in various DCFC configurations.
The carbonaceous materials obtained from renewable resources (e. g., biomass and organic waste) are sustainable fuels due to abundant, cheap and natural characteristics [30]. In recent decades, many researchers have focused on the applications of biochar in the environment and catalysis fields [31,32,33,34]. Biomass and municipal waste are also suggested as raw materials to produce clean energy in DCFCs [35,36,37]. Bio-char is suitable in DCFC for electric energy generation because the biochar obtained by pyrolysis is amorphous, which is conducive to the exposure of carbon fuel surface active sites. Another feature is the high porosity of this kind of biochar, promoting gas transportation in the reaction process [38]. Furthermore, some natural metal ions disperse uniformly in biochar, which are natural catalysts for the carbon gasification reaction [39]. It is worth mentioning that the efficiency of biomass-fueled DCFC is generally 50–60%, while it is up to 80% if heat and electricity co-generation is applied [40].
There are three types of DCFC; these are categorized according to the electrolyte material, i.e., hydroxide, carbonate and solid oxide. Of these, the molten carbonate direct carbon fuel cell (MC-DCFC) was one of the earliest cells, and it has been widely studied [41]. The molten carbonate also remains stable compared to hydroxide-based cells, even in an environment rich in CO2, which helps to prevent electrolyte damage [42, 43]. Recently, an investigation was done on hybrid direct carbon fuel cells (HDCFCs), using mixed solid carbon and carbonate in the anode [21]. This overcomes corrosion issues associated with the molten carbonate fuel cell (MCFC) and yields slightly faster kinetics compared to the solid oxide fuel cell (SOFC) system [21].
Some reviews have been done in DCFC, with the earliest reviews of DCFC done by Howard [44] and Liebhafsky [45]. Later, Cao et al. [46, 47] and Giddey et al. [48] summarized the fundamental electrochemical performance and the developments of DCFC technology. The reaction mechanism of direct carbon oxidation and the conversion was explained by Cooper et al. [49] and Gür [13, 50]. Then, Rady et al. [51] reviewed the performance of various fuels used in MCFCs and SOFCs, and Zhou et al. [52] published a review paper that discussed the anode used in DCFCs. Recently, Jiang et al. [53] presented an overview of the impact of different parameters on the resistance and power output and the electrochemical behavior of DCFCs, and also summarized the challenges associated with developing DCFCs. Glenn et al. [54] reviewed the carbon electro-catalysis mechanism of alkali metal molten carbonates in DCFCs.
This paper focuses on providing a broad and timely summary of the carbonate-based DCFC: MC-DCFC, CO32− + mixed ionic–electronic conductors (MIEC) and CO32− + SOFC. The configurations of three cell systems and some possible reactions related to carbonate are shown in Fig. 1. This review starts with an introduction to molten carbonate species. Then, the fundamental mechanisms of different types of DCFC are provided, with the focus being on the functions of carbonate: (1) transport ions as an electrolyte; (2) catalyze carbon oxidation as a catalyst; (3) enlarge the reaction area, that is, the anode/ electrolyte reaction interface zone. The review concludes with a synopsis of some issues regarding corrosion, compatibility and wettability, and a possible future outlook regarding the utilization of carbonate in DCFC.

Schematic of a DCFC in the presence of carbonate, including three cell systems (MC-DCFC, CO32− + MIEC and CO32− + SOFC) and some possible reactions. The primary reaction between carbonate and carbon is shown in light blue; the electro-catalytic reaction of carbonate with carbon is shown in dark blue; and the possible side reaction is shown in green
Species of molten carbonate in DCFC
Molten carbonate is commonly used in MCFC, and has received wide attention in DCFC [4, 49]. It shows good compatibility with CO2 when used as an electrolyte [55, 56], and provides excellent ionic conductivity at a relatively low temperature [57]. Furthermore, the molten carbonate in the anode chamber can significantly enlarge triple-phase boundaries (TPBs), which favors ion diffusion to the electrochemical reaction sites [21]. Many studies have shown that doping alkali metal ions in molten carbonate can effectively accelerate the reaction rate of carbon gasification in a CO2 gas stream [58,59,60]. In particular, potassium salt delivers high catalytic activity for the carbon gasification reaction [61].
The operating temperature of DCFCs is reduced by changing the carbonate composition while maintaining cell performance, which is another positive factor when using molten carbonate as the electrolyte [62, 63]. A suitable amount of other carbonates or oxides can also further reduce the melting temperature of the carbonate [64]. For example, adding an appropriate amount of Ru2CO3 and SrCO3 to a binary carbonate reduces the surface tension of molten carbonate, which, in turn, increases the solubility of gas and reduces the melting temperature [65]. Therefore, using carbonate with different components in DCFC is discussed in detail.
Binary
Binary carbonate eutectic has been widely used as an electrolyte material in FCs. It is well known that a lower melting point is obtained with a binary carbonate eutectic (Li2CO3–Na2CO3, Li2CO3–K2CO3, K2CO3–Na2CO3) than with a single carbonate [66, 67]. Of the three binary carbonates, Li–K carbonate has the lowest melting point (below 550 °C). Therefore, it is more suitable for use as an electrolyte for the cell. However, an early study suggested that a Li–Na carbonate eutectic exhibited certain benefits in comparison with a Li–K carbonate eutectic, such as a higher ionic conductivity (the ionic conductivity of 43.5 mol% Li2CO3-31.5 mol% Na2CO3-25 mol% K2CO3, 52 mol% Li2CO3-48 mol% K2CO3 and 72 mol% Li2CO3-28 mol% Na2CO3 at 600 °C is 1.20 S cm−1, 1.15 S cm−1, and 1.79 S cm−1, respectively) [68]. The Li–Na carbonate eutectic has a lower dissolution rate of in the cathode (NiO) than that of Ni generated by NiO reduction, and it is quickly oxidized to nickel oxide [69].
The cell life of different carbonate components is predicted based on the evaporated carbonate lost. Li–Na carbonate has a greater projected lifetime of 10.3 × 105 h at 600 °C, compared to the 6.4 × 105 h for Li-Na–K carbonate and the 6.3 × 105 h for Li–K carbonate [70]. Therefore, Li–Na carbonate has often been used as an electrolyte in MCFC to substitute the Li–K carbonate. Nevertheless, there may be a risk of a rapid decrease in cell voltage with Li–Na carbonate at atmospheric pressure and low temperature (≤ 600 °C) because of the relatively low solubility of oxygen in this eutectic [71]. Na–K carbonate eutectic showed the worst cell performance of the various binary carbonate-based DCFCs, probably due to its high melting temperature [72].
One investigation showed that, when using sawdust biofuel, HDCFC with a Li–K carbonate eutectic as the medium in anode showed excellent cell performance (789 mW cm−2) at 750 °C [46]. The low melting point of Li–K carbonate is one of the influencing factors on excellent cell performance because the viscosity of its solution can be reduced when the temperature reaches the melting point, thereby improving the diffusion of solid fuel in the medium. And also, Li–K carbonate is a good catalyst for gasification reactions [22]. Table 1 provides the melting temperatures of the common carbonates.
Ternary
A ternary carbonate eutectic of lithium, sodium and potassium carbonate as the carbon oxidation medium is a potential choice in DCFCs. Initially, the ternary carbonate eutectic was selected as an electrolyte in DCFC due to its good compatibility with unexpected pollutants, such as sulfur and ash contents in coal [10]. Furthermore, compared with the binary carbonate eutectic mixed DCFC, the ternary system (Li2CO3–Na2CO3–K2CO3) has the lowest melting point [73]. (See Table 1.)
Vutetakis et al. [74] employed a ternary carbonate of Li2CO3–K2CO3–Na2CO3 with a weight ratio of 32.1:34.5:33.4 in DCFC with coal as the fuel, and found that it was capable of reducing the cell operating temperature to 500 °C. Jiang et al. [75] also applied a ternary carbonate of Li2CO3, Na2CO3 and K2CO3 in a mole ratio of 43.5:31.5:25 in coal-fueled HDCFC. The cell generated a maximum power density of 50.02 mW cm−2 with an open-circuit voltage (OCV) of 1.1 V at 700 °C.
Carbonate in DCFC
In DCFC, carbonate can contact carbon particles in the electrolyte or anode compartment directly, to catalyze the reaction of carbon oxidation. It can also be used as an electrolyte for ion transportation in MC-DCFC and CO32− + MIEC. The difference is that, in the molten state, carbonate has a catalytic effect on carbon oxidation, due to carbonate mixing with carbon particles in the former system, but carbonate combines with MIEC materials is a solid electrolyte form in the latter. By introducing carbonate in the HDCFC anode, cell performance is improved, possibly due to the catalytic effect on carbon oxidation and the enlargement of the reaction interface. Therefore, it is critical to discuss the role of carbonate in DCFC. Table 2 summarizes some configurations used in carbonate-based DCFCs.
MC-DCFC
MCFC is a commercial fuel cell. Its scale is gradually expanded from kilowatt to megawatt [84]. Besides gaseous fuel, solid carbon is also used as fuel in this cell system. Some progress has been made with developing different cell structures and materials of MC-DCFC, followed by different electrode reaction mechanisms.
Configuration
Baur et al. [85] replaced all the molten hydroxide electrolytes with molten carbonate electrolytes, which opened up the study of MCFCs. In the 1970s, researchers from SRI International first dispersed a carbon particle in a molten Pb using a molten alkali metal carbonate as the electrolyte [86, 87]. Following the first trial, the researchers used coal as the anode in an MC-DCFC with Li–Na–K ternary carbonate electrolyte [10]. Some reports claimed that some alkali metal salts (such as Li2CO3) are regarded as a catalytic agent for accelerating carbon gasification [88, 89], which further suggests the possibility of using carbonate in DCFC. Some researchers from Lawrence Livermore National Laboratory in the United States (LLNL, USA) then made significant progress in MC-DCFC, particularly in terms of the materials related to cathode catalysts, aerogels and xerogel carbon anodes [82,83,84,85,94].
The unconventional tilted design was invented by Cooper et al. [95] using molten carbonate (32 mol% Li2CO3-68 mol% K2CO3) as the electrolyte (See Fig. 2a). The cell cathode is composed of a lithiated NiO, while the anode is made of the carbon-carbonate slurry mixture and the Ni current collector. The key aspect of this device is the electrolyte bed of ZrO2 fabric filled with molten salt that conducts ions while preventing a short circuit of the electrodes. This device presents a 5–45° inclined angle, which favors a continuous fuel supply and facilitates the discharge of excess electrolyte to prevent the electrolyte from flooding the cathode. The cell surface area was expanded from the conventional 2–60 cm2 with no significant polarization loss [48].

Different configurations of MC-DCFC: a Unconventional tilted MC-DCFC favoring the continuous supply of fuel that ensures there is no corrosion of the cathode; b Planar MC-DCFC with the mixture of carbon and carbonate located above the anode to ensure low cell resistance; c MC-DCFC with a stirring rod in the molten carbonate to ensure uniform distribution of carbon particles and improve the mass transfer process inside the cell; d FBEDCFC using the bubbling gas to agitate a mixture of carbon and molten carbonate to accelerate mass and heat transfer; e TMC-DCFC with a closed-end structure that ensures there is no short circuit and that there is a continuous solid fuel supply. Reproduced from refs. [41, 95, 96, 98, 99]
Later, researchers presented another carbon/air molten carbonate cell, as illustrated in Fig. 2b. The composite electrode composed of foam nickel and stainless steel made a great contribution to the stability of the cell structure [41]. This design was called ‘planar MC-DCFC’ due to the mixture of carbon and carbonate located above the anode where the redox reactions occur. A low cell resistance (the resistance of carbon aerogel and the anode current collector was 0.4 Ω cm2) was obtained in this configuration, and therefore improved cell performance.
