
Abstract
Chronic liver injury results in hepatic fibrosis, which is characterized by extensive deposition of extracellular matrix proteins mainly produced by fibrogenic myofibroblasts (MFs) which are not present in the normal liver. Hepatic MFs are a heterogeneous population which is mainly composed of activated hepatic stellate cells (aHSCs), portal fibroblasts (aPFs) and bone marrow-derived circulating fibrocytes, and/or mesenchymal stem cells. While the contribution of aHSCs and aPFs to liver-resident MFs is well documented, the role of fibrocytes in pathogenesis of liver fibrosis is still controversial. It remains unknown if fibrocytes serve as a significant source of MFs or mainly mediate pro-fibrogenic signals to the neighboring cells in response to chronic liver injury. The current review will summarize the up-to-date understanding of the fibrocyte functions, and discuss the similarities and differences of fibrocyte activation in liver and other parenchymal organs. Specifically, here we provide an overview on (1) the role of cytokines and growth factors in recruitment of fibrocytes to the damaged organ; (2) the role of circulating fibrocytes as possible biomarker of fibrogenic diseases; and (3) the role of human Serum amyloid P in pharmacological inhibition of fibrocyte and macrophage recruitment to fibrosing organs as a potential novel strategy for anti-fibrotic therapy.
Similar content being viewed by others
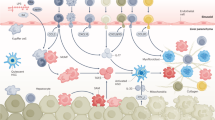

Introduction
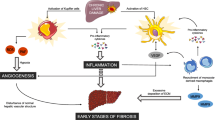
Liver fibrosis is the common outcome of numerous chronic diseases, including viral hepatitis (i.e., hepatitis B and C), alcoholic liver disease, and nonalcoholic steatohepatitis. It is caused by dysregulation of the normal healing process and scar formation, and results in excessive production of extracellular matrix (ECM), mostly collagen type I [1]. Myofibroblasts (MFs), which are not present in normal liver, serve as a major source of ECM in all clinical and experimental liver fibrosis. MFs are characterized by stellate shape, expression of abundant intracellular filaments [a-smooth muscle actin (a-SMA), vimentin], high contractility, and secretion of ECM. Thus, activation and proliferation of hepatic MFs are the key mechanisms in the development of liver cirrhosis [2].
Liver MFs activated in response to injury comprise different cellular populations dependent on the etiology of underlying liver injury [3]. Until now, hepatic stellate cells (HSCs), portal fibroblasts (PFs), and, to a lesser extent, mesothelial cells (MCs) have been identified as resident hepatic cell populations contributing to the generation of MFs during liver fibrosis [3–6]. Liver epithelial cells (e.g., hepatocytes, cholangiocytes, and their progenitors) have been previously suggested to contribute to the intrahepatic source of fibrogenic MFs via epithelial-to-mesenchymal transition (EMT) process during liver fibrosis. However, most recent cell fate mapping studies of mature hepatocytes, cholangiocytes, and their precursors have dismissed this concept [7–9]. In addition, bone marrow (BM)-derived fibrocytes and mesenchymal progenitors have been shown to migrate to the damaged liver and differentiate into hepatic MFs during liver fibrosis [10••, 11]. Here we summarize the current understanding of the fibrocyte biology, their origin and functions, migration, and phenotypical changes occurring in response to fibrogenic injury.
Definition of Fibrocytes
Chronic liver injury causes irreversible damage to hepatocytes. Apoptotic hepatocytes secrete factors that facilitate recruitment and activation of inflammatory cells to the injured liver. Fibrocytes comprise one population of BM-derived cells that are recruited to fibrotic liver [10••, 12]. Fibrocytes were first described by Bucala et al. in 1994 as collagen-producing leukocytes capable of antigen presentation, which could be identified by the simultaneous expression of CD45 and collagen type I [13, 14]. Fibrocytes possess dual characteristics of fibroblasts (expression of collagen type I, fibronectin, and vimentin) and hematopoietic cells (CD45, CD34, MHCII, CD11b, Gr1, Ly6c, CD54, CD80, CD86, CCR2, CCR1, CCR7, and CCR5) [15, 16], and they secrete growth factors that promote deposition of ECM. Under physiological conditions, fibrocytes are primarily located in the BM (comprising 0.1 % of mononuclear cells), but rapidly proliferate in the BM in response to fibrogenic insult, then egress the BM into the blood stream and migrate to the damaged liver [16]. In fibrotic liver, or in response to stimulation with transforming growth factor-β1(TGF-β1), fibrocytes downregulate expression of hematopoietic markers and differentiate into α-SMA+ MFs [16, 17]. Due to their ability to give rise to MFs, fibrocytes were implicated in the pathogenesis of skin, pancreatic, cardiac, lung, kidney, and liver fibrosis [10••, 18–22, 23•, 24–26].
The unique ability of fibrocytes to simultaneously express collagen Type I and CD45-distinguishing fibrocytes from the liver-resident MFs or other hematopoietic cells has been recently developed [10••]. Based on these unique characteristics of fibrocytes, a novel method has been recently developed to track down fibrocytes migration in response to injury or stress. This method utilizes transgenic reporter Collagen-α1(I)-GFP mice in which every cell producing collagen Type I upregulates expression of GFP. Therefore, specific labeling of BM-derived CD45+Collagen-α1(I)+ fibrocytes in real time can be achieved in BM chimeric mice generated by transplantation of the Collagen-α1(I)-GFP+ (Col-GFP) BM into lethally irradiated wild-type recipient mice. The expression of Col-GFP was observed in fibrocytes but not in other hematopoietic cells (such as activated macrophages [27]). Resulted BM chimeric Col-GFP → wt mice were suggested to serve as a useful tool to monitor fibrocyte transmigration from the BM to the peripheral tissues under physiological conditions and in response to fibrogenic injury. Col-GFP → wt mice were successfully used to monitor fibrocyte flux into fibrotic liver, but could be also used to compare the contribution of fibrocytes to fibrogenesis of other organs and tissues. Thus, other studies have used this approach and methodology to visualize fibrocytes recruited to fibrosing kidney [26].
