
Abstract
Introduction
There is a substantial unmet need for effective therapies to treat patients with refractory dry eye disease (DED). The goal of this open-label pilot study was to investigate the efficacy and safety of repository corticotropin injection (RCI; Acthar® Gel; Mallinckrodt Pharmaceuticals) in subjects with DED, most of whom did not experience adequate response to standard-of-care therapies.
Methods
Adults with moderate or severe-acute DED received 80 U of subcutaneous RCI twice weekly for 12 weeks. Primary efficacy outcomes were improvements in corneal fluorescein staining of superficial punctate keratitis (SPK) lesions and Symptom Assessment in Dry Eye (SANDE) scores. Secondary outcomes included changes in Schirmer’s test scores, conjunctival lissamine green staining, erythema, intraocular pressure (IOP), and best corrected visual acuity (BCVA). Adverse events (AEs) were assessed continuously throughout the study.
Results
Fifteen subjects received at least 1 dose of RCI, and 12 subjects completed the study. Compared to baseline (day 1), significantly fewer fluorescein-stained SPK lesions were detected at day 14 (p = 0.0250) and day 84 (p = 0.0240) after RCI treatment. Mean SANDE scores progressively declined from 62.0 at baseline to 46.9 at day 84. Erythema (p = 0.0046), conjunctival lissamine green staining of SPK lesions (p = 0.0317), and IOP (p = 0.0052) were all significantly improved after 12 weeks of RCI therapy. Schirmer’s test scores and BCVA showed no significant changes throughout the study. No ocular AEs or deaths occurred, and no new safety signals were identified for RCI.
Conclusions
These results suggest that RCI may be a safe and effective treatment for moderate and severe DED.
Trial registration
ClinicalTrials.gov identifier: NCT03287635.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Dry eye disease (DED) affects approximately 6.8% of adults in the USA, many of whom do not adequately respond to standard-of-care therapies, such as artificial tears, topical steroids, topical T-cell inhibitors (e.g., cyclosporine and lifitegrast), and intense pulsed light. |
This open-label pilot study evaluated the efficacy and safety of repository corticotropin injection (RCI) in the treatment of severe-acute and moderate-to-severe DED that is inadequately responsive to standard-of-care therapies. |
What was learned from this study? |
Primary efficacy endpoint assessments showed a significant reduction in corneal fluorescein staining of superficial punctate keratitis (SPK) lesions at days 14 and 84 of RCI treatment compared with baseline, and Symptom Assessment in Dry Eye (SANDE) scores progressively declined from a mean of 62.0 at baseline to 46.9 at day 84. |
From baseline to day 84 of RCI treatment, significant reductions were observed in erythema, intraocular pressure, and conjunctival lissamine green staining of SPK lesions; RCI was generally well tolerated with few adverse events reported. |
This open-label pilot study provides preliminary evidence supporting the efficacy and safety of RCI treatment for moderate-to-severe or severe-acute DED in patients who were treatment-naive or had inadequate disease control with standard-of-care therapies. |
Introduction
Dry eye disease (DED) is a highly prevalent, chronic, and multifactorial disease that can diminish quality of life and incur significant socioeconomic costs [1, 2]. DED is characterized by ocular surface irritation, foreign body sensation, erythema, and visual disturbances. Causes of DED include medications, hormonal or environmental factors, contact lenses, topical eye drops, use of electronic devices, and ocular surgery [2, 3]. DED results from hyperosmolarity or alterations of immune homeostasis of the ocular surface and can be related to aqueous deficiency, meibomian gland dysfunction, goblet cell loss, or any combination of the three [1, 3]. DED may be an ocular manifestation of systemic diseases like rosacea, allergies, or autoimmune disorders [1].