A schematic of the MC-DCFC prepared by Li et al. [96] is shown in Fig. 2c. The method applied was similar to that used by Vutetakis et al. [74]. This MC-DCFC consists of three electrodes: a working electrode (WE); a gold counter electrode (CE); and a 12 mm diameter alumina sheath that serves as the reference electrode (RE). An Inconel stirring bar is also introduced into the molten carbonate electrolyte to ensure the uniform distribution of carbon particles and improve mass transfer process inside the cell. However, the power density in this configuration is not good enough because the reaction area (fuel/electrode/electrolyte contact area) is severely limited. In this case, the reaction area is only a few square centimeters in the 250 g melt carbonate bath [97].
In an effort to extend the formation zone of TPBs, a fluidized bed cell with a three-dimensional (3D) electrode was adopted by Gür and Huggins [100]. Zhang et al. [98, 101] showed another one with a self-designed fluidized bed electrode anode, as shown in Fig. 2d. In this design, the bubbling gas is applied in DCFC to ensure mass and heat transfer.
Recently, a tubular DCFC with a closed-end structure was conceived by Ido et al. [99], using carbonate as the electrolyte (See Fig. 2e). This design can effectively protect the short circuit between the electrodes using carbon powder. Additionally, the continuous solid fuel supply can be realized through calcinating the anode nickel particles outside the DCFC.
Mechanisms
In the MC-DCFC system, the solid carbon fuel can be oxidized directly to CO2 (as given in Eq. 1). Thereafter, it is circulated to the cathode compartment through the molten carbonate electrolyte to achieve mass balance (as given by Eq. 2). The reactions are detailed below [102, 103].
The total reaction is shown in Eq. (3).
The above equations also indicate that \({\text{P}}_{{\text{CO}}_{\text{2}}}\) could influence the open-circuit voltage (OCV) of the cell, as shown in Eq. (4) [41].
The possible oxidation reactions of carbon in MC-DCFC and the gas produced are shown in Fig. 3, which shows that: the decomposition of carbonate can also produce CO2; it has an influence on the DCFC performance (Eq. 5) [104]. One investigation showed a noticeable increase in over-potential at a higher current density, owing to the mass transfer process being prevented, but it was easy for the released CO2 gas to make contact with the carbon and ions at the anode again, with a long-term discharge recorded [105].

Schematic of the MC-DCFC system: CO2 and O2 gain electrons to produce carbonate ion at the cathode; carbonate ion and carbon generate CO or CO2 and electrons at the anode. At the same time, carbonate ion is likely to decompose into O2− and CO2
Generally, it requires a high operating temperature to enhance the anode reaction rate. Early experimental results show that the predominant product with a carbon anode is CO above 700 °C and this is dependent on the reversal of the Boudouard reaction (as given by Eq. 6). Expected energy from reaction (Eq. 1) is halved, because only two equivalent charges are obtained from one mole of carbon without a significant voltage change at 750 °C.
Other reactions besides the Boudouard reaction are possible, such as partial oxidation of carbon, as shown in Eqs. (7) and (8) [106].
The main product of the carbon anode is identified as CO2 through off-gas analysis [107]. The electrochemical formation of CO is not an insurmountable problem in DCFC, as this reaction in the anode compartment occurs only in the process of cell operation [74]. The OCV value can be diminished or even eliminated if pure CO2 is blown into the anode compartment under the cell when operating above 700 °C, or when the CO2 residue in the open-circuit condition is minimal (Table 2).
Cooper et al. [41, 108] proposed that the mechanisms of carbon oxidation in MCFC are the same as in the Hall process, as given by Eqs. (9–14), while MCFC also forms oxygen ions. The oxygen ions are formed by the ready decomposition of the molten carbonate at the DCFC operating temperature (as given by Eq. 5), which triggers the subsequent oxidation reactions of carbon.
The above reactions indicate that oxidation of carbon monoxide is key to the whole carbon oxidation process, because carbon is easily oxidized to CO with the one-electron transfer. The combination of carbon and O2− is affected by the number and concentration of carbon active sites, depending on the carbon surface area. Li et al. [96] used different carbon sources as the fuel and proposed that the redox reaction rate in carbonate slurry is mainly determined by the crystallinity and surface properties, especially the surface area and the quantity of the carbon surface functional group. These results also verify the rationality of Cherepy's electrochemical mechanism to a certain extent.
Recently, more attention has been paid to tailoring the cell structure to improve the performance of MC-DCFC. Lee et al. [109] proposed that the addition of Gd2O3 to a Ni anode improved cell performance due to the enlarged TPBs and the reduced charge-transfer resistance. They also concluded that the Ni: Gd2O3 = 1:5 anode was an optimal value between the wettability and the electronic conductivity. Then, Lee et al. [78] also reported that the addition of lanthanum strontium cobalt ferrite (LSCF) and MIEC to the Ni anode, at a molar ratio of 1:1, showed a better power density of 111 mW cm−2 at 700 °C compared to the single Ni anode. The excellent power output is attributed to the decreased ohmic and charge-transfer resistance and the expansion of TPBs. Additionally, Bie et al. [110] designed a novel syringe-type anode, which ensured extended region TPBs by pressing the carbon powder into the molten electrolyte, and preventing carbon oxidation.
CO3 2− + MIEC
It is well known that ceria-based oxide is a typical transition metal oxide with mixed electronic and ionic conductivity (MIEC). Its doped oxides, including Gd-doped ceria (GDC) and Sm-doped ceria (SDC), have been applied to an intermediate temperature (IT) SOFC ranging from 400 to 700 °C [111]. The SDC-carbonate composite electrolyte shows excellent conductivity of 10–2 to 1.0 S cm−1 in the range of 400–700 °C, which is better than a pure GDC or SDC (5 × 10–3–10–2 S cm−1), and similar to the traditional solid oxide electrolyte—yttrium-stabilized zirconia (YSZ) at 1000 °C [112, 113]. Additionally, this composite electrolyte is not subject to corrosion issues in the normal MCFC. This type of composite electrolyte has also now been popularized in DCFC and has shown acceptable performance.
The cell performance was improved when using the composite electrolyte of SDC and carbonate because molten carbonate with mobility can expand the TPB at the anode. Therefore, reduced electrode polarization resistance was often achieved. It was also found that the redox reaction of carbon fuel was enhanced when using doped ceria materials. A diagram of the electrochemical process between the electrolyte and the electrode is presented in Fig. 4. It shows that two electrochemical mechanisms might be responsible for forming carbonate ions and oxygen ions in the cathode chamber filled with O2 and CO2 (Eqs. 2 and 15).

Schematic of DCFC with a composite electrolyte: CO2 and O2 receive electrons and produce carbonate ion; only O2 receives electrons and produces O2− at the cathode. Then, the O2− and carbonate ions are transferred to the anode through a composite electrolyte, and react with carbon to produce CO, CO2 and electrons
In the composite electrolyte of SDC-carbonate, it is apparent that the charged species are carbonate ions and oxygen ions and that carbonate ions are transferred in the molten electrolyte, while oxygen ions are the conducting species within the SDC [114, 115]. The carbon particles in the anode cavity, part of which is immersed in molten carbonate, combine with carbonate ions, while others are in contact with the electrolyte of SDC directly and react with oxygen ions, which simultaneously release CO or CO2 and generate electrons (Eqs.1, 7, 16 and 17). CO can also be produced from Eq. (6) through the Boudouard reaction, with further oxidation by oxygen ions [116].
Despite many studies done on the molten carbonate system to explore the oxidation reactions of carbon, the reaction mechanism is still not fully understood [117].
Varieties of carbonaceous fuels used in DCFC with the doped ceria-carbonate composite electrolyte have been reported in literature. Elleuch et al. [79] used low-cost solid carbon-containing coal coke, petroleum coke and almond shell carbonization as fuel for DCFC with an SDC-NiO anode, an SDC-(66 mol% Li2CO3-33 mol% Na2CO3) electrolyte and LixNi1-xO-SDC as the cathode. The results showed that the carbonized-almond shell with more oxygen-containing functional groups has good cell performance at 700 °C, with a power output of 127 mW cm−2. The researchers further explored the electrochemical oxidation of carbon using graphite in the same cell [113]. They found that cell performance was improved with a maximum power density of 59 mW cm−2 at 700 °C when adjusting the anode environment (N2 atmosphere). In CO2 atmosphere, the value reached 37 mW cm−2 [113].
Recently, a dual 3D ceramic textile electrode was integrated into a GDC-carbonate composite electrolyte-supported DCFC, and thus, the TPB region was expanded [118]. Bian et al. [81] developed a unique electrolyte-supported DCFC consisting of a GDC-(67 mol% Li2CO3-33 mol% Na2CO3) electrolyte, a NiO-GDC anode and a Sm0.5Sr0.5CoO3-GDC cathode. It exhibited unprecedented cell performance at 600 °C, with a power output of 392 mW cm−2 when using graphitic fuel, due to the enhanced charge and mass transfer on the electrode below 600 °C [81]. High fuel utilization of 87.3% was also achieved, as the carbon fuel could quickly reach the TPBs via the flowing molten carbonate in the cell operation process.
CO3 2− + SOFC
Recently, HDCFC was combined with SOFC technology and MCFC technology to provide a new way for carbon fuel to reach the reaction region [21]. This method ensures that the molten carbonate is incorporated into the anode cavity, which significantly expands the reaction region from a two-dimensional to a three-dimensional region, and this accelerates the mass transfer to the solid anode/electrolyte. The carbonate serves as a medium to promote the complete oxidation of carbon [23, 119]. In this system, the solid oxide electrolyte separates the anode from the cathode and prevents the diffusion of carbonate to the cathode with no risk of carbonate corrosion on the cathode. Another feature is that no need for the CO2 cycle. The cathode is exposed to air, which simplifies the cell structure.
The first HDCFC was demonstrated by SRI International [120], and was further developed by the University of St Andrews [21, 121, 122], Contained Energy [123] and Technical University of Denmark [124, 125]. An early tubular HDCFC using a Pt cathode and YSZ electrolyte was designed by the University of St Andrews, with the anode nickel mesh placed in a mixture of carbonate and carbon [126]. However, it is difficult to observe the reaction at the anode in this configuration, as the active zones of the electrolyte/electrode are uncertain in a closed-cell above the melting point of the lithium and potassium carbonate [126, 127]. The researchers at the University of St Andrews also developed a planar button cell with better sealing for improved gas purification [128]. The electrochemical reaction mechanism in the anode was also investigated in detail [21, 122]. To date, the highest power density was obtained by Jiang et al. [15] using pyrolyzed medium-density fiberboard in HDCFC, which reached 878 mW cm−2 at 750 °C.
Reaction mechanism
HDCFC is based on two typical fuel cells—SOFC and MCFC—in which: the solid oxide electrolyte (which includes YSZ, GDC or SDC) separates the electrode chambers; the molten carbonate electrolyte has fluidity at a high temperature and expands the oxidation reaction area [21]. The oxygen ions reduced from the oxygen molecule are transmitted from the cathode to the anode compartment through the solid oxide electrolyte (as given by Eq. 15). In the anode compartment, carbon particles may be completely oxidized to CO2 or partially oxidized to CO (Eqs. 16 and 17) [129, 130]. The OCV value would be 1.02 V, if Eq. 16 was the only anode reaction, regardless of temperature.
An OCV of approximately 1.5 V was observed by a current study, which is higher than the theoretical voltage with working temperatures of 550–700 °C in HDCFC [119]. A higher OCV value indicates that other reactions are also underway apart from the direct electrochemical oxidation of carbon to CO2. Figure 5 shows that some reactions and some gas production occur in the anode compartment of HDCFC, which is full of CO2 and CO at the same time. In the anode chamber filled with nitrogen, the reaction processes of carbon (both electrochemical and chemical) are complicated. In the anode, both oxygen ions and carbonate ions are active species of electrochemical oxidation in the slurry of carbon/carbonate [21]. When the number of oxygen ions is sufficiently high, the CO2 could be converted to carbonate ions, which would result in a slow decline of CO2 activity in the molten carbonate, as indicated by Eq. (18). With continued consumption of the oxygen ions, the carbonate ions oxidize carbon to CO2 or CO, as indicated by Eqs. (7 and 8). Carbonate ions then regenerate from oxygen ions and CO2 as indicated by Eq. (18), which maintains the electric charge balance in the molten carbonate solution. Some reports have claimed that the low activity of CO2 could be due to it being dissolved in molten carbonate by physical or chemical methods [131, 132], which may increase the Nernst potential. Nevertheless, the presence of molten carbonate, which facilitates the flow of carbon particles to the anode chamber and expands the TPBs, is expected to act as an electrochemical mediator and accelerate the oxidation reaction kinetics of the carbon particles [13, 41].

