
Abstract
Purpose
Monogenic diabetes (MD) is caused by a mutation in a single gene and accounts for approximately 2.5–6% of all diabetes cases. Maturity-onset diabetes of the young (MODY) is the most common form of MD. To date, 14 different genes have been identified and associated with the presence of MODY phenotype. However, the number of potential candidate genes with relevance to beta cell function and glucose metabolism is increasing as more research is published. The aim of the study was to identify potentially causative variants in selected candidate genes in patients with a clinical diagnosis of MD.
Methods
Targeted Next-Generation Sequencing (tNGS) on Illumina NextSeq 550 platform involving Agilent SureSelectQXT Target Enrichment protocol for 994 patients with suspected MD was performed. In the next step, the sequencing data of 617 patients with no pathogenic variants in main MD-related genes were reanalysed for the presence of causative variants in six candidate genes (MTOR, TBC1D4, CACNA1E, MNX1, SLC19A2, KCNH6). The presence of the selected variants was confirmed by Sanger sequencing.
Results
Seven heterozygous possibly damaging variants were identified in four candidate genes (MTOR, TBC1D4, CACNA1E, MNX1). Five changes were assessed as novel variants, not previously described in available databases. None of the described variants were present among patients previously diagnosed with MODY diabetes due to causative, pathogenic variants in known MODY-related genes.
Conclusions
The results obtained seem to confirm the effectiveness of the NGS method in identifying potentially causative variants in novel candidate genes associated with MODY diabetes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Monogenic diabetes (MD), including maturity-onset diabetes of the young (MODY), neonatal diabetes mellitus (NDM) and syndromic forms of diabetes, are rare diseases caused by pathogenic/ likely pathogenic variants of a single gene. It is estimated that monogenic forms of diabetes represent around 2.5–6% of all diabetes cases [1, 2]. MODY diabetes is mainly diagnosed in adolescents or adults and is the most common type of MD [3,4,5]. Classically, MODY diabetes is characterised by mild/progressive hyperglycaemia, autosomal dominant inheritance, early onset of diabetes, absence of ketoacidosis and autoantibodies characteristic for autoimmune forms of diabetes, preserved insulin secretion, normal BMI and positive family history of diabetes [2, 6, 7].
To date, variants in at least 14 different genes have been identified as a cause of MODY diabetes. They include: GCK, HNF1A, HNF4A, HNF1B, INS, NEUROD1, PDX1, PAX4, ABCC8, KCNJ11, KLF11, CEL, BLK and APPL1 genes. Pathogenic variants in the glucokinase gene (GCK), hepatocyte nuclear factor 1α (HNF1A) and hepatocyte nuclear factor 4 alpha (HNF4A) are most commonly found in patients with suspected MD [8].
In the latest large studies conducted among Polish patients with a suspected diagnosis of MD, the highest number of heterozygous causative variants was confirmed in the GCK and HNF1A genes [9, 10], which is consistent with the results obtained in the similar studies in France [11], Russia [12], Lithuania [13], North America [14] and Canada [15]. Another study conducted among 45 Polish patients with long-term T1DM without advanced complications, identified 9 patients as carriers of 10 variants in 4 genes associated with MODY diabetes (ABCC8, GCK, HNF1A, HNF1B), confirming that MODY cases are frequently misdiagnosed as type 1 or type 2 diabetes [16].
However, still a significant number of patients remain undiagnosed and genetic background of their disease is unknown (the so-called MODY-X cases). In individual cases, other genes may also be involved in the aetiology of MODY diabetes [17, 18]. Candidate genes are still identified based on their involvement in carbohydrate metabolism and insulin signalling [19].
Next-generation sequencing (NGS) is a powerful method, that may be used to discover novel causative genes in the MODY-X cases. Correct genetic diagnosis is important for determining the appropriate treatment for patients with diabetes [17, 19].
In the present study, NGS analysis was performed in a cohort of Polish patients with the MODY clinical phenotype to increase the possibility of identifying the pathogenic variant in the novel candidate genes that we selected: MTOR, TBC1D4, CACNA1E, MNX1, SLC19A2, KCNH6.
Materials and methods
Subjects
The study group consisted of 994 subjects (F/M; 48.9/ 51.1%), referred to the Genetic Outpatient Clinic of the Centre for Rare Diseases in Lodz, Poland (between 2016 and 2022) with suspected MD [10, 19]. The median age of the study participants at diagnosis was 12 years.