According to the National Health and Wellness Survey, approximately 16 million Americans (6.8% of the US adult population) have been diagnosed with DED, although up to 120 million may also have signs and symptoms [2, 4]. Inflammation may progress DED from an acute to a chronic phase, which can result in permanent damage of corneal epithelial cells, goblet cells, and corneal nerves [3]. These nerves participate in the neural feedback arc, and damage may lead to a lack of symptoms, over-watering, irreversible damage to the ocular surface, and blindness [2, 5]. Chronic inflammatory processes play a pivotal role in DED pathogenesis, characterized by infiltration of immune cells into the cornea, conjunctiva, and lacrimal glands, as well as elevated levels of cytokines in tears (e.g., interleukin [IL]-1, IL-6, and IL-17) [3, 6].
Traditional treatments for DED have included over-the-counter artificial tears, warm compresses, oral omega-3 pills, topical non-steroidal anti-inflammatory drugs, topical steroids, topical T-cell inhibitors (e.g., cyclosporine and lifitegrast), meibomian gland expression, and intense pulsed light [2]. Many patients with DED continue to exhibit signs or remain symptomatic after one or more of these therapies. Therefore, alternative treatments are needed for these patients with refractory disease.
Repository corticotropin injection (RCI; Acthar® Gel; Mallinckrodt Pharmaceuticals, Hampton, NJ, USA) is a naturally sourced complex mixture of porcine adrenocorticotropic hormone (ACTH) analogs (a major component of which is ACTH1–39) and other pituitary peptides, supplied as a sterile preparation in 16% gelatin to provide a prolonged release after intramuscular or subcutaneous injection [7]. RCI may reduce inflammation in part by stimulation of the adrenal cortex to produce endogenous cortisol. However, recent data suggest that RCI may also function via non-steroidogenic mechanisms and provides effective treatment for patients with other inflammatory conditions, such as rheumatoid arthritis and systemic lupus erythematosus, who do not adequately respond to corticosteroids [8,9,10]. RCI is currently approved by the US Food and Drug Administration (FDA) for the treatment of severe, acute, and chronic allergic and inflammatory processes involving the eye and its adnexa including but not limited to keratitis, iritis, iridocyclitis, diffuse posterior uveitis and choroiditis, optic neuritis, chorioretinitis, and anterior segment inflammation [7].
RCI binds and activates all five melanocortin receptors (MCRs) [11]. MCRs differ in both their distribution throughout the body and their affinity for agonists. With the exception of MC2R, which is located in the adrenal cortex and is responsible for the steroid-dependent actions of RCI, the other MCRs are widely distributed in various cell types throughout the body [11]. Recent studies have demonstrated a direct immunomodulatory effect of RCI on immune cells. In vitro studies have shown that RCI exerts dose-dependent inhibitory effects on immunoglobulin G production and B-cell proliferation, distinct from corticosteroid signaling pathways [12, 13]. A study using a murine multiple sclerosis model reported that RCI can directly dampen the response of T-cells and macrophages [14].
Given the multiple mechanisms by which RCI may function and the lack of highly effective treatments for DED, the objective of this open-label pilot study was to evaluate the efficacy and safety of RCI in the treatment of severe-acute and moderate-to-severe DED that was inadequately responsive to standard-of-care therapies.
Methods
Study Design
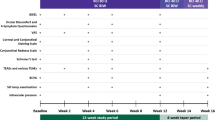
This was a single-site, prospective, open-label, phase 4 pilot study designed to examine the efficacy and safety of RCI in the treatment of severe–acute or moderate-to-severe DED in patients with inadequate response to standard-of-care therapies. Enrolled subjects were treated with 80 U of subcutaneous RCI twice weekly for 12 weeks. Study visits were performed at screening (1–7 days before the baseline day 1 visit), day 1 (visit 1 [baseline]), day 7 (visit 2), day 14 (visit 3), and day 84 (visit 4). The study protocol was approved by the Western Institutional Review Board (No.: 20180950) centrally and conducted in accordance with the Declaration of Helsinki and requirements of clinical trials registration (ClinicalTrials.gov identifier: NCT03287635). Written informed consent from patients was required before enrollment to participate in the trial and to allow their de-identified information to be published.