Schematic diagram of the SOFC + MCFC system: O2 gains electrons and then produces O2− at the cathode; O2− then passes through a solid oxide electrolyte and arrives at the electrolyte/anode interface, where it reacts with carbon to produce CO, CO2 and electrons
In addition, the non-electrochemical reaction of the Boudouard reaction that occurs at 750 °C has a strong influence on the entire anode reaction, which consumes carbon through a chemical reaction and causes a sharp decrease in current density [28]. But in terms of long-term stability, the influence of current density is not obvious [29]. However, the CO produced by the reverse Boudouard reaction can also generate electricity through electrochemical oxidation, which contributes to higher-power output [133].
Deleebeeck et al. [124] compared the effects of different anode gases on cell performance and found that introducing pure CO2 could reduce the mass transfer limitation by facilitating the Boudouard reaction or preventing the carbonate decomposition. In addition, introducing N2 leads to a high OCV, due to the movement of CO2 or CO [134].
Recently, Li et al. [135] also found that introducing CO2 into the anode chamber could improve cell performance through the electrochemical reaction (Eqs. 19 and 20) at 700–800 °C. However, poor performance by filing CO2 was obtained at 650 °C, as a low temperature is unfavorable for the reverse Boudouard reaction.
Given this prior research, Lee et al. [136] designed three-fuel cells that provided different contacts with the anode and analyzed the oxidation of carbon fuel by observing the current density using different voltages. The various means of contact method between the carbon and anode are presented in Fig. 6a. Cell I, cell II and cell III indicate: direct contact, physically separated, in contact with the carbonate medium. The current density of the cell at 800 °C, when using a different voltage load, is shown in Fig. 6b. It shows that the values of iI and iII were lower than that of iIII at a given voltage. Therefore, adding carbonate increases the current density significantly, proving that the carbonate ions dominate the oxidation reaction (See Fig. 6c.

Reproduced from ref. [136]
The relationship between the different contact modes of the anode and the cell performance of DCFC: a three different contact methods for carbon fuel and the anode; b comparison of current density using different voltage levels at 800 °C; and c relative contribution value of various reaction mechanisms to the total current.
Recently, Jiang et al. [137] designed an experiment to investigate the possible reaction active sites at the anode chamber in HDCFC. The configuration of the HDCFC design is shown in Fig. 7a. The current collection wire could be moved to allow easy adjustment of the space between the current collector and the layer at the anode (L), to further ensure that the reactions occur in the area containing the carbon and carbonate. Figure 7b and c show the AC impedance spectra at 700 °C when using an anode current collector of Au and Pt and different Ls. This demonstrates that the ohmic resistance was determined by the location of the current collection with unchanged polarization resistance. The unchanged polarization resistance with the current collection position also indicates a possible extension from a 2D anode in the TPBs to a 3D area filled with carbon and carbonate for all instances of carbon oxidation.

Reproduced from ref. [137]
The electrochemical reaction zone in HDCFC with a NiO-YSZ anode, YSZ electrolyte, LSM-YSZ cathode and 62 mol% Li2CO3-38 mol%K2CO3: a configuration of the HDCFC design with removable current collection wires; b AC impedance spectrum at 700 °C with Au/NiO-YSZ/YSZ/LSM-YSZ; c AC impedance spectrum at 700 °C with Pd/YSZ/LSM-YSZ.
Apart from the temperature, the carbon oxidation process at the anode also depends on the electrode and electrolyte. Jiang et al. [122] further explored the reaction mechanism of the anode by exchanging the carbonate content in the HDCFC system with a configuration of NiO-YSZ/YSZ/LSM, while using 62 mol% Li2CO3-38 mol% K2CO3 in the anode chamber. They observed reduced polarization resistance of the cell with a carbonate content of 20 mol% or 50 mol%. A different result was found when more carbonate was applied, and higher polarization resistance was achieved when the carbonate increased to 80 mol%. This finding demonstrates that carbon particles are prevented from reaching the electrode by a high concentration of molten carbonate, which has a limited effect on various carbon oxidation reactions.
Some corrosion and sealing issues are significant concerns when using high carbonate concentrations. Cantero-Tubilla et al. [72] proposed that the liquid phase was related to the carbonate content, and therefore the rate of movement of carbon fuel slowed in the presence of high carbonate concentrations. The melt carbonate could result in a concomitant increase in polarization resistance and, consequently, a significant decline in electrochemical output.
Chemical catalysis
McKee et al. [133,134,135,136,137,138,139,140] used a definite redox cycle and postulated that alkali metal carbonates (Li2CO3, Na2CO3, K2CO3, etc.) produce catalytic activity, which is helpful in the process of gasification of carbon under an oxygen or carbon dioxide atmosphere. In a flowing oxygen atmosphere, carbon-induced decomposition of metal carbonates (M2CO3) into metal oxides (M2O), as shown Eq. (21) [138]:
At a higher temperature, M2O is further oxidized to peroxide or higher oxide (M2O1+n), and eventually reduced to M2O by carbon, which completes the redox cycle—see Eqs. (22, 23) [139].
Nevertheless, it is unlikely that the reaction of the oxide-peroxide cycle will take place in a flowing carbon dioxide atmosphere because the conversion of M2CO3 to M2O is difficult. Instead, M2CO3 is reduced to the elemental state, while C is oxidized to CO:
Then, the metal element generates metal oxide in the carbon dioxide atmosphere:
After that, carbonate is formed by the metal oxide and carbon dioxide, which completes a cycle reaction [138]:
Nagase et al. [141] proposed that some metal carbonates also had an influence on the reverse Boudouard reaction in an inert atmosphere, which acted as the catalytic media in the process of electrochemical oxidation or gasification of carbon.
Recently, Li et al. [22] studied the reaction mechanism of the HDCFC anode filled with N2. These researchers found that more CO was produced using eutectic salts (Li2CO3–K2CO3) as the catalyst media, than the absence of carbonate, which therefore improved cell performance. Two reactions produced the carbon dioxide (Eq. 27) and the carbon monoxide (Eq. 28), with both converting M2CO3 to M2O:
Lastly, more CO and electrons were generated by series of reactions (Eqs. 29 and 30) than in an oxygen atmosphere:
Issues with the presence of carbonate
Even though DCFC is being developed continuously, some issues, like materials and technology, must be addressed. Most molten carbonates are corrosive and reactive, which raises widespread concern about the thermal corrosion and chemical stability of cell materials and the chemical composition of solid fuels. Besides the ash of solid carbon fuels, the wettability of carbonates also affects cell performance. These problems ultimately lead to a significant reduction in conversion efficiency and long-term stability.
Corrosion
The highly corrosive action of molten carbonate has prevented progress from being made with MCFC technology. From 1996 to 2006, the cell lifetime was greatly increased from just a few months to 2 years [142]. However, when various carbonates are used as the electrolyte contacted by cell components, it still results in thermal corrosion attacks (such as oxidation, carburization and flux reactions) at high operating temperatures [143]. The corrosion issue gives rise to severe challenges in improving the chemical and physicochemical stability of electrode materials for MCFC. Although some new power generation devices are emerging, stability has always been an issue when developing new technologies.
At present, nickel is one of the most widely used electrode materials because of its low price and good performance in conductivity and electro-catalytic ability. However, Ni and NiO are easily dissolved into the molten carbonate, which causes an internal short-circuit in MCFC or a short lifespan [144]. It was reported that the Ni/NiO solubility in carbonate could be reduced by adding some oxides (e.g., SrO or MgO [140,141,142,143,144,145,146,147,148]) or carbonates (e.g., CaCO3, BaCO3, or SrCO3 [146, 149]) to alkali carbonate, due to the change in the pH value of the solution.
Doping a rare-earth metal [150] or a rare-earth metal oxide (such as La2O3, Y2O3, and Yb2O3) [146,147,148,149,150,151,152,153] is another practical approach to protect the nickel or nickel oxide electrode against dissolution in molten carbonate. For example, a Ni–Ce cathode showed more durability (136 mW cm−2 during 2100 h) than a commercial Ni cathode material (but the voltage decreased from 120 to 108 mW cm−2 in 1000 h) [150]. Further, Liu et al. [154] observed that adding the rare-earth metal Dy to a mixture of Li2CO3 and K2CO3 in a mole ratio of 62:38 could reduce the NiO/ Ni passive anodic current, inhibiting the outward diffusion of Ni at 650 °C, and eventually increase the resistant ability to corrosion.
In terms of HDCFC, the solid oxide electrolyte separates the two electrodes and prevents the cathode from carbonate corrosion. Nevertheless, the chemical compatibility between the electrolyte and the carbonate is essential because of the contact-attack. Suski et al. [155] reported on a double-cell with YSZ and an electrolyte of 53% Li2CO3-47% Na2CO3, with the OCV of O2 + Ar/YSZ/Au at TPBs being similar to the OCV value of the reference electrode over 1000 h of operation. The long operation hours indicate that the kinetics of the chemical reaction between YSZ and Li2CO3–Na2CO3 is very slow. However, another report used XRD analysis to determine that YSZ immersed in Li2CO3–K2CO3 at 700 °C for 10 days was converted to lithium zirconate, but with no change K2CO3–Na2CO3 [126].
Jiang et al. [15] did a more detailed investigation of YSZ corrosion. First, they confirmed that the YSZ of 5–10 μm did not change for 13 h in an atmosphere of 97–3% H2O. Then, several corrosion experiments were done on the YSZ electrolyte in molten carbonate of 62 mol% Li2CO3-38 mol% K2CO3 when using different atmospheres at 700 °C for 10 h. Figure 8 shows the corrosion of the YSZ surface when using different atmospheres. With the air atmosphere, the grain boundaries of YSZ are destroyed and new particles appear, as shown in Fig. 8b, which may be due to the formation of Li2ZrO3, K2ZrO3 or LiKZrO3 [151,152,153,154,155,156,157,158]. Some wrinkles appear on the surface of YSZ and the grain boundaries become inconspicuous in a reducing atmosphere, as shown in Fig. 8c, but less significantly than in the air test. There is no significant change after testing performance using 5% H2–95% Ar, as shown in Fig. 8d. The insignificant change is possible because of the formation of Li2ZrO3 being prevented in a reducing atmosphere [159].

Reproduced from ref. [15]
SEM images of the YSZ surface with the corrosion test in Li-K carbonate at 700 °C for 10 h: a before the corrosion test; b in air; c in Ar; d in 5% H2/Ar.
The researchers used the basicity model of Lux-Flood to explain the experimental result. The formation of oxides and superoxides was more likely to occur in an oxygen environment, which was the primary reason for corrosion [43]. It is common to purge a carrier gas (e.g., N2 or CO2) in the anode chamber. The electrolyte material, YSZ, is either in the reducing environment at the interface of the electrolyte/anode or in the oxidizing environment at the interface of the electrolyte/cathode. Therefore, it is necessary to investigate the stability of YSZ in both the reducing atmosphere and the oxidizing atmosphere to simulate both operating conditions [159, 160]. According to Xu et al. [161], YSZ is a good choice of electrolyte for HDCFC, due to the reducing atmosphere of the anode chamber.
The doped cerium oxide-based material is a typical solid electrolyte in DCFC that shows good stability below 650 °C, due to the complete microstructure being maintained at 650 °C and 10% H2–90%N2 for 1000 h [162]. Xu et al. [161] further explored the stability of SDC in 62 mol% Li2CO3-38 mol% K2CO3 in air, and found that YSZ easily reacted with K2CO3 to form Li2ZrO3 at 700 °C after 10 days of testing. And, there is no new phase formation in the SDC sample when using the same conditions. This suggests that SDC is more resistant to corrosion than YSZ, when using air. Although there was no impurity formation of SDC at 700 °C after 10 days in air, cracks appeared at the grain boundaries [163]. This suggests that crystal boundaries might be the most preferred site for corrosion development [163], as shown in Fig. 9. In addition, the resistance result also showed an insignificant increase in the ohmic resistance of the SDC electrolyte at 650 °C during the 70-h testing, which may be due to the slow corrosion rate along the grain boundary. As mentioned, the corrosion of molten carbonate can be ameliorated by different methods, but cannot be avoided. Therefore, it is vital to develop nascent corrosion-resistant materials, to maximize the advantages of carbonate for DCFC devices.