The inclusion clinical criteria for the study were as follows: presence of hyperglycaemia (H) or diabetes mellitus (DM) recognised according to the WHO definition, positive family history of diabetes, preserved insulin secretion and absence of autoantibodies and ketoacidosis at clinical diagnosis. The following parameters were analysed for all patients: HbA1c value (%) and fasting C-peptide value (ng/mL) at the time of clinical diagnosis of H/DM, the presence of T1DM-specific antibodies and body mass index (BMI). The clinical characteristics of the study participants are shown in Table 1.
The project was approved by the Bioethics Committee of the Medical University of Lodz (RNN/148/13/KE). Patients gave written informed consent for participation in the study.
Molecular analysis
Genomic DNA was isolated from peripheral blood samples using an automated Maxwell system (Maxwell® RSC Blood DNA Kit, Promega, Madison, USA). The quantitative and qualitative assessment of the extracted DNA was performed on a NanoPhotometer® N80 (Implen, USA). The library was prepared using the Agilent SureSelectQXT Target Enrichment protocol with a custom gene panel according to the manufacturer’s instructions. Into the wells of a 96-well plate, 5 µl each of 10ng/µl DNA samples were transferred and subjected to enzymatic fragmentation. Then adapters were added to the ends of the fragments in a single reaction. The libraries were purified using AMPure XP magnetic beads. Then, the adaptor-tagged gDNA libraries were repaired and PCR-amplified. The products were again purified using AMPure XP magnetic beads. The concentration and size of the resulting product were assessed using an Agilent TapeStation and a dedicated D1000 ScreenTape and reagent kit. In the next step, the prepared gDNA samples were hybridised to the target-specific Capture Library. Then the hybridised DNA was captured using streptavidin-coated beads and enriched DNA libraries were PCR- amplified using appropriate pair of dual indexing primers. The resulting products were again purified using AMPure XP magnetic beads. The concentrations and sizes of the indexed DNA libraries were assessed using an Agilent TapeStation and a dedicated HS1000 ScreenTape and reagent kit. The indexed samples were pooled, diluted and combined with PhiX control. Targeted next generation sequencing (NGS) was performed on Illumina NextSeq 550 platform, (2 × 150 bp).
Initially, a 15-gene NGS panel for MD including HNF4A, GCK, HNF1A, PAX4, HNF1B, NEUROD1, APPL1, KLF11, CEL, BLK, PDX1, ABCC8, KCNJ11, INS and WFS1 was analysed. In the next step, the sequencing data of patients with no pathogenic variants in the 15 studied genes were reanalysed for the presence of causative variants in six candidate genes: MTOR, TBC1D4, CACNA1E, MNX1, SLC19A2, KCNH6. The candidate genes were selected based on their involvement in carbohydrate metabolism and insulin signalling [20,21,22,23,24,25].
Bioinformatic analysis
Alignments and variant calling were performed using BWA Enrichment v2.1 BaseSpace application (Illumina, San Diego, USA). Variant annotations and filtering were carried out in Illumina Variant Studio Software (Illumina, San Diego, USA). Minor allele frequency (MAF) < 0.01 for the European population in the GnomAD and ExAC database was used to filter significant variants in the candidate genes. The potential damaging role of the variants was determined in silico using web-based software, such as PROVEAN and Mutation Taster. The pathogenicity of detected genetic variants was evaluated according to the guidelines of the American College of Medical Genetics and Genomics (ACMG) [26].
Variant confirmation
The presence of selected variants was confirmed by bidirectional Sanger sequencing on a 3500 Series Genetic Analyzer (Applied Biosystems, Waltham, MA, USA). Sequence analysis was performed using Sequencher 5.0 software.
Statistical analysis
The normality of linear values distribution was assessed using the Shapiro-Wilk test. Medians (Me) and interquartile ranges (IQR 25-75%) were used to present continuous variables. Nominal values were presented as a percentage (%) for the study group. Statistical analysis was performed using STATISTICA 13.3 software (Statsoft, Tulsa, OK, USA).
Results
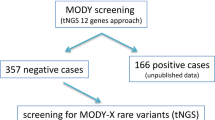
A total of 994 patients with suspected MD were included in the study. Pathogenic and likely pathogenic variants responsible for causing MD were identified in 377 patients and were found in eight different MD-related genes: ABCC8, GCK, HNF1A, HNF1B, HNF4A, KCNJ11, PDX1, WFS1. Subsequently, reanalysis of the sequencing data revealed seven heterozygous possibly causative candidate gene variants in the negative patient cohort. Three of them were in the MTOR gene, one in the TBC1D4, two in the CACNA1E and one in the MNX1 gene. No pathogenic or likely pathogenic variant was identified in the SLC19A2 and KCNH6 genes that could potentially cause MODY diabetes in the study group (Fig. 1).