Inclusion and Exclusion Criteria
Subjects were patients aged 18 to 85 years. Generally healthy subjects could be enrolled who had persistent signs and symptoms of DED after 90 days of a stable standard-of-care treatment regimen, including IPL, cyclosporine, lifitegrast, loteprednol, artificial tears, and/or warm compresses. Two subjects who had severe–acute signs and symptoms without prior DED treatments were also included. Subjects continued any stable, active DED treatments throughout the study. Other inclusion criteria included at least 40 mm on the Symptom Assessment in Dry Eye (SANDE) scale and at least five superficial punctate keratitis (SPK) lesions on at least one cornea with an erythema grade of ≥ 1 in the nasal or temporal conjunctiva of one eye.
Exclusion criteria included those with a history of ocular or systemic viral/fungal disease, tuberculosis, scleroderma, peptic ulcer disease, cirrhosis, or thyroid disease. Patients were excluded who were immunocompromised or had uncontrolled hypertension or diabetes, hypersensitivity or a contraindication to the investigational drug, topical or nasal vasoconstrictor use within 14 days, pregnancy or lactation, unwillingness to abstain from eyelash growth medications, history of herpetic keratitis, febrile illness within 1 week, ocular surgery within 12 weeks, participation in any study within the preceding 30 days, severe disease other than DED, or an uncontrolled medical condition that could confound safety or efficacy assessments or limit compliance.
Assessments
At each study visit except day 7, assessments included Early Treatment Diabetic Retinopathy Study (ETDRS) best corrected visual acuity (BCVA) measurements, slit-lamp examination, intraocular pressure (IOP) measurement, corneal fluorescein staining, conjunctival lissamine staining, Schirmer’s testing, and SANDE scoring. Day 7 was a safety visit for assessment of adverse events (AEs) with slit-lamp biomicrospcopy and measurements of IOP and ETDRS BCVA; however, SANDE scoring was also assessed on day 7. Dilated fundus examinations were performed at day 1 (visit 1) and day 84 (visit 4). AEs were evaluated continuously throughout the duration of the study.
Fluorescein and lissamine staining and quantification were performed according to previously established protocols [15, 16]. Briefly, corneal fluorescein staining was performed by adding 1 drop of proparacaine to a fluorescein strip, holding the strip to the inferior fornix for 10 s, then allowing 60 s to elapse prior to examination. For conjunctival lissamine staining, 1 drop of lissamine green was applied to each eye, the excess was blotted, and the stained SPK lesions were tallied after 60 s had elapsed.
To assess erythema and AEs, slit-lamp biomicrospcopy was performed using 8 × 2-mm slit-lamp beams. The beam was administered right to left to right, thoroughly examining the structures of the anterior segment: eyelids, lashes, conjunctiva, cornea, iris, anterior chamber, and lens. Erythema was graded using the Cornea and Contact Lens Research Unit scale [17]. Any abnormal result was recorded, and its pathology was evaluated.
For the anesthetized Schirmer’s test, the rounded and notched end of the test strip was folded, 1 to 2 drops of proparacaine or tetracaine were instilled, and 5 min elapsed. The subjects were instructed to look up and away from the strip during placement, then to close their eyes for the duration of testing. The rounded end of the strip was positioned toward the temporal one-third of the lower lid of both eyes. After 5 min, strips were removed, and the distances to the leading edges of moisture were measured.
IOP was measured at each study visit using Goldman applanation tonometry with pressure recorded (mmHg) according to the investigators’ standard protocol [18]. Dilated ophthalmoscopy or wide-field fundus imaging occurred on day 1 and day 84 and included assessment of the optic nerve for pallor and/or cupping.
Participants were trained on day 1 how to administer the study medication by specialists contracted by Mallinckrodt Pharmaceuticals. The study coordinator dispensed study medications and the temperature recorder with instructions for proper administration of RCI.