Reproduced from ref. [161]
Cross section of the SDC for the corrosion test in Li-K carbonate at 700 °C in an air atmosphere for 10 days: a before the corrosion test; b after the corrosion test.
Ash
Solid carbon fuels come from a wide range of sources and have a unique constituency. The molten carbonate is likely to change chemically due to interaction with some ash components, including heavy metals and their oxides, sulfides and chlorides, which ultimately lead to cell failure [74]. However, some metals in ash have a positive effect on cell performance. For example, Cao, MgO and Fe2O3 have a catalytic effect on the oxidation reaction of solid carbon, which increases the current density [24]. A mixture of FemOn, Li2O, K2O and CaO is the catalyst for the Boudouard reaction [26] and it can improve the reaction activity and electrochemical performance of the fuel [164]. Therefore, studying the effect of impurities in the ash on cell materials is key to improving the performance of DCFC.
It has been reported that coal with a low ash content shows better cell performance than high ash [29, 160,161,162,163,164,165,166,167,168]. Ju et al. [169] compared the electrochemical performance of different grades of coal, and found that ash-free coal (low ash content) exhibited the most prolonged stability (300 min at 50 mA cm−2 and 900 °C), while the raw coal was under operation for 50 min before failure due to blockage of the anode channel by ash and thus prevention of the reaction. The high ash content in raw coal is considered detrimental to the short-term stability with a Ni-YSZ anode, as the ash blocks the anode and causes contact between the reaction interface and the carbon interface during cell operation [167].
One study reported that when using Al2O3, SiO2 and TiO2 from coal ash, it is easy to generate a passivating membrane on the electrode interface during operation of the cell, which leads to passivation of the electrode [74]. In particular, SiO2 is likely to react with carbonate electrolytes, to generate CO2 and M2SiO3 (where M = Li, Na, K) in MCFC, leading to additional electrochemical reactions [170]. Tulloch et al. [171] reported that coal with about 70 wt% SiO2 ash showed a significant decrease in current density for MC-DCFC, as SiO2 inhibits the oxidation behavior of carbon.
Recently, different pretreatment methods have been proposed to remove unwanted impurities and enhance cell performance [167,168,169,170,171,172,173,174,175]. Eom et al. [172] reported that using HCl to pre-treat coal could ensure maximum reduction of ash content and the sensitivity of cell performance to surface silicon content may be reduced when the temperature is higher than 733 °C because Li2SiO3 can be completely decomposed at 733 °C [176]. Xie et al. [174] modified bituminous coal with acetic acid, and effectively removed ash of about 84 wt% Si and 64 wt% Al, to optimize cell performance.
It has been reported that some impurities in ash may promote an electrode reaction, e.g., CaO, MgO and Fe2O3 could improve the current density slightly by acting as a catalyst in MC-DCFC [24, 177]. Cai et al. [39] employed orchid leaf char in SO-DCFC with yttrium-stabilized zirconia electrolyte and an Ag-GDC electrode. The researchers concluded that the natural Ca in biochar derived from orchid tree leaves exerted a catalytic effect on the reverse Boudouard reaction and enhanced the performance of DCFC. Then, they loaded 5% Ca on wheat straw char in the same cell configuration and recorded a higher-power performance (258 mW cm−2) compared to that of unloading char (197 mW cm−2) at 700 °C [178]. Hao et al. [36] further confirmed the positive effect of calcite and magnesium calcite in magazine waste paper, which indicated that more amorphous carbon exhibited a higher degree of carbon oxidation also catalyzed the gasification reaction of carbon fuel and delivered a higher-power density.
Recently, a DCFC supported by an SDC-(67% Li-33% Na)2CO3 composite molten carbonate showed excellent cell performance (378 mW cm−2) at an operating temperature of 750 °C. The enhanced cell performance was because the KCl in raw reed ash increases the degree of structural disorder of biochar during the pyrolysis process, which leads to the high oxidation activity of reed char [179].
Wetting
Complete wetting of the carbon particles in eutectic carbonate is critical for the charge transfer at the TPBs zone [180, 181], as this is a dominant factor in ensuring uninterrupted round-the-clock power generation by the DCFC. During the operating process, the eutectic carbonate is completely diffused and penetrates the carbon pores. Therefore, the potential of a stable cell can be reached [49]. When measuring the contact angle of a graphite rod in 62 mol% Li2CO3-38 mol% K2CO3 at 520–560 °C, Chen et al. [182] found that wetting of carbon in carbonate is driven by capillary force, as well as being largely determined by CO bubbles produced from the reverse Boudouard reaction. The OCV increases from 0.5 to 0.81 V during the process of wetting, which contributes to the adsorption of CO on the surface. Therefore, no matter what carbonate is employed in DCFC, as long as the solid carbon is completely soaked in the molten carbonate to form the interface where the carbon oxidation reaction occurs, the electrons can be continuously generated and transferred.
The wettability of solid carbon fuel in a molten carbonate is enhanced by pre-treatment to increase the surface area and the pore volume of the carbon particles [183]. The effect of base or acid pre-treatment on the electrochemical activity of carbon in lithium and potassium carbonate has also been examined [183], with HF, HNO3 and NaOH are the common acid and base materials used. Of all the samples, activated carbon pre-treated with HF exhibited the highest electro-oxidation activity, with an increased current of approximately 50 mA cm−2. The enhanced current is because HF pre-treatment increases the porosity and surface area of the carbon particles, forming a double electrical layer that facilitates electron transfer of carbon during anodization.
It has also been shown that carbon particle wettability relies on carbonate composition. Watanabe et al. [184] indicated that the surface tension of solid carbon could be reduced by lowering the amount of sodium carbonate and thus improving its wettability. The test results showed that carbon particles had a better wetting effect in a ternary carbonate of 12.2 mol% Li2CO3-25 mol% Na2CO3-62.8 mol% K2CO3 compared to 12.2 mol% Li2CO3-45 mol% Na2CO3-42.8 mol% K2CO3, and that a stable OCV of 0.63 V could be sustained for 45 min.
In terms of DCFC with molten carbonate, the degree of stirring is also an important variable that affects carbon particle wetting. Li et al. [96] observed that mass transfer has been significantly improved at 400 rpm, and that current density can be further increased in the whole electrode potential range when the stirring rate is increased up to 600 rpm. Improved current density when increasing the stirring rate aligns with the earlier report of Vutetakis et al. [74]. However, the performance of DCFC will be greatly reduced if the stirring rate exceeds a specific value, because the fuel splash phenomenon leads to a fuel shortage [4]. Whether the stirring improves cell performance or not depends on other factors, such as cell operating temperature and electrolyte viscosity [185].
Carbonate has good ionic conductivity but poor electronic conductivity at a high temperature. Although carbon particles with different sizes are dispersed in molten carbonate, the electrochemical oxidation of carbon occurs only in TPBs, thus the percolation limit of carbon particles must be considered, to ensure efficient and continuous power generation [186]. Using highly conductive carbon fuels such as graphite could improve electronic conductivity, but the chemical activity of graphite is very low. At present, molten metal anodes can solve this problem and ensure good conductivity [187]. However, the corrosion issue remains unresolved.
Outlook
With the increase in environmental concern and the high energy demand, DCFC may provide a clean electrochemical device for generating electricity from solid carbon. The introduction of carbonate in the DCFC system is conducive to improving cell performance of open-circuit voltage and output power. The carbonate accelerates ion transfer as a medium or is a catalyst for carbon oxidation and gasification reaction.
At present, research on basic theory is still an essential aspect of the development of DCFC. Although various DCFC systems that use carbonate are being developed rapidly, the specific reaction mechanism with carbonate involved in the whole-cell system still lacks sufficient proof of experiments due to the complexity of reactions inside the cell. Therefore, new experimental configurations should be designed. Alternatively, a combination of a theoretical model and experimental tests should be considered, which would be beneficial to cell performance if determined what substances affect the electrode reaction process.
The issue of material corrosion in molten carbonate has not been solved satisfactorily. Despite a plethora of literatures that have been published to solve the problem of corrosion of carbonate in MC-DCFC, there is still no detailed research on the corrosion of carbonate to the solid-state electrolyte for emerging HDCFCs. It is unknown whether adding some carbonates, oxides or rare-earth elements has a positive or negative effect on cell performance. It has been confirmed that the addition of carbonate to the anode favors expanding TPBs, because the fluidity of molten carbonate could promote the transfer of carbon fuel to the anode, resulting in a dramatic improvement of electrochemical reactions. However, carbonate attacks the solid electrolyte. Although SDC has better corrosion resistance than YSZ, the corrosion issue cannot be avoided. Provided that corrosion occurs during the entire cell operation, the cell will degrade quickly and cannot generate power over the long term. Hence, more attention needs to be focused on the carbonate corrosion to other cell components (including solid-state electrolytes), instead of only common electrode materials.
It is well known that the variety of solid carbonaceous fuels is one of the most significant advantages of DCFC. Nonetheless, the carbonate component in DCFC easily reacts with the inorganic salts of fuels ash—especially coal and biomass fuels with a high ash content—which shows various effects on cell performance. For example, a large amount of Fe, Mg and Ca promotes gasification of carbon, while Si and Al have a negative effect on the cell performance of DCFC with carbonate. It is difficult to identify the effect of each inorganic salt on cell performance because there is often more than one salt in the fuel.
Removing ash from non-pure carbon fuels such as biomass should also be considered seriously. Pre-treatment should be attempted as an essential treatment method applied to DCFC (including heat treatment, acid or base washing and air plasma, etc.).
Other issues worth mentioning include the effect of pre-treatment on the properties of the fuel itself and the reactivity of the pretreated fuel with carbonates to eventually explore the feasibility of using different renewable carbon-based materials in the cell.
While many cell designs have been developed to provide better continuous power generation, the fluidized bed cell can provide a more convenient feeding mode. Thus, designing different cell configurations is still key to the commercialization of DCFC.