Details of molecular diagnosis in patients with suspected monogenic diabetes
Details of the identified variants are contained in Table 2. Among the seven variants identified in the candidate genes, four were missense, one frameshift, one in- frame deletion and one start-lost mutations. Five changes were assessed as novel variants not previously described in available databases. Moreover, high conservativeness in the studied variants (except p.His196_Ala198del in MNX1 gene) was confirmed among five species (G.gorilla, M.musculus, C. lupus, O. cuniculus, S. scrofa). The selected variants were identified in a total of seven patients. The clinical characteristics of these patients are presented in Table 3.
The median age of clinical diagnosis of hyperglycaemia or diabetes in these patients was 13 years and was 1 year lower than the median age at the time of genetic testing (Me = 14.0 years). Five out of the seven patients (71%) had a positive family history of diabetes. The HbA1c level at clinical diagnosis was different among patients (IQR 25-75% − 5.5–6.9%). A decreased C-peptide level − 0.7 ng/ml (normal range: 0.9-4.0 ng/ml) was observed in two cases. Three patients were treated with insulin therapy, while four were exclusively on a diet with low glycaemic index (IG) at the time of the genetic diagnosis.
None of the described variants were present among 377 patients who were previously diagnosed with MODY diabetes due to changes in known MODY-related genes, which confirms that the selected changes are not the methodological errors or artifacts. In addition, the presence of selected variants was confirmed by bidirectional Sanger sequencing in all patients.
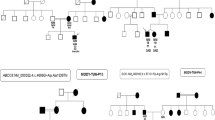
The analysis of segregation in the family was only possible for the c.6187delC variant in the MTOR gene. A heterozygous c.6187delC variant was identified in a 14-year-old female patient with diabetes diagnosed at the age of 13 and in her father with suspected type 2 diabetes, while it was not present in her asymptomatic mother (Figs. 2 and 3).

Pedigree of family with heterozygous c.6187delC variant in the MTOR gene. Genotype is shown underneath each symbol. Squares represent male family members, and circles represent female sex. Blue-filled symbols denote patients with diabetes, an arrow denotes the proband in the family

Sanger sequencing chromatogram showing the heterozygous c.6187delC variant in exon 44 of the MTOR gene identified in the proband and in her father (a-proband, b-father, c-mother)
Discussion
This is the first study to analyse the sequence of the MTOR, TBC1D4, CACNA1E, SLC19A2 and KCNH6 genes in Polish patients with suspected MODY-X diabetes using the NGS method. Seven heterozygous possibly causative variants in candidate genes were identified in 7 of 617 previously undiagnosed patients with a positive family history, preserved insulin secretion and absence of autoantibodies characteristic for autoimmune diabetes. No potentially pathogenic variant associated with diabetes was detected in the remaining patients. According to the results, it is difficult to identify a specific type of monogenic diabetes in the above patients. Therefore, in such cases, whole exome sequencing (WES) could be an important complement to the diagnostics [17, 27].
In our study group, three of the selected variants are present in the MTOR gene encoding mammalian target of rapamycin (mTOR), which play an important role in regulating cell growth as well as lipid and glucose metabolism. Numerous studies have shown that MTOR is an important gene in the insulin signalling pathway and susceptibility to type 2 diabetes mellitus [20, 28, 29]. mTOR exists in two structurally and functionally distinct multiprotein complexes – mTORC1 and mTORC2. The effects of mTOR on glucose homeostasis are complex and depend on the level of mTORC1 activity. Activation of mTORC1 in β-cells increases insulin secretion and thus reduces blood glucose levels. However, a sustained activation of mTORC1 leads to exhaustion of insulin secreting capacity by β-cells and to impaired glucose homeostasis. There are several reports that treatment with mTOR inhibitors in various cancers is associated with a high incidence of hyperglycaemia and new-onset diabetes [30]. These findings could lead to the hypothesis that both gain-of-function and loss-of-function MTOR variants may cause hyperglycaemia or diabetes. All changes indicated in MTOR gene in this study are novel variants, not previously described in available databases (dbSNP, ClinVar, HGMD). There are no data on the frequency of these variants in the gnomAD and ExAC databases. Unfortunately, detailed clinical characteristics of the patient with the identified p.Ser916Asn variant were not available. However, the other two patients with the p.Gln2063ArgfsTer3 and p.Phe871Cys variants in the MTOR gene had a positive family history, normal C-peptide and HbA1c level, absence of autoantibodies characteristic for autoimmune diabetes and were exclusively on a nutrition therapy. The examination of the proband’s family confirmed the co-segregation of the c.6187delC (p.Gln2063ArgfsTer3) variant with diabetes, which may confirm variant pathogenicity. Based on the above, we could suppose that the MTOR gene is associated with rare cases of MODY-X.