Study Endpoints
The primary efficacy endpoints were improvements in participant dry eye comfort using the validated SANDE questionnaire and the number of fluorescein-stained SPK lesions on the cornea after 12 weeks of RCI treatment. Secondary endpoints were changes in conjunctival staining with lissamine green and Schirmer’s test scores.
The primary safety endpoint was to evaluate the occurrence, severity, and treatment-relatedness of AEs and serious AEs (SAEs) over 12 weeks of RCI therapy. Whether AEs were treatment-related was assessed by the primary investigator after each occurrence for the duration of the study. Slit-lamp examinations, IOP and ETDRS BCVA measurements, and dilated fundoscopy were performed at each visit to assess erythema and ocular AEs.
Statistical Analyses
Subjects who completed the 12-week RCI treatment without significant protocol deviations or rescue medications comprised the per protocol (PP) analysis set. The PP set was used for the primary efficacy analysis. All subjects who received a single dose of RCI were included in the safety set and assessed for AEs. For sensitivity analysis, the full analysis set (FAS) was used, comprising all enrolled subjects who received at least one dose of RCI and completed at least one post-baseline visit with efficacy assessments. Descriptive statistics were used to characterize the categorical and continuous data presented in this study.
To evaluate the primary efficacy endpoints, the statistical hypothesis was that SANDE scores or the average number of observed corneal-fluorescein stained SPK lesions at the final visit (day 84) would be less than at baseline (day 1). Additional secondary outcomes evaluated included changes from baseline to the final visit in conjunctival lissamine green staining, erythema, Schirmer’s test scores, and IOP, with the hypotheses being that these scores would decrease from baseline to final visit, except for Schirmer’s test scores, which may increase. All safety and efficacy endpoint values were compared using 2-sided paired t tests with a significance threshold of p < 0.05.
Results
The study was conducted between 1 July 2018 and 4 March 2020. Patient disposition is summarized in Fig. 1. Twenty-one patients were screened, and 19 ultimately were enrolled in the trial. Fifteen subjects in the safety set received at least one dose of RCI, whereas the 14 subjects who attended at least one subsequent visit were included in the FAS. The 12 subjects who completed the trial composed the PP set. Subjects in the PP set were all Caucasian, were predominately women (75%), and had a mean age of 51.5 ± 13.7 years with an age range of 27 to 71 years. Ten of these subjects had DED that was inadequately responsive to standard-of-care therapies; the remaining two subjects presented with previously untreated severe DED.

CONSORT diagram showing patient disposition. AE Adverse event, RCI repository corticotropin injection
Two screened patients were discovered to have exclusionary pre-existing conditions (e.g., scleroderma, glaucoma). Three patients withdrew consent at the first visit due to concerns regarding pre-existing diabetes and/or hypertension. One patient withdrew consent upon advice from a pharmacist that the shingles vaccine might be less effective while taking the study drug. These six subjects had signed an informed consent, but none received RCI. After initiation of RCI, two participants withdrew from the trial due to AEs. One subject was removed from the study after initiating a prohibited medication.
The primary efficacy endpoint of a reduction in corneal fluorescein staining of SPK lesions was met: 19/24 eyes (79%) from 12 RCI-treated subjects showed improvement. Significantly fewer SPK lesions were detected at day 14 and day 84 than at baseline (Fig. 2a). The other primary efficacy assessment showed that mean SANDE scores progressively declined between baseline and the final visit (day 84) from a mean of 62.0 to 46.9 (Fig. 2b). Rescue medication was not required in any subject.