References
Grove, W.R.: XXIV On voltaic series and the combination of gases by platinum. Phil. Mag. 14(86–87), 127–130 (1839). https://doi.org/10.1080/14786443908649684
Behling, N., Managi, S., Williams, M.: Updated look at the application of solid particles in fuel cell technology. 2019 SME Annual Conference and Expo and CMA 121st National Western Mining Conference (2019)
Williams, M.C., Horita, T., Yamaji, K., Yokokawa, H.: An application of solid particles in fuel cell technology. KONA Powder Part. J. 25, 153–161 (2007). https://doi.org/10.14356/kona.2007014
Ahn, S.Y., Eom, S.Y., Rhie, Y.H., Sung, Y.M., Moon, C.E., Choi, G.M., Kim, D.J.: Utilization of wood biomass char in a direct carbon fuel cell (DCFC) system. Appl. Energ. 105, 207–216 (2013). https://doi.org/10.1016/j.apenergy.2013.01.023
Edison, T.A.: Process of and apparatus for generating electricity. US Patent No 460, 122 (1891)
Jacques, W.W.: Method of converting potential energy of carbon into electrical energy. US Patent No 555511 A (1896)
Srinivasan, S.: Fuel cells for extraterrestrial and terrestrial applications. J. Electrochem. Soc. 136(2), 41C (1989). https://doi.org/10.1149/1.2096647
Meibuhr, S.: Review of united states fuel-cell patents issued from 1860 to 1947. Electrochim. Acta. 11(9), 1301–1308 (1966). https://doi.org/10.1016/0013-4686(66)87029-9
Haber, F., Bruner, L.: Das Kohlenelement, eine Knallgaskette. Zeitschrift für Elektrochemie und angewandte physikalische Chemie 10(37), 697–713 (1904). https://doi.org/10.1002/bbpc.19040103702
Weaver, R.D., Leach, S.C., Bayce, A.E., Nanis, L.: Direct electrochemical generation of electricity from coal. SRI International Corp. (1979)
Weaver, R.D., Tietz, L., Cubicciotti, D.: Direct use of coal in a fuel cell: Feasibility investigation. SRI International Corp. (1975)
Gür, T.M.: Utilization modes for solid carbon conversion in fuel cells. ECS Trans. 25(2), 1099 (2009). https://doi.org/10.1149/1.3205637
Gür, T.M.: Mechanistic modes for solid carbon conversion in high temperature fuel cells. J. Electrochem. Soc. 157(5), B751 (2010). https://doi.org/10.1149/1.3357050
Nürnberger, S., Bußar, R., Desclaux, P., Franke, B., Rzepka, M., Stimming, U.: Direct carbon conversion in a SOFC-system with a non-porous anode. Energy Environ. Sci. 3(1), 150–153 (2010). https://doi.org/10.1039/b916995d
Jiang, C., Ma, J., Bonaccorso, A.D., Irvine, J.T.S.: Demonstration of high power, direct conversion of waste-derived carbon in a hybrid direct carbon fuel cell. Energy Environ. Sci. 5(5), 6973–6980 (2012). https://doi.org/10.1039/c2ee03510c
Hackett, G.A., Zondlo, J.W., Svensson, R.: Evaluation of carbon materials for use in a direct carbon fuel cell. J. Power Sources 168(1), 111–118 (2007). https://doi.org/10.1016/j.jpowsour.2007.02.021
Dicks, A.L.: The role of carbon in fuel cells. J. Power Sources 156(2), 128–141 (2006). https://doi.org/10.1016/j.jpowsour.2006.02.054
Lan, R., Tao, S.: A simple high-performance matrix-free biomass molten carbonate fuel cell without CO2 recirculation. Sci. Advs. 2(8), e1600772 (2016). https://doi.org/10.1126/sciadv.1600772
Dudek, M., Tomczyk, P.: Composite fuel for direct carbon fuel cell. Catal. Today 176(1), 388–392 (2011). https://doi.org/10.1016/j.cattod.2010.11.029
Liu, C., Pu, J., Chen, X., Ma, Z., Ding, X., Zhou, J., Wang, S.: Influence of anode’s microstructure on electrochemical performance of solid oxide direct carbon fuel cells. Inter. J. Hydrogen Energy 45(20), 11784–11790 (2020). https://doi.org/10.1016/j.ijhydene.2020.02.119
Nabae, Y., Pointon, K.D., Irvine, J.T.S.: Electrochemical oxidation of solid carbon in hybrid DCFC with solid oxide and molten carbonate binary electrolyte. Energy Environ. Sci. 1(1), 148–155 (2008). https://doi.org/10.1039/b804785e
Li, S., Jiang, C., Liu, J., Tao, H., Meng, X., Connor, P., Hui, J., Wang, S., Ma, J., Irvine, J.T.S.: Mechanism of enhanced performance on a hybrid direct carbon fuel cell using sawdust biofuels. J. Power Sources 383(15), 10–16 (2018). https://doi.org/10.1016/j.jpowsour.2018.02.040
Jiang, C., Cui, C., Ma, J., Irvine, J.T.S.: Insight into graphite oxidation in a NiO-based hybrid direct carbon fuel cell. Inter. J. Hydrogen Energy 45(17), 10559–10568 (2020). https://doi.org/10.1016/j.ijhydene.2019.08.208
Li, X., Zhu, Z., De Marco, R., Bradley, J., Dicks, A.: Evaluation of raw coals as fuels for direct carbon fuel cells. J. Power Sources 195(13), 4051–4058 (2010). https://doi.org/10.1016/j.jpowsour.2010.01.048
Lee, C.-G., Kim, W.-K.: Oxidation of ash-free coal in a direct carbon fuel cell. Inter. J. Hydrogen Energy 40(15), 5475–5481 (2015). https://doi.org/10.1016/j.ijhydene.2015.01.068
Tang, Y., Liu, J.: Effect of anode and Boudouard reaction catalysts on the performance of direct carbon solid oxide fuel cells. Inter. J. Hydrogen Energy 35(20), 11188–11193 (2010). https://doi.org/10.1016/j.ijhydene.2010.07.068
Jewulski, J., Skrzypkiewicz, M., Struzik, M., Lubarska-Radziejewska, I.: Lignite as a fuel for direct carbon fuel cell system. Inter. J. Hydrogen Energy 39(36), 21778–21785 (2014). https://doi.org/10.1016/j.ijhydene.2014.05.039
Fuente-Cuesta, A., Jiang, C., Arenillas, A., Irvine, J.T.S.: Role of coal characteristics in the electrochemical behaviour of hybrid direct carbon fuel cells. Energy Environ. Sci. 9(9), 2868–2880 (2016). https://doi.org/10.1039/c6ee01461e
Jiang, C., Ma, J., Arenillas, A., Bonaccorso, A.D., Irvine, J.T.: Comparative study of durability of hybrid direct carbon fuel cells with anthracite coal and bituminous coal. Inter. J. Hydrogen Energy 41(41), 18797–18806 (2016). https://doi.org/10.1016/j.ijhydene.2016.04.047
König, A., Ulonska, K., Mitsos, A., Viell, J.R.: Optimal applications and combinations of renewable fuel production from biomass and electricity. Energy Fuel 33(2), 1659–1672 (2019). https://doi.org/10.1021/acs.energyfuels.8b03790
Li, M., Xiao, H., Zhang, T., Li, Q., Zhao, Y.: Activated carbon fiber derived from sisal with large specific surface area for high-performance supercapacitors. ACS Sustain. Chem. Eng. 7(5), 4716–4723 (2019). https://doi.org/10.1021/acssuschemeng.8b04607
Rybarczyk, M.K., Li, Y., Qiao, M., Hu, Y.-S., Titirici, M.-M., Lieder, M.: Hard carbon derived from rice husk as low cost negative electrodes in Na-ion batteries. J. Energy Chem. 29, 17–22 (2019). https://doi.org/10.1016/j.jechem.2018.01.025
Tiwari, J.N., Dang, N.K., Sultan, S., Thangavel, P., Jeong, H.Y., Kim, K.S.: Multi-heteroatom-doped carbon from waste-yeast biomass for sustained water splitting. Nat. Sustain. 3(7), 556–563 (2020). https://doi.org/10.1038/s41893-020-0509-6
Xu, C., Chen, J., Li, S., Gu, Q., Wang, D., Jiang, C., Liu, Y.: N-doped honeycomb-like porous carbon derived from biomass as an efficient carbocatalyst for H2S selective oxidation. J. Hazard Mater. 403(5), 123806 (2021). https://doi.org/10.1016/j.jhazmat.2020.123806
Yu, J., Zhao, Y., Li, Y.: Utilization of corn cob biochar in a direct carbon fuel cell. J. Power Sources 270(15), 312–317 (2014). https://doi.org/10.1016/j.jpowsour.2014.07.125
Hao, W., Mi, Y.: Evaluation of waste paper as a source of carbon fuel for hybrid direct carbon fuel cells. Energy 107(15), 122–130 (2016). https://doi.org/10.1016/j.energy.2016.04.012
Wang, C., Lü, Z., Li, J., Cao, Z., Wei, B., Li, H., Shang, M., Su, C.: Efficient use of waste carton for power generation, tar and fertilizer through direct carbon solid oxide fuel cell. Renew. Energy 158, 410–420 (2020). https://doi.org/10.1016/j.renene.2020.05.082
Elleuch, A., Boussetta, A., Yu, J., Halouani, K., Li, Y.: Experimental investigation of direct carbon fuel cell fueled by almond shell biochar: Part I. Physico-chemical characterization of the biochar fuel and cell performance examination. Inter. J. Hydrogen Energy 38(36), 16590–16604 (2013). https://doi.org/10.1016/j.ijhydene.2013.08.090
Cai, W., Zhou, Q., Xie, Y., Liu, J., Long, G., Cheng, S., Liu, M.: A direct carbon solid oxide fuel cell operated on a plant derived biofuel with natural catalyst. Appl. Energy 179(2), 1232–1241 (2016). https://doi.org/10.1016/j.apenergy.2016.07.068
Ali, A., Raza, R., Shakir, M.I., Iftikhar, A., Alvi, F., Ullah, M.K., Hamid, A., Kim, J.-S.: Promising electrochemical study of titanate based anodes in direct carbon fuel cell using walnut and almond shells biochar fuel. J. Power Sources. 434, 12 (2019). https://doi.org/10.1016/j.jpowsour.2019.05.085
Cherepy, N.J., Krueger, R., Fiet, K.J., Jankowski, A.F., Cooper, J.F.: Direct conversion of carbon fuels in a molten carbonate fuel cell. J. Electrochem. Soc. 152(1), A80–A87 (2005). https://doi.org/10.1149/1.1836129
Cisneros, S., Sánchez, C.: Kinetic characterization and molten KOH fuel cell simulation for dissolved coal oxidation on nickel anodes. J. Electrochem. Soc. 161(14), F1330–F1339 (2014). https://doi.org/10.1149/2.1181412jes
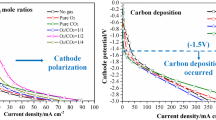
Bie, K., Zhou, H., Fu, P., Liu, Y., Yue, F.: Investigation of the cathode polarization and carbon deposition in a molten carbonate direct carbon fuel cell. J. Appl. Electrochem. 49(6), 585–597 (2019). https://doi.org/10.1007/s10800-019-01307-0
Howard, H.: Direct generation of electricity from coal and gas (fuel cells). Chem. Coal Util. 2, 1568–1585 (1945)
Liebhafsky, H.A., Cairns, E.J.: Fuel cells and fuel batteries. Guide to their research and development (1968)
Cao, D., Sun, Y., Wang, G.: Direct carbon fuel cell: Fundamentals and recent developments. J. Power Sources 167(2), 250–257 (2007). https://doi.org/10.1016/j.jpowsour.2007.02.034
Cao, T., Huang, K., Shi, Y., Cai, N.: Recent advances in high-temperature carbon–air fuel cells. Energy Environ. Sci. 10(2), 460–490 (2017). https://doi.org/10.1039/c6ee03462d
Giddey, S., Badwal, S.P.S., Kulkarni, A., Munnings, C.: A comprehensive review of direct carbon fuel cell technology. Prog. Energy Combust. 38(3), 360–399 (2012). https://doi.org/10.1016/j.pecs.2012.01.003
Cooper, J.F., Selman, R.: Electrochemical oxidation of carbon for electric power generation: a review. J. Electrochem. Soc. 19(14), 15–25 (2009). https://doi.org/10.1149/1.3220176