Another variant was identified in the TBC1D4 gene encoding the TBC1 domain family member 4 protein. This protein play an important role in glucose homeostasis by regulating trafficking of the glucose transporter 4 (GLUT4), which is insulin-dependent and important for removing glucose from the bloodstream into skeletal muscle and fat tissues [31]. The heterozygous start-loss variant c.2T > C (p.Met1?) identified in our patient has already been reported in the dbSNP database and is classified as likely pathogenic according to the ACMG guidelines. In silico analysis showed that this variant leads to the loss of the first methionine, thus precluding the translation initiation of the corresponding protein. Other potentially damaging variants of TBC1D4 were previously reported to cause a higher risk of type 2 diabetes and insulin resistance [31, 32]. Moltke et al. identified a common nonsense variant (p.R684Ter) in the Greenlandic population in TBC1D4, which in homozygous carriers causes insulin resistance and increases the risk of type 2 diabetes. However, heterozygous carriers had only slightly higher plasma glucose concentration 2 h after an oral glucose load [32]. We suppose that rare heterozygous variants with more deleterious effects could be involved in MODY, while those with a smaller effect might have a role in type 2 diabetes. Similarly, variants of the KCNJ11 and HNF4A genes could cause MODY or type 2 diabetes, depending on how the variant affects the protein function [33]. However, this is only a hypothesis and we cannot confirm that the variant in TBC1D4 was directly causative and correlated with diabetes in our patient.
Next, two novel variants in the CACNA1E gene were identified. The nonsynonymous p.Tyr1469His variant is located in the 31st exon of the CACNA1E gene. Both predictive programs used indicate the damaging nature of this change. The second variant (p.Ala324Asp) is located in the seventh exon. Predictive programs show conflicting information about the pathogenicity of this variant. Both variants were classified as variants of uncertain significance (VUS) according to the ACMG guidelines. CACNA1E was selected as a candidate gene because of their previously reported role in the risk of type 2 diabetes, insulin resistance, impaired insulin secretion and regulation of β-cell function [22, 34]. The generation of β cell electrical activity that results from the functional interaction of the ATP-sensitive beta cell K + channel (encoded by the KCNJ11 gene) and voltage-dependent Ca2 + channels (VDCCs) is required to insulin secretion. Human pancreatic β cells express several types of VDCCs including the CaV1.2 and CaV2.3. The CaV2.3 channel, encoded by CACNA1E gene is responsible for sustained second-phase insulin release. The absence of the CaV2.3 channel in mice causes fasting hyperglycaemia and reduced glucose clearance. Holkmvist et al. concluded that the CaV2.3 channel could play an important role in regulating insulin secretion, thus CACNA1E is an relevant candidate gene in type 2 diabetes [35]. The presence of two potentially causative CACNA1E variants in two patients from our cohort seems to suggest that this gene may also play a role in monogenic diabetes. In addition, patients with the p.Ala324Asp variant had slightly decreased C-peptide levels (0.7 ng/ml), which could indicate a defect in insulin secretion caused by a change in the CACNA1E gene. Thus, by analogy, it can be concluded that rare severe variants in the CACNA1E gene may cause monogenic diabetes, as is the case with the HNF1β, GCK or KCNJ11 genes, while common single nucleotide alterations are associated with an increased risk of type 2 diabetes [36].
The last variant was identified in the first exon of the MNX1 gene which encodes a homeobox transcription factor described as important for pancreatic beta cell differentiation and development [23, 37]. The p.His196_Ala198del variant identified in the present study has been previously reported in the dbSNP and ClinVar databases as VUS. The prevalence of this variant in the gnomAD database is 0.0000208 for the European population. Therefore, it can be concluded that this is a very rare variant, classified as VUS according to the ACMG criteria. Heterozygous loss of function variants in MNX1 cause Currarino syndrome, a rare clinical condition that is characterised by presacral mass, sacral agenesis and an anorectal anomaly [23]. Several homozygous variants in the MNX1 gene have already been described in the literature as associated with the phenotype of persistent neonatal diabetes mellitus (PNDM). All of these patients were diagnosed in infancy [23, 37, 38]. In contrast, a heterozygous variant was identified in our patient, who was diagnosed with diabetes at the age of 53. It is possible that, similarly to the GCK and RFX6 genes, biallelic variants cause a NDM, while the heterozygous variants are associated with MODY diabetes [3, 39].