Primary efficacy endpoint analyses of fluorescein staining of SPK lesions (a) and SANDE scores (b) after treatment with RCI. All data are presented as the mean ± 95% confidence intervals. Fluorescein SPK staining was not performed at day 7. RCI Repository corticotropin injection, SANDE Symptom Assessment in Dry Eye, SPK superficial punctate keratitis
Erythema was significantly reduced from baseline to day 84 (Fig. 3a). Conjunctival lissamine green staining of SPK lesions was significantly lower at day 84 than at baseline in 21/24 (88%) eyes from 12 subjects (Fig. 3b). IOP also decreased significantly from baseline to day 84 (Fig. 3c). Schirmer’s test scores did not show any significant change throughout the study (Fig. 4a). ETDRS BCVA was higher by day 84, but the change was not statistically significant (Fig. 4b). Dilated fundoscopy of the optic nerve and slit-lamp biomicroscopy showed no derangements of the optic nerve or the anterior segment, respectively (data not shown).

Erythema score (a), number of lissamine-stained SPK lesions (b), and IOP (c) after RCI treatment. All data are presented as the mean ± 95% confidence intervals. Erythema and lissamine SPK staining were not assessed at day 7. IOP Intraocular pressure, mm HG millimeters of mercury, RCI repository corticotropin injection, SPK superficial punctate keratitis

Schirmer’s test score (a) and BCVA (b) after treatment with RCI. All data are presented as the mean ± 95% confidence intervals. Schirmer’s score was not assessed at day 7. BCVA Best corrected visual acuity, logMAR logarithm of the minimum angle of resolution
RCI was generally well tolerated, with few AEs reported (Table 1). AEs leading to discontinuation were observed in two patients. Although in both of these patients the AEs were re-emergences of pre-existing conditions (i.e., heart palpitations or dysfunctional uterine bleeding), a relationship to the study drug could not be ruled out for the heart palpitations. The dysfunctional uterine bleeding was determined to be an SAE unrelated to the study drug. One patient reported insomnia that was not resolved by dosing earlier in the day but was resolved with an over-the-counter sleep aid. Three patients reported insomnia as a pre-existing condition. There were no reported ocular AEs.
Discussion
Despite RCI being an FDA-approved medication for the treatment of inflammatory ophthalmic diseases, its safety and efficacy in the treatment of DED has not been well established. Treatments for DED have traditionally consisted of topical medications. Currently, only topical cyclosporine, loteprednol and lifitegrast, varenicline nasally and intense pulsed light are FDA-approved first-line therapies for DED. Injectable therapies for DED are a relatively new addition to the therapeutic landscape, although at-home injectable medications are used increasingly for osteoporosis, inflammatory conditions, and autoimmune diseases.
This study, the first to investigate the effect of RCI in the treatment of DED, met its primary endpoint of significant reduction in SPK lesions detected with corneal fluorescein staining. The other primary endpoint of SANDE score reduction, a subjective measure of improvement, showed progressive mean improvements from baseline to day 84. Secondary endpoints also indicated that RCI can be effective in the treatment of DED: lissamine-stained SPK lesions significantly improved with RCI, whereas Schirmer’s test score did not. Notably, the mean Schirmer’s test score was 16 mm at baseline, with a maximum of 35 mm in one subject, which is considered normal for patients with DED. Schirmer’s test scores may vary widely in patients with DED, who often underproduce or overproduce aqueous tears; hence, Schirmer’s test was chosen as a secondary endpoint.
RCI was found to be well tolerated in this study, with few AEs reported. Two subjects experienced AEs leading to discontinuation, but these were re-emergences of pre-existing conditions. The dysfunctional uterine bleeding was an SAE determined to be unrelated to RCI treatment, whereas the heart palpitations AE was unlikely to be related to RCI, but could not be ruled out. No new safety signals for RCI were identified. Secondary safety assessments showed no significant reductions in BCVA or dilated fundoscopy of the optic nerve, but did show significant decreases in IOP and erythema at day 84. While glucocorticoid use is associated with increased IOP, recent studies have shown that RCI induces relatively low endogenous glucocorticoid production in animals [19] and humans [20, 21]. RCI is also known to exert direct immunomodulatory effects on immune cells [12, 13]. Therefore, it is likely that the non-steroidogenic effects of RCI contribute to the observed reduction in IOP. The positive results of this study were expected based on data published from studies of other refractory inflammatory diseases, such as systemic lupus erythematosus [9, 10] and rheumatoid arthritis [8], which demonstrated the safety and efficacy of RCI.