Gür, T.M.: Critical review of carbon conversion in “carbon fuel cells.” Chem. Rev. 113(8), 6179–6206 (2013). https://doi.org/10.1021/cr400072b
Rady, A.C., Giddey, S., Badwal, S.P.S., Ladewig, B.P., Bhattacharya, S.: Review of fuels for direct carbon fuel cells. Energy Fuel 26(3), 1471–1488 (2012). https://doi.org/10.1021/ef201694y
Zhou, W., Jiao, Y., Li, S.D., Shao, Z.: Anodes for carbon-fueled solid oxide fuel cells. ChemElectroChem 3(2), 193–203 (2016). https://doi.org/10.1002/celc.201500420
Jiang, C., Ma, J., Corre, G., Jain, S.L., Irvine, J.T.S.: Challenges in developing direct carbon fuel cells. Chem. Soc. Rev. 46(10), 2889–2912 (2017). https://doi.org/10.1039/c6cs00784h
Glenn, M.J., Allen, J.A., Donne, S.W.: Carbon electro-catalysis in the direct carbon fuel cell utilising alkali metal molten carbonates: a mechanistic review. J. Power Sources 453, 227662 (2020). https://doi.org/10.1016/j.jpowsour.2019.227662
Tanimoto, K., Yanagida, M., Kojima, T., Tamiya, Y., Matsumoto, H., Miyazaki, Y.: Long-term operation of small-sized single molten carbonate fuel cells. J. Power Sources 72(1), 77–82 (1998). https://doi.org/10.1016/S0378-7753(97)02673-6
Morita, H., Kawase, M., Mugikura, Y., Asano, K.: Degradation mechanism of molten carbonate fuel cell based on long-term performance: long-term operation by using bench-scale cell and post-test analysis of the cell. J. Power Sources 195(20), 6988–6996 (2010). https://doi.org/10.1016/j.jpowsour.2010.04.084
Glugla, P., De Carlo, V.: The specific conductance of molten carbonate fuel cell tiles. J. Electrochem. Soc. 129(8), 1745 (1982). https://doi.org/10.1149/1.2124263
McKee, D.W., Spiro, C.L., Kosky, P.G., Lamby, E.J.: Catalysis of coal char gasification by alkali metal salts. Fuel 62(2), 217–220 (1983). https://doi.org/10.1016/0016-2361(83)90202-8
McKee, D.W., Spiro, C.L., Kosky, P.G., Lamby, E.J.: Eutectic salt catalysts for graphite and coal char gasification. Fuel 64(6), 805–809 (1985). https://doi.org/10.1016/0016-2361(85)90014-6
McKee, D.: Rare earth oxides as carbon oxidation catalysts. Carbon 23(6), 707–713 (1985). https://doi.org/10.1016/0008-6223(85)90232-5
Yu, X., Shi, Y., Wang, H., Cai, N., Li, C., Ghoniem, A.F.: Using potassium catalytic gasification to improve the performance of solid oxide direct carbon fuel cells: experimental characterization and elementary reaction modeling. J. Power Sources 252(2), 130–137 (2014). https://doi.org/10.1016/j.jpowsour.2013.12.021
Janz, G.J., Lorena, M.R.: Solid-liquid phase equilibria for mixtures of lithium, sodium, and potassium carbonates. J. Chem. Eng. Data 6(3), 321–323 (1961). https://doi.org/10.1021/je00103a001
Weaver, R., Leach, S., Nanis, L.: Electrolyte management for the coal air fuel cell. IECEC Conference (1981)
Kouchachvili, L., Ikura, M.: Performance of direct carbon fuel cell. Inter. J. Hydrogen Energy 36(16), 10263–10268 (2011). https://doi.org/10.1016/j.ijhydene.2010.10.036
Kojima, T., Yanagida, M., Tanimoto, K., Tamiya, Y., Matsumoto, H., MiyazakiI, Y.: The surface tension and the density of molten binary alkali carbonate systems. Electrochemistry 67(6), 593–602 (1999). https://doi.org/10.5796/electrochemistry.67.593
Posypaiko, V., Alekseeva, E., Vasina, N.: Diagrammy plavkosti solevykh sistem. Troynyye sistemy [Charts of salt systems' fusion. Triple systems]. Moscow: Chemistry (1977)
Licht, S.: Stabilization of STEP electrolyses in lithium-free molten carbonates. arXiv preprint arXiv:1209.3512. (2012)
Ang, P., Sammells, A.: Influence of electrolyte composition on electrode kinetics in the molten carbonate fuel cell. J. Electrochem. Soc. 127(6), 1287 (1980). https://doi.org/10.1149/198110.0341PV
Belhomme, C., Devynck, J., Cassir, M.: New insight in the cyclic voltammetric behaviour of nickel in molten Li2CO3–Na2CO3 at 650 °C. Electroanal. Chem. 545(27), 7–17 (2003). https://doi.org/10.1016/S0022-0728(03)00060-3
Maru, H.C., Dullea, J., Ong, E., Pigeaud, A., Sampath, V., Selman, J.R.: Fuel cell research on second-generation molten carbonate system. Institute of Gas Technology, Chicago (1976)
Morita, H., Komoda, M., Mugikura, Y., Izaki, Y., Watanabe, T., Masuda, Y., Matsuyama, T.: Performance analysis of molten carbonate fuel cell using a Li/Na electrolyte. J. Power Sources 112(2), 509–518 (2002). https://doi.org/10.1016/S0378-7753(02)00468-8
Cantero-Tubilla, B., Xu, C., Zondlo, J.W., Sabolsky, K., Sabolsky, E.M.: Investigation of anode configurations and fuel mixtures on the performance of direct carbon fuel cells (DCFCs). J. Power Sources 238(2), 227–235 (2013). https://doi.org/10.1016/j.jpowsour.2013.03.072
Mamantov, G., Mamantov, C.: Advances in molten salt chemistry 5. Elsevier Science Publishers Science and Technology Div (1983)
Vutetakis, D., Skidmore, D., Byker, H.: Electrochemical oxidation of molten carbonate-coal slurries. J. Electrochem. Soc. 134(12), 3027–3035 (1987). https://doi.org/10.1149/1.2100334
Jiang, C., Ma, J., Arenillas, A., Irvinea, J.T.S.: Application of ternary carbonate in hybrid direct coal fuel cells. ECS Trans. 59(1), 281–288 (2014). https://doi.org/10.1149/05901.0281ecst
Eom, S., Cho, J., Ahn, S., Sung, Y., Choi, G., Kim, D.: Comparison of the electrochemical reaction parameter of graphite and sub-bituminous coal in a direct carbon fuel cell. Energ Fuel 30(4), 3502–3508 (2016). https://doi.org/10.1021/acs.energyfuels.5b02904
Li, C., Yi, H., Lee, D.: On-demand supply of slurry fuels to a porous anode of a direct carbon fuel cell: attempts to increase fuel-anode contact and realize long-term operation. J. Power Sources 309, 99–107 (2016). https://doi.org/10.1016/j.jpowsour.2016.01.080
Lee, E.-K., Park, S.-A., Jung, H.-W., Kim, Y.-T.: Performance enhancement of molten carbonate-based direct carbon fuel cell (MC-DCFC) via adding mixed ionic-electronic conductors into Ni anode catalyst layer. J. Power Sources 386(15), 28–33 (2018). https://doi.org/10.1016/j.jpowsour.2017.03.078
Elleuch, A., Boussetta, A., Halouani, K., Li, Y.: Experimental investigation of direct carbon fuel cell fueled by almond shell biochar: Part II. Improvement of cell stability and performance by a three-layer planar configuration. Inter. J. Hydrogen Energy 38(36), 16605–16614 (2013). https://doi.org/10.1016/j.ijhydene.2013.07.061
Elleuch, A., Halouani, K., Li, Y.: Investigation of chemical and electrochemical reactions mechanisms in a direct carbon fuel cell using olive wood charcoal as sustainable fuel. J. Power Sources. 281(1), 350–361 (2015). https://doi.org/10.1016/j.jpowsour.2015.01.171
Bian, W., Wu, W., Orme, C.J., Ding, H., Zhou, M., Ding, D.: Dual 3D ceramic textile electrodes: fast kinetics for carbon oxidation reaction and oxygen reduction reaction in direct carbon fuel cells at reduced temperatures. Adv. Funct. Mater. 30(19), 1–9 (2020). https://doi.org/10.1002/adfm.201910096
Hao, W., He, X., Mi, Y.: Achieving high performance in intermediate temperature direct carbon fuel cells with renewable carbon as a fuel source. Appl. Energy 135(15), 174–181 (2014). https://doi.org/10.1016/j.apenergy.2014.08.055
Hao, W., Mi, Y.: A direct carbon fuel cell with a CuO–ZnO–SDC composite anode. RSC Adv. 6(55), 50201–50208 (2016). https://doi.org/10.1039/c6ra04949d
He, W.: Adaptation of a one megawatt molten carbonate fuel cell system. Energy Convers. Manage. 38(7), 659–663 (1997). https://doi.org/10.1016/S0196-8904(96)00080-5
Baur, E., Brunner, R.: Über die eisenoxyd-kathode in der kohle-luft-kette. Zeitschrift für Elektrochemie und angewandte physikalische Chemie 43(9), 725–727 (1937). https://doi.org/10.1002/bbpc.19370430902
Anbar, M.: Methods and apparatus for the pollution-free generation of electrochemical energy. US Patent No 3,741,809 (1973)
Anbar, M., McMillen, D.F., Weaver, R.D., Jorgensen, P.J.: Method and apparatus for electrochemical generation of power from carbonaceous fuels. US Patent No 3,741,809 (1976)
McKee, D.W.: Gasification of graphite in carbon dioxide and water vapor—the catalytic effects of alkali metal salts. Carbon 20(1), 59–66 (1982). https://doi.org/10.1016/0008-6223(82)90075-6
Dunks, G.B.: Electrochemical studies of graphite oxidation in sodium carbonate melt. Inorg. Chem. 23(7), 828–837 (1984). https://doi.org/10.1021/ic00175a008
Cooper, J.F., Krueger, R., Cherepy, N.: Fuel cell apparatus and method thereof. US Patent No 6815105B2 (2004)
Luo, Z., Wang, S., Liao, Y., Zhou, J., Gu, Y., Cen, K.: Research on biomass fast pyrolysis for liquid fuel. Biomass Bioenergy 26(5), 455–462 (2004). https://doi.org/10.1016/j.biombioe.2003.04.001
Huang, J., Mao, Z., Yang, L., Peng, R.: SDC-Carbonate composite electrolytes for low-temperature SOFCs. Electrochem. Solid State 8(9), A437–A440 (2005). https://doi.org/10.1149/1.1960139
Cooper, J.F., Cherepy, N., Krueger, R.L.: Tilted fuel cell apparatus. US Patent No 6878479B2 (2005)
Cooper, J.F.: Reactions of the carbon anode in molten carbonate electrolyte. Presentations in Direct Carbon Fuel Cell Workshop, NETL (2003)
Cooper, J.F.: Direct conversion of coal and coal-derived carbon in fuel cells. The 2nd International Conference on Fuel Cell Science, Engineering and Technology (2004)
Li, X., Zhu, Z.H., Roland, D.M., Andrew, D., John, B., Liu, S., Lu, G.Q.: Factors that determine the performance of carbon fuels in the direct carbon fuel cell. Ind. Eng. Chem. Res. 47(23), 9670–9677 (2008). https://doi.org/10.1021/ie800891m
Li, X., Zhu, Z., Roland, D.M., John, B., Andrew, D.: Modification of coal as a fuel for the direct carbon fuel cell. J. Phys. Chem. A 114(11), 3855–3862 (2010). https://doi.org/10.1021/jp9062719
Zhang, J., Zhong, Z., Shen, D., Xiao, J., Fu, Z., Zhang, H., Zhao, J., Li, W., Yang, M.: Characteristics of a fluidized bed electrode for a direct carbon fuel cell anode. J. Power Sources 196(6), 3054–3059 (2011). https://doi.org/10.1016/j.jpowsour.2010.11.130
Ido, A., Kawase, M.: Development of a tubular molten carbonate direct carbon fuel cell and basic cell performance. J. Power Sources 449(15), 227483 (2020). https://doi.org/10.1016/j.jpowsour.2019.227483
Gür, T.M., Huggins, R.A.: Direct electrochemical conversion of carbon to electrical energy in a high temperature fuel cell. J. Electrochem. Soc. 139(10), L95 (1992). https://doi.org/10.1149/1.2069025