Unfortunately, our study has several limitations. Due to the lack of available DNA from relatives, it was not possible to perform a family segregation analysis for all variants, and the inference is based only on in silico prediction and descriptions in available databases. Similarly, we did not have the BMI values, C-peptide level and HbA1c level for one patient with the MTOR gene variant, which could have enriched the clinical characteristics of patients with the MTOR gene changes. Another limitation of the present study is that no functional studies have been conducted in order to explore the biological relevance of the variants on glucose metabolism. Furthermore, the final confirmation of the effect of selected candidate genes on the MODY disease phenotype requires testing on a larger group of carriers, performing a family segregation analysis and functional studies.
In conclusion, the results obtained in this study seem to confirm the effectiveness of the NGS method in identifying potentially pathogenic variants in novel candidate gene related to carbohydrate metabolism and insulin signalling.
The use of multigene panel testing based on next-generation sequencing increases the chance of accurately diagnosing extremely rare forms of MODY, and thus implementing appropriate treatment in patients. However, still a significant number of patients remain undiagnosed, and in such cases, WES or WGS methods can play a key role in understanding the genetic basis of the disease. In the future, it is planned to conduct further studies on a larger group of patients with suspected MD, obtain biological material from family members and conduct a family segregation analysis to definitively confirm or exclude the influence of selected candidate genes on the MODY disease phenotype.
References
Yang YS, Kwak SH, Park KS. Update on monogenic diabetes in Korea. Diabetes Metab J. 2020;44(5):627–39. https://doi.org/10.4093/dmj.2020.0214.
Greeley SAW, Polak M, Njølstad PR, Barbetti F, Williams R, Castano L, Raile K, Chi DV, Habeb A, Hattersley AT, Codner E. ISPAD Clinical Practice Consensus Guidelines 2022: the diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes. 2022;23(8):1188–211. https://doi.org/10.1111/pedi.13426.
De Franco E. From Biology to genes and back again: Gene Discovery for monogenic forms of Beta-cell dysfunction in diabetes. J Mol Biol. 2020;432(5):1535–50. https://doi.org/10.1016/j.jmb.2019.08.016.
De Franco E, Flanagan SE, Houghton JA, Lango Allen H, Mackay DJ, Temple IK, Ellard S, Hattersley AT. The effect of early, comprehensive genomic testing on clinical care in neonatal diabetes: an international cohort study. Lancet. 2015;386(9997):957–63. https://doi.org/10.1016/S0140-6736(15)60098-8.
Peixoto-Barbosa R, Reis AF, Giuffrida FMA. Update on clinical screening of maturity-onset diabetes of the young (MODY). Diabetol Metab Syndr. 2020;12:50. https://doi.org/10.1186/s13098-020-00557-9. PMID: 32528556; PMCID: PMC7282127.
McDonald TJ, Ellard S. Maturity onset diabetes of the young: identification and diagnosis. Ann Clin Biochem. 2013;50(Pt5):403–15. https://doi.org/10.1177/0004563213483458.
Breidbart E, Deng L, Lanzano P, Fan X, Guo J, Leibel RL, LeDuc CA, Chung WK. Frequency and characterization of mutations in genes in a large cohort of patients referred to MODY registry. J Pediatr Endocrinol Metab. 2021;34(5):633–8. https://doi.org/10.1515/jpem-2020-0501. PMID: 33852230; PMCID: PMC8970616.
Mirshahi UL, Colclough K, Wright CF, Wood AR, Beaumont RN, Tyrrell J, Laver TW, Stahl R, Golden A, Goehringer JM, Geisinger-Regeneron DEHR, Frayling C, Hattersley TF, Carey AT, Weedon DJ, Patel MN. Reduced penetrance of MODY-associated HNF1A/HNF4A variants but not GCK variants in clinically unselected cohorts. Am J Hum Genet. 2022;109(11):2018–28. Epub 2022 Oct 17. PMID: 36257325; PMCID: PMC9674944.
Małachowska B, Borowiec M, Antosik K, Michalak A, Baranowska-Jaźwiecka A, Deja G, Jarosz-Chobot P, Brandt A, Myśliwiec M, Stelmach M, Nazim J, Peczyńska J, Głowińska-Olszewska B, Horodnicka-Józwa A, Walczak M, Małecki MT, Zmysłowska A, Szadkowska A, Fendler W, Młynarski W. Monogenic diabetes prevalence among polish children-summary of 11 years-long nationwide genetic screening program. Pediatr Diabetes. 2018;19(1):53–8. https://doi.org/10.1111/pedi.12532. Epub 2017 Apr 24. PMID: 28436179.