Functional Properties of Fibrocytes
Contribution of Fibrocytes to Liver Fibrosis
It has been reported that contribution of BM-derived fibrocytes to collagen production during the progression of organ fibrosis may vary depending on the etiologies and/or duration of tissue injury, the sites of affected organs, and the stages of tissue fibrosis [28]. Several studies have implicated fibrocytes in the pathogenesis of fibrogenic diseases in lungs, skin, and kidneys, where they contribute to 5–25 % of collagen-producing cells [12].
In experimental liver fibrosis, fibrocytes are recruited to the liver in response to cholestatic (BDL) and hepatotoxic (CCl4) liver injury, and could be accounted for up to 5 % of hepatic collagen-producing cells, suggesting their functional involvement in the pathogenesis of liver fibrosis [29••, 30••]. Unlike BDL-induced liver injury in which recruitment of fibrocytes is limited to fibrotic liver, BM-derived CD45+Col+ fibrocytes were detected in livers, kidneys, and lungs in CCl4-treated mice, suggesting that CCl4 induces a systemic effect. In addition, CD45+Col+ cells were also detected in lymphoid organs such as spleen [29••], but did not cause fibrosis of spleen, indicating that the function of fibrocyte-like cells may not be limited to ECM deposition.
Although fibrocytes are not the major sources of collagen type I deposition in fibrotic liver caused by cholestatic or toxic liver injury because of the lower numbers of fibrocytes compared with other collagen-expressing cells in fibrotic liver such as activated hepatic stellate cells (aHSCs)/MFs [3], this conclusion may not extend to some genetic defects causing liver fibrosis in mice. In another model of cholestatic liver disease caused by genetic mutation of multidrug resistance gene (Mdr2 −/−, Abcb4 −/− mice) resulting in sclerosing cholangitis, BM-derived fibrocytes, portal fibroblasts, and HSCs were demonstrated to be involved in biliary fibrogenesis in Abcb4 −/− mice. Abcb4 deficiency results in a significant flux of fibrocytes to the liver, which has been implicated to pathogenesis in this injury and may significantly contribute to liver fibrosis in these mice [31]. Although the nature of these differences remains unresolved, this phenomenon can be in part explained by differential expression of multiple genes in Abcb4 −/− mice (versus wild-type mice), causing a significant amplification of immunoregulatory function of fibrocytes in these Abcb4 −/− mice [32, 33]. Notably, the specific model (i.e., the etiology of tissue injury and timeframe) and the method of analysis (i.e., applied surrogate parameters reflecting fibrosis or fibrogenesis, respectively) are important factors for understanding of the role of BM-derived cells in pathogenesis of fibrosis [31].
Immune Functions of Fibrocytes
Fibrocytes were originally described by Dr. Bucala as Collagen Type I-expressing CD45+ leukocytes that are capable of antigen presentation. Fibrocytes were shown to function as antigen-presenting cells and prime T cell responses in fibrotic liver [34, 35]. Fibrocytes also display many functions that could influence chronic inflammatory responses [36]. Isolated fibrocytes respond to Interleukin-1 beta (IL-1β) by increasing secretion of IL-6, IL-8, and IL-10, and by increasing expression of Intercellular adhesion molecule-1 (ICAM-1) which would be expected to recruit and activate leukocytes [37]. Of note, it has been found that fibrocytes from asthmatic patients produce pro-inflammatory mediators when stimulated with IL-17A, and produce collagen when stimulated with TH2 cytokines [38]. Despite of extensive studies, the phenotype of hepatic fibrocytes is not well defined, majorly because of difficulties of fibrocyte isolation from fibrotic lesions. Therefore, it remains unknown if fibrocytes can also contribute to myeloid population in fibrotic liver, or mainly differentiate into fibrogenic MFs.
In addition, several independent studies have suggested that fibrogenesis of parenchymal organs is associated with recruitment of fibrocytes to spleen. These fibrocytes were also detected in spleen in mice with kidney fibrosis [39]. Migration of fibrocytes to spleen was reported in mice with BDL- and CCl4-induced liver fibrosis. In spleen, fibrocytes were strictly located in the marginal zone surrounding the white pulp. More detailed analysis of fibrocyte recruitment to spleen had suggested that TGF-β and lipopolysaccharide (LPS) both trigger massive flux of CD45+Col+ cells in the spleen [30••]. Spleen has been suggested to serve as a major reservoir of fibrocytes, where they may undergo activation and/or differentiation. Interestingly, surgical removal spleen in CCl4-injured mice resulted in an increased flux of fibrocytes to the damaged liver but did not cause (at least under these specific experimental conditions) increased differentiation of fibrocytes into fibrogenic myofibroblasts [30••]. Splenic fibrocytes express the same surface markers as liver fibrocytes (CD45+ Col+ CD11b+ F4/80+ Gr-1+ MHC II+), and also have a capability to differentiate into MFs when cultured on plastic or stimulated with TGF-β1 [30••]. The biological significance of recruitment of CD45+Col+ fibrocytes to spleen in acute liver injury or infection has been suggested but still remains unresolved. However, recent studies have suggested that splenic fibrocytes may mediate immune and inflammatory responses rather than differentiate into MFs. Splenic CD45+Col+ fibrocytes were shown to facilitate innate antimicrobial responses in response to septic shock and infection with Listeria monocytogenes, and present antigens to naïve CD8+ T cells to induce their proliferation. Although splenic fibrocyte-like cells lacked phagocytic activity, they responded to bacteria by releasing extracellular traps formed by DNA fibers containing the antimicrobial peptide cathelicidin [30••]. These extracellular traps are a mechanism recently described in neutrophils, eosinophils, and macrophages to entrap and kill bacteria [40–42]. Interestingly, stimulated by macrophage colony-stimulating factor (MCSF), fibrocytes downregulate Collagen Type I expression and differentiate in fully functional F4/80+ CD68+ macrophages with high phagocytic activity. Furthermore, culturing of CD45+Col+ fibrocytes in the presence of granulocyte macrophage colony-stimulating factor (GM-CSF) results in upregulation of CD11c and downregulation of Collagen Type I in these cells, suggesting that fibrocytes may acquire a phenotype similar to that observed in dendritic cells (DCs). Therefore, splenic CD45+Col+ fibrocytes may represent a population non-terminally differentiated myeloid cells that can be rapidly mobilized to the site of injury and respond to infection or other insults by providing an immediate innate immune defense, stimulating adaptive immunity as APCs, and regulating inflammatory responses in situ by differentiating into macrophages, granulocytes and/or dendritic cells [30••, 36]. Such a variety of responses to different stimuli may indicate that CD45+Col+ splenic fibrocytes possess progenitor properties. In addition, it remains unknown if fibrocytes are composed of myeloid progenitors of a mixed phenotype or at different stages of hematopoietic development. Consistent with this notion, intravenous (i.v.) adoptive transfer of CD45+Col+ fibrocytes into CCl4-injured mice resulted in engraftment of the transferred population in spleen with an expansion of Col-CD11b+ and Gr1+ progeny [30••].