Zhang, J., Jiang, X., Piao, G., Yang, H., Zhong, Z.: Simulation of a fluidized bed electrode direct carbon fuel cell. Inter. J. Hydrogen Energy 40(8), 3321–3331 (2015). https://doi.org/10.1016/j.ijhydene.2014.12.090
Kruesi, W., Fray, D.: Fundamental study of the anodic and cathodic reactions during the electrolysis of a lithium carbonate-lithium chloride melt using a carbon anode. J. Appl. Electrochem. 24(11), 1102–1108 (1994). https://doi.org/10.1007/BF00241307
Weaver, R.D., Nanis, L.: Electrochemical oxidation of carbon in a molten carbonate coal-air fuel cell. J. Electrochem. Soc. 1981, 316–333 (1981). https://doi.org/10.1149/198109.0316PV
Glenn, M.J., Allen, J.A., Donne, S.W.: Carbon gasification from a molten carbonate eutectic. Energy Technol-ger. (2019). https://doi.org/10.1002/ente.201900602
Watanabe, H., Umehara, D., Hanamura, K.: Impact of gas products around the anode on the performance of a direct carbon fuel cell using a carbon/carbonate slurry. J. Power Sources 329, 567–573 (2016). https://doi.org/10.1016/j.jpowsour.2016.08.122
Kaklidis, N., Strandbakke, R., Arenillas, A., Menéndez, J.A., Konsolakis, M., Marnellos, G.E.: The synergistic catalyst-carbonates effect on the direct bituminous coal fuel cell performance. Inter. J. Hydrogen Energy 44(20), 10033–10042 (2019). https://doi.org/10.1016/j.ijhydene.2019.02.038
Tamaru, S., Kamada, M.: Brennstoffketten, deren Arbeitstemperatur unterhalb 600 ℃ liegt. Zeitschrift für Elektrochemie und angewandte physikalische Chemie 41(2), 93–96 (1935). https://doi.org/10.1002/bbpc.19350410207
Thonstad, J.: The electrode reaction on the C, CO2 electrode in cryolite-alumina melts-I Steady state measurements. Electrochim. Acta. 15(10), 1569–1580 (1970). https://doi.org/10.1016/0013-4686(70)80079-2
Lee, E.-K., Chun, H.H., Kim, Y.-T.: Enhancing Ni anode performance via Gd2O3 addition in molten carbonate-type direct carbon fuel cell. Inter. J. Hydrogen Energy 39(29), 16541–16547 (2014). https://doi.org/10.1016/j.ijhydene.2014.03.180
Bie, K., Fu, P., Liu, Y., Muhammad, A.: Comparative study on the performance of different carbon fuels in a molten carbonate direct carbon fuel cell with a novel anode structure. J. Power Sources (2020). https://doi.org/10.1016/j.jpowsour.2020.228101
Eguchi, K., Setoguchi, T., Inoue, T., Arai, H.: Electrical properties of ceria-based oxides and their application to solid oxide fuel cells. Solid State Ion. 52(1–3), 165–172 (1992). https://doi.org/10.1016/0167-2738(92)90102-U
Zhu, B.: Functional ceria–salt-composite materials for advanced ITSOFC applications. J. Power Sources 114(1), 1–9 (2003). https://doi.org/10.1016/s0378-7753(02)00592-x
Elleuch, A., Yu, J., Boussetta, A., Halouani, K., Li, Y.: Electrochemical oxidation of graphite in an intermediate temperature direct carbon fuel cell based on two-phases electrolyte. Inter. J. Hydrogen Energy 38(20), 8514–8523 (2013). https://doi.org/10.1016/j.ijhydene.2012.11.070
Li, Y., Rui, Z., Xia, C., Anderson, M., Lin, Y.S.: Performance of ionic-conducting ceramic/carbonate composite material as solid oxide fuel cell electrolyte and CO2 permeation membrane. Catal Today 148(3–4), 303–309 (2009). https://doi.org/10.1016/j.cattod.2009.08.009
Rui, Z., Anderson, M., Lin, Y.S., Li, Y.: Modeling and analysis of carbon dioxide permeation through ceramic-carbonate dual-phase membranes. J. Membr. Sci. 345(1–2), 110–118 (2009). https://doi.org/10.1016/j.memsci.2009.08.034
Lahijani, P., Zainal, Z.A., Mohammadi, M., Mohamed, A.R.: Conversion of the greenhouse gas CO2 to the fuel gas CO via the boudouard reaction: a review. Renew. Sust. Energy Rev. 41, 615–632 (2015). https://doi.org/10.1016/j.rser.2014.08.034
Jia, L., Tian, Y., Liu, Q., Xia, C., Yu, J., Wang, Z., Zhao, Y., Li, Y.: A direct carbon fuel cell with (molten carbonate)/(doped ceria) composite electrolyte. J. Power Sources 195(17), 5581–5586 (2010). https://doi.org/10.1016/j.jpowsour.2010.03.016
Wu, W., Zhang, Y., Ding, D., He, T.: A high-performing direct carbon fuel cell with a 3D architectured anode operated below 600℃. Adv. Mater. 30(4), 1704745 (2018). https://doi.org/10.1002/adma.201704745
Nabae, Y., Pointon, K.D., Irvine, J.T.S.: Ni/C slurries based on molten carbonates as a fuel for hybrid direct carbon fuel cells. J. Electrochem. Soc. 156(6), B716–B720 (2009). https://doi.org/10.1149/1.3110862
Alexander Lipilin, Iouri Balachov, Lawrence Dubois, Angel Sanjurjo, Michael McKubre, Steven Crouch-Baker, Marc Hornbostel, Tanzella, F.: Liquid anode electrochemical cell. US Patent No 2007/0269688 A1 (2007)
Jain, S.L., Barry Lakeman, J., Pointon, K.D., Marshall, R., Irvine, J.T.S.: Electrochemical performance of a hybrid direct carbon fuel cell powered by pyrolysed MDF. Energy Environ. Sci. 2(6), 687–693 (2009). https://doi.org/10.1039/b820836k
Jiang, C., Irvine, J.T.S.: Catalysis and oxidation of carbon in a hybrid direct carbon fuel cell. J. Power Sources 196(17), 7318–7322 (2011). https://doi.org/10.1016/j.jpowsour.2010.11.066
Ruflin, J., Perwich, A.D., II., Brett, C., Berner, J.K., Lux, S.M.: Direct carbon fuel cell: a proposed hybrid design to improve commercialization potential. J. Power Sources 213, 275–286 (2012). https://doi.org/10.1016/j.jpowsour.2012.04.048
Deleebeeck, L., Hansen, K.K.: HDCFC performance as a function of anode atmosphere (N2-CO2). J. Electrochem. Soc. 161(1), F33–F46 (2013). https://doi.org/10.1149/2.027401jes
Deleebeeck, L., Ippolito, D., Hansen, K.K.: Enhancing hybrid direct carbon fuel cell anode performance using Ag2O. Electrochim. Acta. 152, 222–239 (2015). https://doi.org/10.1016/j.electacta.2014.11.064
Pointon, K., Lakeman, B., Irvine, J., Bradley, J., Jain, S.: The development of a carbon–air semi fuel cell. J. Power Sources 162(2), 750–756 (2006). https://doi.org/10.1016/j.jpowsour.2005.07.023
Jain, S., Lakeman, B., Pointon, K.D., Irvine, J.T.: Carbon content in a direct carbon fuel cell. Ecs. Trans. 7(1), 829–836 (2007). https://doi.org/10.1149/1.2729172
Jain, S.L., Nabae, Y., Lakeman, B.J., Pointon, K.D., Irvine, J.T.S.: Solid state electrochemistry of direct carbon/air fuel cells. Solid State Ion. 179(27–32), 1417–1421 (2008). https://doi.org/10.1016/j.ssi.2008.01.078
Lee, A.C., Mitchell, R.E., Gür, T.M.: Thermodynamic analysis of gasification-driven direct carbon fuel cells. J. Power Sources 194(2), 774–785 (2009). https://doi.org/10.1016/j.jpowsour.2009.05.039
Lee, A.C., Mitchell, R.E., Gür, T.M.: Modeling of CO2gasification of carbon for integration with solid oxide fuel cells. AIChE J. 55(4), 983–992 (2009). https://doi.org/10.1002/aic.11713
White, S., Twardoch, U.: The solubility and electrochemistry of alkali metal oxides in the molten eutectic mixture of lithium carbonate-sodium carbonate-potassium carbonate. J. Appl. Electrochem. 19(6), 901–910 (1989). https://doi.org/10.1007/BF01007939
Peeters, D., Moyaux, D., Claes, P.: (1999) Solubility and solvation of carbon dioxide in the molten Li2CO3/Na2CO3/K2CO3 (43.5: 31.5: 25.0 mol%) eutectic mixture at 973 K II. Theoretical Part. Eur. J. Inorg. Chem. 4, 589–592 (1999). https://doi.org/10.1002/(SICI)1099-0682(199904)1999:4
Nakagawa, N., Ishida, M.: Performance of an internal direct-oxidation carbon fuel cell and its evaluation by graphic exergy analysis. Ind. Eng. Chem. Res. 27(7), 1181–1185 (1988). https://doi.org/10.1021/ie00079a016
Hemmes, K., Cassir, M.: A theoretical study of the carbon/carbonate/hydroxide (electro-) chemical system in a direct carbon fuel cell. J. Fuel Cell Sci. Technol. 8(5), 1 (2011). https://doi.org/10.1115/1.4003750
Li, S., Pan, W., Wang, S., Meng, X., Jiang, C., Irvine, J.T.: Electrochemical performance of different carbon fuels on a hybrid direct carbon fuel cell. Inter. J. Hydrogen Energy 42(25), 16279–16287 (2017). https://doi.org/10.1016/j.ijhydene.2017.05.150
Lee, J.Y., Song, R.H., Lee, S.B., Lim, T.H., Park, S.J., Shul, Y.G., Lee, J.W.: A performance study of hybrid direct carbon fuel cells: Impact of anode microstructure. Inter. J. Hydrogen Energy 39(22), 11749–11755 (2014). https://doi.org/10.1016/j.ijhydene.2014.05.145
Ma, J., Zhang, B., Hou, X., Gong, J., Yu, H., Xu, R., Jiang, C.: The function of carbonate in a hybrid direct carbon fuel cell. Solid State Ion. 344, 115094–115099 (2020). https://doi.org/10.1016/j.ssi.2019.115094
McKee, D., Chatterji, D.: The catalytic behavior of alkali metal carbonates and oxides in graphite oxidation reactions. Carbon 13(5), 381–390 (1975). https://doi.org/10.1016/0008-6223(75)90006-8
McKee, D.W.: Mechanisms of the alkali metal catalysed gasification of carbon. Fuel 62(2), 170–175 (1983). https://doi.org/10.1016/0016-2361(83)90192-8
McKee, D., Chatterji, D.: The catalyzed reaction of graphite with water vapor. Carbon 16(1), 53–57 (1978). https://doi.org/10.1016/0008-6223(78)90116-1
Nagase, K., Shimodaira, T., Itoh, M., Zheng, Y.: Kinetics and mechanisms of the reverse Boudouard reaction over metal carbonates in connection with the reactions of solid carbon with the metal carbonates. Phys. Chem. Chem. Phys. 1(24), 5659–5664 (1999). https://doi.org/10.1039/A906687J
Selman, J.R.: Molten-salt fuel cells—technical and economic challenges. J. Power Sources 160(2), 852–857 (2006). https://doi.org/10.1016/j.jpowsour.2006.04.126
Kulkarni, A., Giddey, S.: Materials issues and recent developments in molten carbonate fuel cells. J. Solid State Electr. 16(10), 3123–3146 (2012). https://doi.org/10.1007/s10008-012-1771-y
Kudo, T., Hisamitsu, Y., Kihara, K., Mohamedi, M., Uchida, I.: X-ray diffractometric study of in situ oxidation of Ni in Li/K and Li/Na carbonate eutectic. J. Power Sources 104(2), 272–280 (2002). https://doi.org/10.1016/S0378-7753(01)00962-4
Doyon, J.D., Gilbert, T., Davies, G., Paetsch, L.: NiO solubility in mixed alkali/alkaline earth carbonates. J. Electrochem. Soc. 134(12), 3035 (1987). https://doi.org/10.1149/1.2100335
Scaccia, S.: Investigation on NiO solubility in binary and ternary molten alkali metal carbonates containing additives. J. Mol. Liq. 116(2), 67–71 (2005). https://doi.org/10.1016/j.molliq.2004.07.078