Zmysłowska A, Jakiel P, Gadzalska K, Majos A, Płoszaj T, Ben-Skowronek I, Deja G, Glowinska-Olszewska B, Jarosz-Chobot P, Klonowska B, Kowalska I, Mlynarski W, Mysliwiec M, Nazim J, Noczynska A, Robak-Kontna K, Skala-Zamorowska E, Skowronska B, Szadkowska A, Szypowska A, Walczak M, Borowiec M. Next- generation sequencing is an effective method for diagnosing patients with different forms of monogenic diabetes. Diabetes Res Clin Pract. 2022;183:109154. https://doi.org/10.1016/j.diabres.2021.109154.
Donath X, Saint-Martin C, Dubois-Laforgue D, Rajasingham R, Mifsud F, Ciangura C, Timsit J, Bellanné-Chantelot C. Monogenic diabetes Study Group of the Société Francophone du Diabète. Next-generation sequencing identifies monogenic diabetes in 16% of patients with late adolescence/adult-onset diabetes selected on a clinical basis: a cross-sectional analysis. BMC Med. 2019;17(1):132. https://doi.org/10.1186/s12916-019-1363-0. PMID: 31291970; PMCID: PMC6621990.
Glotov OS, Serebryakova EA, Turkunova ME, Efimova OA, Glotov AS, Barbitoff YA, Nasykhova YA, Predeus AV, Polev DE, Fedyakov MA, Polyakova IV, Ivashchenko TE, Shved NY, Shabanova ES, Tiselko AV, Romanova OV, Sarana AM, Pendina AA, Scherbak SG, Musina EV, Petrovskaia-Kaminskaia AV, Lonishin LR, Ditkovskaya LV, Zhelenina LА, Tyrtova LV, Berseneva OS, Skitchenko RK, Suspitsin EN, Bashnina EB, Baranov VS. Whole exome sequencing in russian children with non type 1 diabetes mellitus reveals a wide spectrum of genetic variants in MODY related and unrelated genes. Mol Med Rep. 2019;20(6):4905–14. Epub 2019 Oct 16. PMID: 31638168; PMCID: PMC6854535.
Stankute I, Verkauskiene R, Blouin JL, Klee P, Dobrovolskiene R, Danyte E, Dirlewanger M, Santoni F, Razanskaite-Virbickiene D, Marciulionyte D, Jasinskiene E, Mockeviciene G, Schwitzgebel VM. Systematic genetic study of Youth with Diabetes in a single country reveals the prevalence of diabetes subtypes, Novel candidate genes, and response to Precision Therapy. Diabetes. 2020;69(5):1065–71. https://doi.org/10.2337/db19-0974. Epub 2020 Feb 21. PMID: 32086287.
Bennett JT, Vasta V, Zhang M, Narayanan J, Gerrits P, Hahn SH. Molecular genetic testing of patients with monogenic diabetes and hyperinsulinism. Mol Genet Metab. 2015;114(3):451–8. https://doi.org/10.1016/j.ymgme.2014.12.304. Epub 2014 Dec 20. PMID: 25555642; PMCID: PMC7852340.
Brahm AJ, Wang G, Wang J, McIntyre AD, Cao H, Ban MR, Hegele RA. Genetic confirmation rate in clinically suspected maturity-onset diabetes of the Young. Can J Diabetes. 2016;40(6):555–60. https://doi.org/10.1016/j.jcjd.2016.05.010. Epub 2016 Sep 12. PMID: 27634015.
Hohendorff J, Kwiatkowska M, Pisarczyk-Wiza D, Ludwig-Słomczyńska A, Milcarek M, Kapusta P, Zapała B, Kieć-Wilk B, Trznadel-Morawska I, Szopa M, Zozulińska-Ziółkiewicz D, Małecki MT. Mutation search within monogenic diabetes genes in polish patients with long-term type 1 diabetes and preserved kidney function. Pol Arch Intern Med. 2022;132(2):16143. https://doi.org/10.20452/pamw.16143. Epub 2021 Nov 26. PMID: 34825797.
Johansson S, Irgens H, Chudasama KK, Molnes J, Aerts J, Roque FS, Jonassen I, Levy S, Lima K, Knappskog PM, Bell GI, Molven A, Njølstad PR. Exome sequencing and genetic testing for MODY. PLoS ONE. 2012;7(5):e38050. https://doi.org/10.1371/journal.pone.0038050.
Yalçın Çapan Ö, Aydın N, Yılmaz T, Berber E. Whole exome sequencing reveals novel candidate gene variants for MODY. Clin Chim Acta. 2020;510:97–104. https://doi.org/10.1016/j.cca.2020.07.005. Epub 2020 Jul 6. PMID: 32645390.