Fibrocytes in Pulmonary and Kidney Fibrosis
Studies of the origin of fibrogenic MFs in different parenchymal organs revealed that fibrocytes contribute differently to lung, liver, and kidney fibrosis. Thus, based on recent findings from experimental models of lung fibrosis, fibrocytes constitute ≈25 % of collagen-producing cells in fibrotic lungs [12, 43] (compared to ≈3–5 % in fibrotic liver [10••]), suggesting that the magnitude of fibrocyte differentiation into MFs depends on the organ and the type of injury [44]. Original experiments that were utilizing the BM chimeric mice in which all BM cells were labeled by expression of green fluorescent protein (GFP), and have demonstrated a large percentage of collagen-expressing cells in the bleomycin-injured lung derived from the BM source, presumably fibrocytes [45]. Follow-up studies further confirmed that the amount of collagen deposition in bleomycin-injured lungs directly correlated with the numbers of intrapulmonary-recruited CD45+Col I+CXCR4+ fibrocytes, suggesting that BM-derived fibrocytes migrate to the injured lung where they undergo differentiation into α-SMA+ MFs, and this way contributes to lung fibrosis [23•, 43, 46]. In addition, fibrocytes were shown to promote fibrosis via paracrine pathways via secreting growth factors, proteases, and matricellular proteins that affect resident epithelial and mesenchymal cells following fibrogenic injury [47].
Fibrocytes were also reported to migrate into the injured kidneys [27, 39, 48] It is believed that fibrocytes and mesenchymal cell precursors contribute to approximately 14–15 % of BM-derived cells recruited to fibrotic kidneys [49]. T-helper-2-type (TH2) cytokines appear to play profibrotic role in renal fibrosis, inducing stimulation of fibrocyte differentiation into collagen-producing MFs, while T-helper-1-type (TH1) cytokines were implicated in regulating the opposite effects and inhibit fibrocyte differentiation [50–52]. The precise mechanism by which fibrocytes promote renal fibrosis are not clear [46]. Further studies are needed to clarify if infiltrating fibrocytes can also contribute indirectly to collagen production, perhaps, by inducing the recruitment or activation of other inflammatory or connective tissue cell types [53].
Factors Regulating Fibrocyte Migration to Fibrotic Liver
Cytokines/Chemokines Regulate Fibrocyte Migration into Fibrotic Liver
Chronic liver injury results in release of TGF-β1, the major pro-fibrogenic factor, and other pro-inflammatory cytokines IL-6, IL-1β, MIP-1 and 2, leptin, and IL-17A, which play a critical role in recruitment of inflammatory cells into fibrotic liver and activation/proliferation of hepatic MFs [54]. Moreover, liver injury is associated with increased intestinal permeability and leakage of gut-derived liposaccharides (LPS, a mediator of the TLR4 signaling) into the blood stream. Therefore, it is not surprising that these factors, TGF-β1 and LPS [55] shown to be important for activation of liver-resident MFs, can also mediate fibrocyte migration into the fibrosing liver and regulate their in situ transdifferentiation. In agreement, the adenovirus-based overexpression of TGF-β1 in mouse hepatocytes resulted in a rapid flux of fibrocytes into spleen (72 h) and liver (compared to mock-infected control mice) [29••]. Furthermore, intravenous injection with LPS, which mimics the model of septic shock in mice, was shown to trigger an acute mobilization of fibrocytes into spleen. This observation was further supported by the in vitro experiments that demonstrated that TGF-β1 alone or in combination with LPS can trigger fibrocytes transmigration [29••]. All together, these findings support a concept that TGF-β1 and LPS may synergistically regulate fibrocyte recruitment to fibrosing peripheral tissues. Meanwhile, TGF-β1 and LPS are not the only factors that regulate fibrocyte recruitment. Thus, other specific cytokines and chemokines also regulate fibrocyte recruitment into fibrotic liver. For example, secondary lymphoid chemokine (SLC), which is the ligand for CCR7 [15], was shown to trigger egress of fibrocytes from the BM. In concordance, the gene expression profiling of Col+CD45+ cells revealed that hepatic fibrocytes express and further upregulate CXCR4 and other chemokine receptors CCR3, CCR5, and CCR7 [15, 56] in response to injury, indicating that these receptors may have an important impact on regulation of fibrocyte expansion in the BM, egress from the BM, and trafficking and extravagation from the blood stream into the fibrotic liver. Furthermore, studies using chimeric mice in which wild-type BM was replaced by CCR1−/− or CCR2−/− BM have demonstrated that these two chemokine receptors may regulate fibrocyte migration into fibrotic liver. Thus, the number of CCR2−/− fibrocytes recruited into fibrotic liver was decreased by approximately 50 % compared with the wild-type fibrocytes, suggesting that CCR2 signaling may regulate the egress of fibrocytes from the BM and possibly their extravasation. In addition, recruitment of CCR1−/− fibrocytes was also reduced but to a lesser extent (by 25 % versus wild-type mice) [29••]. We can speculate that synergistic activation of CCR1 and CCR2 receptors may define “organ-specific” recruitment of fibrocytes and modulate their specific migration to the injured organ.