Huang, B., Li, F., Yu, Q.-C., Chen, G., Zhao, B.-Y., Hu, K.-A.: Study of NiO cathode modified by ZnO additive for MCFC. J. Power Sources 128(2), 135–144 (2004). https://doi.org/10.1016/j.jpowsour.2003.10.008
Kim, S.-G., Yoon, S.P., Han, J., Nam, S.W., Lim, T.H., Oh, I.-H., Hong, S.-A.: A study on the chemical stability and electrode performance of modified NiO cathodes for molten carbonate fuel cells. Electrochim. Acta. 49(19), 3081–3089 (2004). https://doi.org/10.1016/j.electacta.2004.01.027
Tanimoto, K., Kojima, T., Yanagida, M., Nomura, K., Miyazaki, Y.: Optimization of the electrolyte composition in a (Li0.52Na0.48)2-2xAExCO3(AE=Ca and Ba) molten carbonate fuel cell. J. Power Sources 131(12), 256–260 (2004). https://doi.org/10.1016/j.jpowsour.2003.11.085
Soler, J., Gonzalez, T., Escudero, M., Rodrigo, T., Daza, L.: Endurance test on a single cell of a novel cathode material for MCFC. J. Power Sources 106(1–2), 189–195 (2002). https://doi.org/10.1016/S0378-7753(01)01041-2
Matsuzawa, K., Mizusaki, T., Mitsushima, S., Kamiya, N., Ota, K.-I.: The effect of La oxide additive on the solubility of NiO in molten carbonates. J. Power Sources 140(2), 258–263 (2005). https://doi.org/10.1016/j.jpowsour.2004.08.041
Daza, L., Rangel, C., Baranda, J., Casais, M., Martınez, M., Alonso, J.: Modified nickel oxides as cathode materials for MCFC. J. Power Sources 86(1–2), 329–333 (2000). https://doi.org/10.1016/S0378-7753(99)00499-1
Ota, K.-I., Matsuda, Y., Matsuzawa, K., Mitsushima, S., Kamiya, N.: Effect of rare earth oxides for improvement of MCFC. J. Power Sources 160(2), 811–815 (2006). https://doi.org/10.1016/j.jpowsour.2006.04.055
Liu, Z., Guo, P., Zeng, C.: Effect of Dy on the corrosion of NiO/Ni in molten (0.62Li, 0.38K)2CO3. J. Power Sources 166(2), 348–353 (2007). https://doi.org/10.1016/j.jpowsour.2007.01.063
Suski, L., Kołacz, J., Mordarski, G., Ruggiero, M.: Determination of open-circuit potentials at gas/electrode/YSZ boundary versus molten carbonate reference electrode at medium temperatures. Electrochim. Acta. 50(14), 2771–2780 (2005). https://doi.org/10.1016/j.electacta.2004.11.023
Lee, J.-A., Lee, H.-C., Heo, Y.-W., Lee, J.-H., Kim, J.-J.: Effect of Li doping on sintering characteristics and microstructural behavior of yttria-stabilized zirconia. Ceram. Int. 42(15), 17339–17346 (2016). https://doi.org/10.1016/j.ceramint.2016.08.030
Kohli, R.: The thermodynamic properties of alkali metal compounds at high temperatures. J. Therm. Anal. Calorim. 41(6), 1571–1576 (1994). https://doi.org/10.1007/BF02549955
Chandrakala, H.N., Ramaraj, B.: Shivakumaraiah, siddaramaiah: influence of lithium potassium zirconate nanoparticles on the electrical properties and structural characteristics of poly(vinyl alcohol) films. J. Phys. Chem. Solids 75(2), 252–258 (2014). https://doi.org/10.1016/j.jpcs.2013.09.025
Pasierb, P., Gajerski, R., Komornicki, S., RÄkas, M.: Reactivity of lithium and barium carbonates with ZrO2:Y2O3 and Nasicon in solid state electrochemical gas sensors. J. Therm. Anal. Calorim. 77, 105–113 (2004). https://doi.org/10.1023/B:JTAN.0000033193.98853.d0
Wade, J.L., Lee, C., West, A.C., Lackner, K.S.: Composite electrolyte membranes for high temperature CO2 separation. J. Membr. Sci. 369(1–2), 20–29 (2011). https://doi.org/10.1016/j.memsci.2010.10.05
Xu, X., Zhou, W., Zhu, Z.: Stability of YSZ and SDC in molten carbonate eutectics for hybrid direct carbon fuel cells. RSC Adv. 4(5), 2398–2403 (2014). https://doi.org/10.1039/c3ra46600k
Badwal, S., Ciacchi, F.T., Drennan, J.: Investigation of the stability of ceria-gadolinia electrolytes in solid oxide fuel cell environments. Solid State Ion. 121(1–4), 253–262 (1999). https://doi.org/10.1016/S0167-2738(99)00044-2
Guo, L.Q., Zhao, X.M., Wang, B.C., Bai, Y., Xu, B.Z., Qiao, L.J.: The initial stage of atmospheric corrosion on interstitial free steel investigated by in situ SPM. Corros. Sci. 70, 188–193 (2013). https://doi.org/10.1016/j.corsci.2013.01.028
Jiao, Y., Tian, W., Chen, H., Shi, H., Yang, B., Li, C., Shao, Z., Zhu, Z., Li, S.-D.: In situ catalyzed boudouard reaction of coal char for solid oxide-based carbon fuel cells with improved performance. Appl. Energy 141, 200–208 (2015). https://doi.org/10.1016/j.apenergy.2014.12.048
Ali, A., Shehzad Bashir, F., Raza, R., Rafique, A., Kaleem Ullah, M., Alvi, F., Afzal, M., Ghauri, M., Belova, L.M.: Electrochemical study of composite materials for coal-based direct carbon fuel cell. Inter. J. Hydrogen Energy 43(28), 12900–12908 (2018). https://doi.org/10.1016/j.ijhydene.2018.05.104
Jiang, C., Ma, J., Ana, A., John, T.S.I.: Hybrid direct carbon fuel cells with different types of mineral coal. ECS Trans. 57(1), 3013–3021 (2013). https://doi.org/10.1149/05701.3013ecst
Jiang, C., Ma, J., Arenillas, A., Irvine, J.T.: Application of ternary carbonate in hybrid direct coal fuel cells. ECS Trans. 59(1), 281–288 (2014). https://doi.org/10.1149/05901.0281ecst
Chien, A.C., Arenillas, A., Jiang, C., Irvine, J.T.S.: Performance of direct carbon fuel cells operated on coal and effect of operation mode. J. Electrochem. Soc. 161(5), F588–F593 (2014). https://doi.org/10.1149/2.025405jes
Ju, H., Eom, J., Lee, J.K., Choi, H., Lim, T.-H., Song, R.-H., Lee, J.: Durable power performance of a direct ash-free coal fuel cell. Electrochim. Acta. 115, 511–517 (2014). https://doi.org/10.1016/j.electacta.2013.10.124
Devyatkin, S.V., Pisanenko, A.D., Shapoval, V.I.: Chemical and electrochemical behavior of carbonate melts containing silicon oxide. Russ. J. Appl. Chem. 75(4), 562–564 (2002). https://doi.org/10.1023/A:101950482
Tulloch, J., Allen, J., Wibberley, L., Donne, S.: Influence of selected coal contaminants on graphitic carbon electro-oxidation for application to the direct carbon fuel cell. J. Power Sources 260(15), 140–149 (2014). https://doi.org/10.1016/j.jpowsour.2014.03.026
Eom, S., Ahn, S., Kang, K., Choi, G.: Correlations between electrochemical resistances and surface properties of acid-treated fuel in coal fuel cells. Energy 140(2), 885–892 (2017). https://doi.org/10.1016/j.energy.2017.09.034
Eom, S., Ahn, S., Choi, G.: Effect of fuel acid treatment on the reduction of electrochemical resistance in a direct carbon fuel cell system. Energy Fuel 35(5), 4493–4501 (2021). https://doi.org/10.1021/acs.energyfuels.0c03714
Xie, H., Zhai, S., Chen, B., Liu, T., Zhang, Y., Ni, M., Shao, Z.: Coal pretreatment and Ag-infiltrated anode for high-performance hybrid direct coal fuel cell. Appl. Energy (2020). https://doi.org/10.1016/j.apenergy.2019.114197
Dudek, M.: On the utilization of coal samples in direct carbon solid oxide fuel cell technology. Solid State Ion. 271(2), 121–127 (2015). https://doi.org/10.1016/j.ssi.2014.09.034
Kim, J.-W., Lee, Y.-D., Lee, H.-G.: Decomposition of Li2CO3 by interaction with SiO2 in mold flux of steel continuous casting. ISIJ Int. 44(2), 334–341 (2004). https://doi.org/10.2355/isijinternational.44.334
Li, C., Yi, H., Eom, S., Choi, G., Choi, T.-Y., Lee, D.: Intrinsic solid-state reaction characteristics of coals and chars in a direct carbon fuel cell: with focus on significance assessment of fuel-borne factors. Energy Fuel 34(4), 4129–4138 (2020). https://doi.org/10.1021/acs.energyfuels.9b04387
Cai, W., Liu, J., Liu, P., Liu, Z., Xu, H., Chen, B., Li, Y., Zhou, Q., Liu, M., Ni, M.: A direct carbon solid oxide fuel cell fueled with char from wheat straw. Inter. J. Hydrogen Energy 43(7), 2468–2477 (2018). https://doi.org/10.1002/er.3968
Xu, L.J., Liu, S.A., Wang, Z.Z., Liu, C.Y., Li, S.G.: Zinc isotopic compositions of migmatites and granitoids from the Dabie Orogen, central China: Implications for zinc isotopic fractionation during differentiation of the continental crust. Lithos 324, 454–465 (2019). https://doi.org/10.1016/j.lithos.2018.11.028
Peng, F., Li, Y., Nash, P., Cooper, J.F., Parulekar, S.J., Selman, J.R.: Direct carbon fuel cells–wetting behavior of graphitic carbon in molten carbonate. Inter. J. Hydrogen Energy 41(41), 18858–18871 (2016). https://doi.org/10.1016/j.matchemphys.2006.06.006
Chen, M., Wang, C., Niu, X., Zhao, S., Tang, J., Zhu, B.: Carbon anode in direct carbon fuel cell. Inter. J. Hydrogen Energy 35(7), 2732–2736 (2010). https://doi.org/10.1016/j.ijhydene.2009.04.051
Hong, S.-G., Selman, J.R.: Wetting characteristics of carbonate melts under MCFC operating conditions. J. Electrochem. Soc. 151(1), A77–A84 (2004). https://doi.org/10.1149/1.1629094
Cao, D., Wang, G., Wang, C., Wang, J., Lu, T.: Enhancement of electrooxidation activity of activated carbon for direct carbon fuel cell. Inter. J. Hydrogen Energy 35(4), 1778–1782 (2010). https://doi.org/10.1016/j.ijhydene.2009.12.133
Watanabe, H., Kimura, A., Okazaki, K.: Impact of ternary carbonate composition on the morphology of the carbon/carbonate slurry and continuous power generation by direct carbon fuel cells. Energy Fuel 30(3), 1835–1840 (2016). https://doi.org/10.1021/acs.energyfuels.5b02224
Ahn, S.Y., Eom, S.Y., Rhie, Y.H., Sung, Y.M., Moon, C.E., Choi, G.M., Kim, D.J.: Application of refuse fuels in a direct carbon fuel cell system. Energy 51(1), 447–456 (2013). https://doi.org/10.1016/j.energy.2012.12.025
Gür, T.M.: Solid fuel utilization in high temperature fuel cells. ECS Trans. 41(12), 29–38 (2019). https://doi.org/10.1149/1.3697426
Jayakumar, A., Küngas, R., Roy, S., Javadekar, A., Buttrey, D.J., Vohs, J.M., Gorte, R.J.: A direct carbon fuel cell with a molten antimony anode. Energy Environ. Sci. (2011). https://doi.org/10.1039/c1ee01863a
Acknowledgements
The researchers appreciate the funding support provided by the Sichuan Science and Technology Program (Grant number 2019YFH0177, 2021YFH0092), the Zigong Science and Technology Program (Grant number 2019YYJC24, 2020YGJC18) and the Talent Introduction Plan of Sichuan University of Science and Engineering (Grant numbers 2016RCL36, 2016RCL37).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cui, C., Li, S., Gong, J. et al. Review of molten carbonate-based direct carbon fuel cells. Mater Renew Sustain Energy 10, 12 (2021). https://doi.org/10.1007/s40243-021-00197-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40243-021-00197-7