Płoszaj T, Antosik K, Jakiel P, Zmysłowska A, Borowiec M. Screening for extremely rare pathogenic variants of monogenic diabetes using targeted panel sequencing. Endocrine. 2021;73(3):752–7. https://doi.org/10.1007/s12020-021-02753-7.
Yang L, Zhang Z, Wang D, Jiang Y, Liu Y. Targeting mTOR Signaling in type 2 diabetes Mellitus and Diabetes Complications. Curr Drug Targets. 2022;23(7):692–710. https://doi.org/10.2174/1389450123666220111115528.
Binsch C, Barbosa DM, Hansen-Dille G, Hubert M, Hodge SM, Kolasa M, Jeruschke K, Weiß J, Springer C, Gorressen S, Fischer JW, Lienhard M, Herwig R, Börno S, Timmermann B, Cremer AL, Backes H, Chadt A, Al-Hasani H. Deletion of Tbc1d4/As160 abrogates cardiac glucose uptake and increases myocardial damage after ischemia/reperfusion. Cardiovasc Diabetol. 2023;22(1):17. https://doi.org/10.1186/s12933-023-01746-2.
Trombetta M, Bonetti S, Boselli M, Turrini F, Malerba G, Trabetti E, Pignatti P, Bonora E, Bonadonna RC. CACNA1E variants affect beta cell function in patients with newly diagnosed type 2 diabetes. The Verona newly diagnosed type 2 diabetes study (VNDS) 3. PLoS ONE. 2012;7(3):e32755. https://doi.org/10.1371/journal.pone.0032755.
Aly HH, De Franco E, Flanagan SE, Elhenawy YI. MNX1 mutations causing neonatal diabetes: review of the literature and report of a case with extra-pancreatic congenital defects presenting in severe diabetic ketoacidosis. J Diabetes Investig. 2022 Dec;31. https://doi.org/10.1111/jdi.13968.
Sun C, Pei Z, Zhang M, Sun B, Yang L, Zhao Z, Cheng R, Luo F. Recovered insulin production after thiamine administration in permanent neonatal diabetes mellitus with a novel solute carrier family 19 member 2 (SLC19A2) mutation. J Diabetes. 2018;10(1):50–8. https://doi.org/10.1111/1753-0407.12556. Epub 2017 May 29. PMID: 28371426.
Yang JK, Lu J, Yuan SS, Asan, Cao X, Qiu HY, Shi TT, Yang FY, Li Q, Liu CP, Wu Q, Wang YH, Huang HX, Kayoumu A, Feng JP, Xie RR, Zhu XR, Liu C, Yang GR, Zhang MR, Xie CL, Chen C, Zhang B, Liu G, Zhang XQ, Xu A. From hyper- to Hypoinsulinemia and Diabetes: Effect of KCNH6 on insulin secretion. Cell Rep. 2018;25(13):3800–3810e6. https://doi.org/10.1016/j.celrep.2018.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. https://doi.org/10.1038/gim.2015.30.
Yalçın Çapan Ö, Aydın N, Yılmaz T, Berber E. Whole exome sequencing reveals novel candidate gene variants for MODY. Clin Chim Acta. 2020;510:97–104. https://doi.org/10.1016/j.cca.2020.07.005.
Asahara SI, Inoue H, Watanabe H, Kido Y. Roles of mTOR in the regulation of pancreatic β-Cell Mass and insulin secretion. Biomolecules. 2022;12(5):614. https://doi.org/10.3390/biom12050614. PMID: 35625542; PMCID: PMC9138643.
Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–93. https://doi.org/10.1016/j.cell.2012.03.017.
Vergès B, Cariou B. mTOR inhibitors and diabetes. Diabetes Res Clin Pract. 2015;110(2):101–8. https://doi.org/10.1016/j.diabres.2015.09.014. Epub 2015 Sep 21. PMID: 26421362.
.Dash S, Sano H, Rochford JJ, Semple RK, Yeo G, Hyden CS, Soos MA, Clark J, Rodin A, Langenberg C, Druet C, Fawcett KA, Tung YC, Wareham NJ, Barroso I, Lienhard GE, O’Rahilly S, Savage DB. A truncation mutation in TBC1D4 in a family with acanthosis nigricans and postprandial hyperinsulinemia. Proc Natl Acad Sci U S A. 2009;106(23):9350–5. https://doi.org/10.1073/pnas.0900909106.