An attempt has been made to monitor fibrocyte trafficking in real time in live mice. For this purpose, chimeric mice were generated by transplantation of collagen-α1(I)–Luciferase BM into lethally irradiated wild-type mice. Following liver injury, migration of fibrocytes into the spleen and liver was monitored and quantified by luminescent signal quenched by luciferin injection. These mice became a useful tool to study the egress of fibrocytes from the BM [29••]. Recruitment of Luciferase+ fibrocytes to the extramedullary tissues was observed in two phases corresponding to the acute phase and regression of fibrosis [29••]. During the acute phase, the number of fibrocytes peaked after 2 weeks CCl4 administration. These findings are in concordance with the dual role of CD11b+monocytes and macrophages in liver fibrosis [57]; the flux of monocytes to the liver in response to acute injury facilitates hepatic fibrosis, while the monocytes recruited to the liver upon cessation of liver injury play a critical role in resorption of fibrous scar and resolution of liver fibrosis. Meanwhile, these monocytes might exhibit different phenotypical characteristics. The role of fibrocytes in resolution of liver fibrosis remains unresolved.
Cytokines and Chemokines Mediate Migration of Fibrocytes to Fibrotic Lung and Kidney
The chemokine receptor/chemokine (CXCR4/CXCL12) biological axis plays an important role in trafficking of circulating fibrocytes into the lungs during the pathogenesis of pulmonary fibrosis [23•]. Administration of specific neutralizing anti-CXCL12 antibodies to bleomycin-exposed mice resulted in markedly reduced fibrocyte extravasation into the lungs, reduced pulmonary collagen deposition, and attenuated lung fibrosis [23•, 43]. Furthermore, CCR2−/− mice are protected from fluorescein isothiocyanate (FITC)-induced lung fibrosis, indicating the important role of CCR2 in the pathogenesis of pulmonary fibrosis. CCL12, but not CCL2 (another ligand for CCR2), is likely to be responsible for initiation of CCR2-mediated signaling that regulates fibrocyte recruitment to fibrotic lungs [22, 58]. Meanwhile, the involvement of CCR2 in mediation of fibrocyte trafficking into fibrotic kidney following ureteral ligation has not been confirmed. In contrast, the impact of CCL21/CCR7 signaling on progressive kidney interstitial fibrosis was demonstrated, and the severity of interstitial fibrosis directly correlated with the recruited number of CD45+/Col1+ cells into damaged kidneys. Remarkably, administration of anti-CCL21 antibody to the injured mice reduced kidney fibrosis by almost 50 % when compared with control mice, and was associated with decreased number of recruited fibrocytes [59]. These experiments clearly underline the important role of fibrocytes in fibrogenesis of parenchymal organs. Since fibrocytes migrate exclusively to the fibrosing organ, this process might be regulated by the combination of specific chemokine/receptors signaling.
S1P Mediates Migration of Fibrocytes to Fibrotic Organs
Sphingosine-1-phosphate (S1P) is a blood-borne lysophospholipid mediator that exerts a variety of activities including the regulation of cell migration, proliferation, and differentiation. S1P3, one of S1P receptors, is expressed on the surface of bone marrow-derived fibrocytes and mediates the recruitment of fibrocytes to fibrotic liver [60]. S1P was shown to play an important role in the progress of liver fibrosis caused by BDL [61] or repeated administration of CCl4 [62] to mediate BM-derived cell migration via S1P3 into the liver. An increased synthesis of S1P was accompanied by elevated S1P3 expression and was observed in both BDL and CCl4 models. In addition, treatment with the S1P3 antagonist, suramin, ameliorated BDL-induced hepatic fibrosis [61, 62]. In S1P2−/− mice, a single dose of CCl4 or dimethylnitrosamine as trigger factor for acute liver injury resulted in an enhanced regenerative response and injury protection when compared to WT mice [63]. Also, chronic administration of CCl4 to S1P2−/− mice resulted in a reduction of fibrotic area in the damaged liver [63], indicating that S1P2 is a critical receptor for the development of liver fibrosis. Growing evidence suggests the role of S1P in vascular barrier protection, regulation of migration and proliferation of fibroblasts, and MF differentiation in pathologic fibrotic response to lung injury [60]. It was also demonstrated that S1P is a key molecule in the direct mediation of renal fibrosis [64].
There is a report suggesting that the contribution of S1P to fibrosis has a “Janus-faced” nature, since S1P can exhibit both pro- and anti-fibrotic effects depending on its site of action. Extracellular S1P promotes fibrotic processes in a S1P receptor-dependent manner, whereas intracellular S1P has an opposite effect and dampens a fibrotic reaction by yet unidentified mechanisms [65]. Therefore, targeting S1P signaling might be a promising strategy for the treatment of fibrotic diseases, but a number of important issues remain to be addressed to fully appreciate the full spectrum of S1P actions in pathological fibrogenesis.