Limitations of this pilot study include its small number of subjects and lack of a placebo comparator group; however, the purpose of this study was to obtain additional safety data and establish the first efficacy data for RCI in the treatment of DED. As a single dosing schedule of RCI was used in this study, it is unclear whether the dosing regimen is optimal and whether different dosing quantities or frequencies would provide an improved response or safety profile. In addition, the 12-week length of this study may not fully reveal AEs or treatment efficacy in response to longer courses of therapy.
The results of this open-label pilot study provide a foundation for a much larger placebo-controlled clinical trial to further elucidate the overall safety and efficacy of RCI in the treatment of DED.
Conclusions
The data from this phase 4, open-label pilot study suggest that RCI may be safe and efficacious for the treatment of patients with moderate-to-severe or severe-acute DED who are treatment-naive or have inadequately controlled DED with standard-of-care therapies. RCI may offer a viable treatment alternative for patients with DED and chronic ocular inflammation that does not adequately respond to traditional therapies.
References
Simsek C, Dogru M, Kojima T, Tsubota K. Current management and treatment of dry eye disease. Turk J Ophthalmol. 2018;48(6):309–13.
Stapleton F, Alves M, Bunya VY, et al. TFOS DEWS II epidemiology report. Ocul Surf. 2017;15(3):334–65.
Javadi MA, Feizi S. Dry eye syndrome. J Ophthalmic Vis Res. 2011;6(3):192–8.
Farrand KF, Fridman M, Stillman IO, Schaumberg DA. Prevalence of diagnosed dry eye disease in the United States among adults aged 18 years and older. Am J Ophthalmol. 2017;182:90–8.
Verjee MA, Brissette AR, Starr CE. Dry eye disease: early recognition with guidance on management and treatment for primary care family physicians. Ophthalmol Ther. 2020;9(4):877–88.
Ru Y, Huang Y, Liu H, et al. alpha-Melanocyte-stimulating hormone ameliorates ocular surface dysfunctions and lesions in a scopolamine-induced dry eye model via PKA-CREB and MEK-Erk pathways. Sci Rep. 2015;5:18619.
Mallinckrodt Pharmaceuticals. Acthar gel. Package insert. 2021. https://www.actharhcp.com. Accessed 20 May 2020
Fleischmann R, Furst DE, Connolly-Strong E, Liu J, Zhu J, Brasington R. Repository corticotropin injection for active rheumatoid arthritis despite aggressive treatment: a randomized controlled withdrawal trial. Rheumatol Ther. 2020;7(2):327–44.
Furie RA, Mitrane M, Zhao E, Becker PM. Repository corticotropin injection in patients with persistently active SLE requiring corticosteroids: post hoc analysis of results from a two-part, 52-week pilot study. Lupus Sci Med. 2017;4(1):e000240.
Askanase AD, Zhao E, Zhu J, Bilyk R, Furie RA. Repository corticotropin injection for persistently active systemic lupus erythematosus: results from a phase 4, multicenter, randomized, double-blind, placebo-controlled trial. Rheumatol Ther. 2020;7(4):893–908.
Huang YJ, Galen K, Zweifel B, Brooks LR, Wright AD. Distinct binding and signaling activity of Acthar Gel compared to other melanocortin receptor agonists. J Recept Signal Transduct Res. 2020;20:1–9.
Benko AL, McAloose CA, Becker PM, et al. Repository corticotrophin injection exerts direct acute effects on human B cell gene expression distinct from the actions of glucocorticoids. Clin Exp Immunol. 2018;192(1):68–81.
Olsen NJ, Decker DA, Higgins P, et al. Direct effects of HP Acthar Gel on human B lymphocyte activation in vitro. Arthritis Res Ther. 2015;17:300.