Moltke I, Grarup N, Jørgensen ME, Bjerregaard P, Treebak JT, Fumagalli M, Korneliussen TS, Andersen MA, Nielsen TS, Krarup NT, Gjesing AP, Zierath JR, Linneberg A, Wu X, Sun G, Jin X, Al-Aama J, Wang J, Borch-Johnsen K, Pedersen O, Nielsen R, Albrechtsen A, Hansen T. A common Greenlandic TBC1D4 variant confers muscle insulin resistance and type 2 diabetes. Nature. 2014;512(7513):190–3. https://doi.org/10.1038/nature13425.
Stankute I, Verkauskiene R, Blouin JL, Klee P, Dobrovolskiene R, Danyte E, Dirlewanger M, Santoni F, Razanskaite-Virbickiene D, Marciulionyte D, Jasinskiene E, Mockeviciene G, Schwitzgebel VM. Systematic genetic study of Youth with Diabetes in a single country reveals the prevalence of diabetes subtypes, Novel candidate genes, and response to Precision Therapy. Diabetes. 2020;69(5):1065–71. https://doi.org/10.2337/db19-0974.
Royer-Bertrand B, Jequier Gygax M, Cisarova K, Rosenfeld JA, Bassetti JA, Moldovan O, O’Heir E, Burrage LC, Allen J, Emrick LT, Eastman E, Kumps C, Abbas S, Van Winckel G, Undiagnosed Diseases Network, Chabane N, Zackai EH, Lebon S, Keena B, Bhoj EJ, Umair M, Li D, Donald KA, Superti-Furga A. De novo variants in CACNA1E found in patients with intellectual disability, developmental regression and social cognition deficit but no seizures. Mol Autism. 2021;12(1):69. https://doi.org/10.1186/s13229-021-00473-3. PMID: 34702355; PMCID: PMC8547031.
Holmkvist J, Tojjar D, Almgren P, Lyssenko V, Lindgren CM, Isomaa B, Tuomi T, Berglund G, Renström E, Groop L. Polymorphisms in the gene encoding the voltage-dependent ca(2+) channel ca (V)2.3 (CACNA1E) are associated with type 2 diabetes and impaired insulin secretion. Diabetologia. 2007;50(12):2467–75. https://doi.org/10.1007/s00125-007-0846-2.
Bonnefond A, Froguel P, Vaxillaire M. The emerging genetics of type 2 diabetes. Trends Mol Med. 2010;16(9):407–16. https://doi.org/10.1016/j.molmed.2010.06.004.
Bonnefond A, Vaillant E, Philippe J, Skrobek B, Lobbens S, Yengo L, Huyvaert M, Cavé H, Busiah K, Scharfmann R, Polak M, Abdul-Rasoul M, Froguel P, Vaxillaire M. Transcription factor gene MNX1 is a novel cause of permanent neonatal diabetes in a consanguineous family. Diabetes Metab. 2013;39(3):276–80. https://doi.org/10.1016/j.diabet.2013.02.007.
Flanagan SE, De Franco E, Lango Allen H, Zerah M, Abdul-Rasoul MM, Edge JA, Stewart H, Alamiri E, Hussain K, Wallis S, de Vries L, Rubio-Cabezas O, Houghton JA, Edghill EL, Patch AM, Ellard S, Hattersley AT. Analysis of transcription factors key for mouse pancreatic development establishes NKX2-2 and MNX1 mutations as causes of neonatal diabetes in man. Cell Metab. 2014;19(1):146–54. https://doi.org/10.1016/j.cmet.2013.11.021.
Filibeli BE, Çatli G, Ayranci İ, Manyas H, Kirbiyik Ö, Dündar B. Childhood-onset mild diabetes caused by a homozygous novel variant in the glucokinase gene. Horm (Athens). 2022;21(1):163–9. https://doi.org/10.1007/s42000-021-00330-1.
Funding
This work was supported by the Grant No. 2015/19/B/NZ5/02243 and 2013/09/B/NZ5/00779 from the National Science Centre, Poland.
Author information
Authors and Affiliations
Contributions
P.J., K.G., E.J. and M.G. performed the experiments. P.J., K.G. and M.B. designed the study. P.J., K.G., E.J., M.G., M.B. and A.Z. analysed the results. T.P., S.S., P.J., K.G. performed bioinformatic analysis. P.J., K.G. and A.Z. wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Consent to participate
All patients signed informed consent forms for participation in the experiment.
Ethical approval
The study protocol was approved by the Bioethics Committee of the Medical University of Lodz, Poland (RNN/148/13/KE).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jakiel, P., Gadzalska, K., Juścińska, E. et al. Identification of rare variants in candidate genes associated with monogenic diabetes in polish mody-x patients. J Diabetes Metab Disord 23, 545–554 (2024). https://doi.org/10.1007/s40200-023-01312-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40200-023-01312-3