Migration of Fibrocytes Changes with Age
Hepatic fibrosis was shown to be accelerated in aged mice, and increased recruitment of fibrocytes to fibrotic livers may account for the exacerbation of liver fibrosis in aged rodents [66]. Egress of CD45+Col+ cells from BM was detected in the absence of injury or stress in aged mice but not in young mice, and their egress from BM was associated with increased expression of chemokine receptors CCR1, CCR2, and CCR7 and adhesion molecules(ICAM1) [29••]. Similar results were observed in the aged lungs, suggesting that egress of fibrocytes from the BM to parenchymal organs is a common phenomenon associated with aging. In a murine model of accelerated aging (senescence-accelerated prone mice, SAMP), it was found that bleomycin-induced fibrosis was significantly higher in SAMP mice compared with age-matched control senescence-accelerated resistant mice. Furthermore, SAMP mice showed higher numbers of fibrocytes and higher levels of stromal cell-derived factor-1, ligand for CXCR4, in the peripheral blood, suggesting that increased mobilization of fibrocytes may be involved in age-related susceptibility to lung fibrosis [67]. In support of these findings, the blood in healthy aged persons contains increased concentrations of CD45+ collagen type 1–positive fibrocytes and high circulating levels of MCP-1 and IL-13 [68]. These data suggest that fibrocytes may be associated with certain aging phenomena. Fibrocytes may have a physiologic role in the maintenance of tissue integrity or a pathologic role in the development of age-associated sequel [46].
Fibrocytes: Possible Clinical Biomarker for Fibrotic Diseases
Liver biopsy, although considered to be the gold standard test for determining the extent and progression of fibrosis, cannot be always applied to all patients. In addition, not only is liver biopsy subject to certain variability, but sampling error also may be important, as evidenced by studies examining liver samples from different regions of the liver [69]. There is a great need for noninvasive biomarkers that can monitor fibrogenesis [70]. Recently, the role of circulating fibrocytes as a marker of liver fibrosis in chronic hepatitis C (HCV) has been evaluated [71]. The percentage of peripheral blood fibrocytes in patients with biopsy-proven chronic hepatitis C stratified according to the Metavir stage of liver fibrosis was analyzed. Higher numbers of fibrocytes circulating in peripheral blood were detected in patients with the F2 and F3 groups compared with those in the F0–F1 group. Patients with the F4 Metavir group showed the highest level of circulating fibrocytes compared to patients in lower stages of liver fibrosis. These data demonstrated that the number of circulating fibrocytes is increased progressively in patients with HCV and correlates with the disease progression, suggesting that circulating fibrocytes not only play a role in the pathogenesis of fibrogenic processes but also can be used as potential, easy-to-perform, inexpensive, and not invasive biomarkers of liver fibrosis to monitor disease progression [71].
Furthermore, fibrocytes were shown to be increased in peripheral blood of patients with idiopathic pulmonary fibrosis (IPF). Fibrocyte numbers were not only significantly elevated in patients with stable IPF, but also further increased during disease exacerbation, and served as a prognostic marker that correlated with early mortality. Taken together, early detection of circulating fibrocytes in blood of patients with IPF may become an independent indicator of the disease activity and a useful clinical marker for IPF progression [43, 72]. In support of this notion, fibrocytes were also detected in patients with kidney fibrosis. Thus, the number of interstitial CD45+/proCol1+ cells was significantly decreased during convalescence induced by glucocorticoid therapy in patients with chronic kidney diseases [48]. Therefore, fibrocytes may be a promising target for anti-fibrotic therapy. These findings have to be further confirmed and validated in larger multicentre trials.
Fibrocytes: Potential Target for Anti-Fibrotic Therapy
Serum amyloid P (SAP), which is a member of the pentraxin family of proteins, is a critical factor in plasma that inhibits fibrocyte differentiation from monocytes ex vivo [73]. The short pentraxin SAP has been recently described to attenuate fibrosis in a number of different organ models, including pulmonary, renal, cardiac, and oral submucous fibrosis, partially through inhibiting the accumulation and activation of fibrocytes and macrophages in the injured organ [74•, 75–78].
SAP in Pulmonary Fibrosis
Based on the original observations demonstrating that SAP inhibits fibrocyte differentiation in vitro, the role of SAP on inhibition of fibrocyte migration and differentiation into fibrogenic myofibroblasts has been studied in vivo. Administration of SAP to bleomycin-injured mice resulted in reduced recruitment of fibrocytes to the damaged lungs and attenuation of lung fibrosis in rats and mice. In addition, delayed administration of SAP to mice with already developed inflammation and fibrosis was also effective to reduce manifestation of bleomycin injury to the lungs [74•]. Although the mechanism by which SAP mediates its functions remains not completely understood, SAP inhibits fibrocyte differentiation, by binding to FcγRs, and the in vivo activity of SAP appears to be dependent on the FcRγ [79]. In mice, deletion of the FcRγ, that is required for FcγRI and FcγRIIIa signaling, significantly reduces sensitivity of fibrocytes to SAP. In concordance, deletion or siRNA-mediated knockdown of FcRγ or FcγRI significantly reduced sensitivity of murine and human cells to SAP, indicating that SAP binding to FcγRI and FcRγ is required for inhibition of fibrocyte differentiation [79, 80]. In TGF-β-driven or bleomycin-induced mouse models of pulmonary fibrosis, SAP alleviates fibrosis, in part, through its effect on macrophages [76, 81]. In these models of pulmonary fibrosis, SAP injections decreased M2 macrophage markers, while increasing the pro-inflammatory M1 macrophage marker CXCL10 on pulmonary macrophages. To elucidate the endogenous function of SAP, Apcs−/− (Amyloid P component, serum) ‘‘SAP knockout mice’’ were used in bleomycin-induced pulmonary inflammation and fibrosis. Compared to the wild-type mice, bleomycin induces a more persistent inflammatory response and increased fibrosis in Apcs−/− mice, indirectly indicating the important roles played by fibrocytes and macrophages [82].