Cusick MF, Libbey JE, Oh L, Jordan S, Fujinami RS. Acthar gel treatment suppresses acute exacerbations in a murine model of relapsing-remitting multiple sclerosis. Autoimmunity. 2015;48(4):222–30.
Tauber J, Schechter BA, Bacharach J, et al. A phase II/III, randomized, double-masked, vehicle-controlled, dose-ranging study of the safety and efficacy of OTX-101 in the treatment of dry eye disease. Clin Ophthalmol. 2018;12:1921–9.
Wolffsohn JS, Arita R, Chalmers R, et al. TFOS DEWS II diagnostic methodology report. Ocul Surf. 2017;15(3):539–74.
Murphy PJ, Lau JS, Sim MM, Woods RL. How red is a white eye? Clinical grading of normal conjunctival hyperaemia. Eye. 2007;21(5):633–8.
Stevens S, Gilbert C, Astbury N. How to measure intraocular pressure: applanation tonometry. Commun Eye Health. 2012;25(79–80):60.
Huang YJ, Galen K, Zweifel B, Brooks LR, Wright AD. Distinct binding and signaling activity of Acthar Gel compared to other melanocortin receptor agonists. J Recept Signal Transduct Res. 2021;41(5):425–33.
Poola N, Due B, Wright D, Brooks LR, Zaman F. Pharmacokinetics and pharmacodynamics of repository corticotropin injection compared with synthetic ACTH1–24 depot and methylprednisolone in healthy subjects. Clin Pharmacol Drug Dev. 2022;11(4):502-15.
Wang X, Pham L, Poola N, Brooks LR, Due B. Comparison of steroidogenic exposure following the administration of repository corticotropin injection with a synthetic ACTH1-24 depot and methylprednisolone in healthy subjects. Clin Pharmacol Drug Dev. 2021;10(7):777–88.
Acknowledgements
The authors would like to thank the participants of this study.
Funding
Sponsorship for this study and the Rapid Service Fee were funded by Mallinckrodt Pharmaceuticals.
Authorship
All authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author contributions
MT, RT, BJ, and RB were all involved in the conception or design of the study, the analysis or interpretation of data, any critical revisions of the manuscript for content and accuracy, and the approval of the final manuscript for publication.
Medical writing, editorial, and other assistance
Professional writing and editorial support were provided by Nestor G. Davila, PhD of MedLogix Communications, LLC, Itasca, Illinois, under the direction of the authors and were funded by a grant from Mallinckrodt Pharmaceuticals.
Disclosures
Melissa Toyos has received speaker and/or consultation fees from Mallinckrodt Pharmaceuticals, Bausch Health, RVL Pharmaceuticals, and Sun Pharmaceutical Industries. She has received research funding from Mallinckrodt Pharmaceuticals, Bausch Health, Novartis, Kala Pharmaceuticals, Lumenis, and Sun Pharmaceutical Industries. Rolando Toyos has received speaker and/or consultation fees from Mallinckrodt Pharmaceuticals, Lumenis, Kala Pharmaceuticals, and Sun Pharmaceutical Industries. He has received research funding from Lumenis. Barbara Jodoin and Ryan Bunch have no conflicts of interest to report.
Compliance with ethics guidelines
The study protocol was approved by the Western Institutional Review Board (no: 20180950) centrally and conducted in accordance with the Declaration of Helsinki and requirements of clinical trials registration (ClinicalTrials.gov identifier: NCT03287635). Written informed consent from patients was required before enrollment to participate in the trial and to allow their de-identified information to be published.
Data availability
The de-identified datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request within 2 years of publication of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Toyos, M., Toyos, R., Jodoin, B. et al. Results from a Prospective, Open-Label, Phase 4 Pilot Study of Repository Corticotropin Injection for Moderate and Severe Dry Eye Disease. Ophthalmol Ther 11, 1231–1240 (2022). https://doi.org/10.1007/s40123-022-00501-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40123-022-00501-2