SAP in Kidney Fibrosis
SAP inhibits fibrosis through FcγR-dependent monocyte-macrophage regulation in vivo [75]. In mouse models of renal fibrosis, SAP injections decreased expression of the M1 markers Mip2a and IL-1β, and the profibrotic M2 markers CCL17 and CCL22 on renal macrophages. These changes were accompanied by a significant increase in the levels of IL-10. In IL-10 and FcRγ knockout mice, the effects of SAP on renal fibrosis were reduced [72]. This is contrast to the role of SAP in pulmonary fibrosis, where it promotes M1 macrophages and decreases M2 macrophages. This inconsistency may be attributed to differences that exist in the milieu of kidneys and lungs, and these observations suggest that SAP has a significant role in regulating macrophage polarization, but the outcome is tissue dependent and at times quite different [80].
The role of SAP in liver fibrosis has not been elucidated. However, studies showed that fibrocytes participate in the progression of liver fibrosis [29••], and macrophage depletion in mice during liver injury was associated with a dramatic loss of MF-like HSCs and resulted in extensive loss of ECM components [57]. If the function of SAP is not only limited to inhibition of fibrocyte differentiation, but also extends to regulation of M1 and M2 macrophages, SAP might also affect development of liver fibrosis. Further studies are required to evaluate the role of SAP in liver fibrosis in vitro and in vivo.
Conclusions
There is emerging evidence that fibrocytes may play an important role in fibrogenesis of parenchymal organs. Fibrocytes were implicated in pathogenesis of liver fibrosis, while fibrocytes recruited to spleen after acute liver injury or infection participate in immediate innate immune defense and in situ regulate inflammatory responses. Both TGF-β1 and LPS play a critical role in liver fibrogenesis, and these factors also appear to trigger fibrocyte recruitment to the injured liver, and promote their differentiation into collagen Type I-producing MFs. Fibrocytes may become a novel target for anti-fibrotic therapy. If proven that hSAP can inhibit fibrocyte recruitment to fibrotic liver and attenuate liver fibrosis, it may serve as a drug of choice for anti-fibrotic therapy. Well-designed clinical trials are needed to confirm and validate for the finding that fibrocytes would be a possible clinical biomarker and potential therapeutic target for fibrotic disease.
References
Recently published papers of particular interest have been highlighted as: • Of importance •• Of major importance
Bataller R, Brenner DA (2005) Liver fibrosis. J Clin Invest 115:209–218
Liu X, Xu J, Brenner DA, Kisseleva T (2013) Reversibility of Liver Fibrosis and Inactivation of Fibrogenic Myofibroblasts. Curr Pathobiol Rep 1:209–214
Brenner DA et al (2012) Origin of myofibroblasts in liver fibrosis. Fibrogenesis Tissue Repair 5:S17
Li Y, Wang J, Asahina K (2013) Mesothelial cells give rise to hepatic stellate cells and myofibroblasts via mesothelial-mesenchymal transition in liver injury. Proc Natl Acad Sci USA 110:2324–2329
Kisseleva T, Brenner DA (2006) Hepatic stellate cells and the reversal of fibrosis. J Gastroenterol Hepatol 21(Suppl 3):S84–S87
Wells RG (2014) The portal fibroblast: not just a poor man’s stellate cell. Gastroenterology 147:41–47
Scholten D et al (2010) Genetic labeling does not detect epithelial-to-mesenchymal transition of cholangiocytes in liver fibrosis in mice. Gastroenterology 139:987–998
Taura K et al (2010) Hepatocytes do not undergo epithelial-mesenchymal transition in liver fibrosis in mice. Hepatology 51:1027–1036
Chu AS et al (2011) Lineage tracing demonstrates no evidence of cholangiocyte epithelial-to-mesenchymal transition in murine models of hepatic fibrosis. Hepatology 53:1685–1695
•• Kisseleva T, et al. (2006) Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J Hepatol 45:429–438. This study demonstrated that the BM-derived fibrocytes did not possess features of HSCs or myofibroblasts, and these cells represented a novel collagen-producing cell type in the liver constituting approx. 5% of the collagen-α1(I)-producing population in the liver after BDL
Fausther M, Lavoie EG, Dranoff JA (2013) Contribution of Myofibroblasts of Different Origins to Liver Fibrosis. Curr Pathobiol Rep 1:225–230
Kisseleva T, Brenner DA (2008) Fibrogenesis of parenchymal organs. Proc Am Thorac Soc 5:338–342
Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A (1994) Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med 1:71–81
Bucala R (2008) Circulating fibrocytes: cellular basis for NSF. J Am Coll Radiol 5:36–39
Abe R, Donnelly SC, Peng T, Bucala R, Metz CN (2001) Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol 166:7556–7562
Quan TE, Cowper S, Wu SP, Bockenstedt LK, Bucala R (2004) Circulating fibrocytes: collagen-secreting cells of the peripheral blood. Int J Biochem Cell Biol 36:598–606
Quan TE, Bucala R (2007) Culture and analysis of circulating fibrocytes. Methods Mol Med 135:423–434
Grieb G, Steffens G, Pallua N, Bernhagen J, Bucala R (2011) Circulating fibrocytes: biology and mechanisms in wound healing and scar formation. Int Rev Cell Mol Biol 291:1–19
Lin WR et al (2012) Bone marrow-derived cells contribute to cerulein-induced pancreatic fibrosis in the mouse. Int J Exp Pathol 93:130–138
Xie X et al (2014) Possible involvement of fibrocytes in atrial fibrosis in patients with chronic atrial fibrillation. Circ J 78:338–344
Strieter RM, Gomperts BN, Keane MP (2007) The role of CXC chemokines in pulmonary fibrosis. J Clin Invest 117:549–556
Moore BB et al (2006) The role of CCL12 in the recruitment of fibrocytes and lung fibrosis. Am J Respir Cell Mol Biol 35:175–181
• Phillips RJ, et al. (2004) Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest 114:438–446. This study demonstrated for the first time, that circulating fibrocytes contribute to the pathogenesis of pulmonary fibrosis and these cells migrates in response to CXCL12 and traffics to the bleomycin-induced fibrotic lungs
Ishida Y et al (2007) Essential roles of the CC chemokine ligand 3-CC chemokine receptor 5 axis in bleomycin-induced pulmonary fibrosis through regulation of macrophage and fibrocyte infiltration. Am J Pathol 170:843–854
Wada T, Sakai N, Matsushima K, Kaneko S (2007) Fibrocytes: a new insight into kidney fibrosis. Kidney Int 72:269–273
Lin SL, Kisseleva T, Brenner DA, Duffield JS (2008) Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol 173:1617–1627
Pilling D, Fan T, Huang D, Kaul B, Gomer RH (2009) Identification of markers that distinguish monocyte-derived fibrocytes from monocytes, macrophages, and fibroblasts. PLoS ONE 4:e7475
Inagaki Y, Higashiyama R (2012) Interplay between bone marrow and liver in the pathogenesis of hepatic fibrosis. Hepatol Res 42:543–548
•• Scholten D, et al. (2011) Migration of fibrocytes in fibrogenic liver injury. Am J Pathol 179:189–198. This study demonstrated that in response to liver injury, fibrocytes migrate from BM to the liver, and their migration is regulated by CCR2 and CCR1 but is compromised with age
•• Kisseleva T, et al. (2011) Fibrocyte-like cells recruited to the spleen support innate and adaptive immune responses to acute injury or infection. J Mol Med (Berl) 89:997–1013. This study demonstrated that splenic CD45 + Col + cells are a population of rapidly mobilized BM-derived fibrocyte-like cells that respond to inflammation or infection to participate in immediate innate immune defense, stimulate adaptive immunity as APCs, and regulate inflammatory responses in situ by terminal differentiation into macrophages or dendritic cells
Roderfeld M et al (2010) Bone marrow transplantation demonstrates medullar origin of CD34+ fibrocytes and ameliorates hepatic fibrosis in Abcb4−/− mice. Hepatology 51:267–276
Nakken KE et al (2007) Multiple inflammatory-, tissue remodelling- and fibrosis genes are differentially transcribed in the livers of Abcb4(−/−) mice harbouring chronic cholangitis. Scand J Gastroenterol 42:1245–1255
Roderfeld M et al (2012) Bone marrow transplantation improves hepatic fibrosis in Abcb4−/− mice via Th1 response and matrix metalloproteinase activity. Gut 61:907–916
Chesney J, Bacher M, Bender A, Bucala R (1997) The peripheral blood fibrocyte is a potent antigen-presenting cell capable of priming naive T cells in situ. Proc Natl Acad Sci USA 94:6307–6312
Balmelli C, Ruggli N, McCullough K, Summerfield A (2005) Fibrocytes are potent stimulators of anti-virus cytotoxic T cells. J Leukoc Biol 77:923–933
Peng H, Herzog EL (2012) Fibrocytes: emerging effector cells in chronic inflammation. Curr Opin Pharmacol 12:491–496
Chesney J, Metz C, Stavitsky AB, Bacher M, Bucala R (1998) Regulated production of type I collagen and inflammatory cytokines by peripheral blood fibrocytes. J Immunol 160:419–425
Bellini A et al (2012) Interleukin (IL)-4, IL-13, and IL-17A differentially affect the profibrotic and proinflammatory functions of fibrocytes from asthmatic patients. Mucosal Immunol 5:140–149
Niedermeier M et al (2009) CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc Natl Acad Sci USA 106:17892–17897
Brinkmann V et al (2004) Neutrophil extracellular traps kill bacteria. Science 303:1532–1535
Yousefi S et al (2008) Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med 14:949–953
Chow OA et al (2010) Statins enhance formation of phagocyte extracellular traps. Cell Host Microbe 8:445–454
Strieter RM, Keeley EC, Hughes MA, Burdick MD, Mehrad B (2009) The role of circulating mesenchymal progenitor cells (fibrocytes) in the pathogenesis of pulmonary fibrosis. J Leukoc Biol 86:1111–1118
Kisseleva T, Brenner DA (2012) The phenotypic fate and functional role for bone marrow-derived stem cells in liver fibrosis. J Hepatol 56:965–972
Hashimoto N, Jin H, Liu T, Chensue SW, Phan SH (2004) Bone marrow-derived progenitor cells in pulmonary fibrosis. J Clin Invest 113:243–252
Herzog EL, Bucala R (2010) Fibrocytes in health and disease. Exp Hematol 38:548–556
Kleaveland KR, Moore BB, Kim KK (2014) Paracrine functions of fibrocytes to promote lung fibrosis. Expert Rev Respir Med 8:163–172
Sakai N et al (2010) Fibrocytes are involved in the pathogenesis of human chronic kidney disease. Hum Pathol 41:672–678
Kalluri R, Neilson EG (2003) Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest 112:1776–1784
Wynn TA (2004) Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol 4:583–594
Shao DD, Suresh R, Vakil V, Gomer RH, Pilling D (2008) Pivotal Advance: Th-1 cytokines inhibit, and Th-2 cytokines promote fibrocyte differentiation. J Leukoc Biol 83:1323–1333
Farris AB, Colvin RB (2012) Renal interstitial fibrosis: mechanisms and evaluation. Curr Opin Nephrol Hypertens 21:289–300
Duffield JS, Lupher M, Thannickal VJ, Wynn TA (2013) Host responses in tissue repair and fibrosis. Annu Rev Pathol 8:241–276
Brenner DA (2009) Molecular pathogenesis of liver fibrosis. Trans Am Clin Climatol Assoc 120:361–368
Seki E et al (2007) TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med 13:1324–1332
Blakaj A, Bucala R (2012) Fibrocytes in health and disease. Fibrogenesis Tissue Repair 5:S6
Duffield JS et al (2005) Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest 115:56–65
Moore BB et al (2001) Protection from pulmonary fibrosis in the absence of CCR2 signaling. J Immunol 167:4368–4377
Sakai N et al (2006) Secondary lymphoid tissue chemokine (SLC/CCL21)/CCR7 signaling regulates fibrocytes in renal fibrosis. Proc Natl Acad Sci USA 103:14098–14103
Takuwa Y, Ikeda H, Okamoto Y, Takuwa N, Yoshioka K (2013) Sphingosine-1-phosphate as a mediator involved in development of fibrotic diseases. Biochim Biophys Acta 1831:185–192
Li C et al (2009) Involvement of sphingosine 1-phosphate (SIP)/S1P3 signaling in cholestasis-induced liver fibrosis. Am J Pathol 175:1464–1472
Li C et al (2009) Homing of bone marrow mesenchymal stem cells mediated by sphingosine 1-phosphate contributes to liver fibrosis. J Hepatol 50:1174–1183
Ikeda H et al (2009) Sphingosine 1-phosphate regulates regeneration and fibrosis after liver injury via sphingosine 1-phosphate receptor 2. J Lipid Res 50:556–564
Shiohira S et al (2013) Sphingosine-1-phosphate acts as a key molecule in the direct mediation of renal fibrosis. Physiol Rep 1:e00172
Schwalm S, Pfeilschifter J, Huwiler A (2013) Sphingosine-1-phosphate: a Janus-faced mediator of fibrotic diseases. Biochim Biophys Acta 1831:239–250
Medeiros MV, Freitas LA, Andrade ZA (1988) Differences in hepatic pathology resulting from bile duct obstruction in young and old rats. Braz J Med Biol Res 21:75–83
Xu J et al (2009) Use of senescence-accelerated mouse model in bleomycin-induced lung injury suggests that bone marrow-derived cells can alter the outcome of lung injury in aged mice. J Gerontol A Biol Sci Med Sci 64:731–739
Mathai SK et al (2010) Circulating monocytes from systemic sclerosis patients with interstitial lung disease show an enhanced profibrotic phenotype. Lab Invest 90:812–823
Regev A et al (2002) Sampling error and intraobserver variation in liver biopsy in patients with chronic HCV infection. Am J Gastroenterol 97:2614–2618
Rockey DC (2013) Translating an understanding of the pathogenesis of hepatic fibrosis to novel therapies. Clin Gastroenterol Hepatol 11:224–231.e1-5
Nunnari G et al (2010) Circulating fibrocytes as a marker of liver fibrosis in chronic hepatitis C. Front Biosci 2:1241–1245
Moeller A et al (2009) Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 179:588–594
Pilling D, Buckley CD, Salmon M, Gomer RH (2003) Inhibition of fibrocyte differentiation by serum amyloid P. J Immunol 171:5537–5546
• Pilling D, et al. (2007) Reduction of bleomycin-induced pulmonary fibrosis by serum amyloid P. J Immunol 179:4035–4044. This study demonstrated that SAP injections led to reduced numbers of fibrocytes in the lungs and reduced fibrosis in rats and mice and that delaying SAP injections until inflammation and fibrosis were already apparent could also reduce symptoms, suggesting the possibility that SAP may be useful as a therapy for pulmonary fibrosis in humans
Castano AP, et al. (2009) Serum amyloid P inhibits fibrosis through Fc gamma R-dependent monocyte-macrophage regulation in vivo. Sci Transl Med 1:5ra13
Murray LA et al (2011) TGF-beta driven lung fibrosis is macrophage dependent and blocked by Serum amyloid P. Int J Biochem Cell Biol 43:154–162
Murray LA et al (2010) Serum amyloid P ameliorates radiation-induced oral mucositis and fibrosis. Fibrogenesis Tissue Repair 3:11
Haudek SB et al (2008) Fc receptor engagement mediates differentiation of cardiac fibroblast precursor cells. Proc Natl Acad Sci USA 105:10179–10184
Crawford JR, Pilling D, Gomer RH (2012) FcgammaRI mediates serum amyloid P inhibition of fibrocyte differentiation. J Leukoc Biol 92:699–711
Cox N, Pilling D, Gomer RH (2014) Serum amyloid P: a systemic regulator of the innate immune response. J Leukoc Biol. doi:10.1189/jlb.1MR0114-068R
Murray LA et al (2010) Serum amyloid P therapeutically attenuates murine bleomycin-induced pulmonary fibrosis via its effects on macrophages. PLoS ONE 5:e9683
Pilling D, Gomer RH (2014) Persistent lung inflammation and fibrosis in serum amyloid P component (APCs−/−) knockout mice. PLoS ONE 9:e93730
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81370544), Beijing Natural Science Foundation (7142043) to Min Cong.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Cong, M., Jia, J. The Role of Fibrocytes in Fibrogenic Liver Diseases. Curr Pathobiol Rep 2, 191–199 (2014). https://doi.org/10.1007/s40139-014-0055-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40139-014-0055-x