
Abstract
Introduction
Dexketoprofen/tramadol 25/75 mg (DKP/TRAM) is a fixed-dose combination of a cyclooxygenase inhibitor and opioid receptor agonist. To better understand the efficacy and safety of DKP/TRAM in the treatment of moderate to severe acute lower back pain (LBP) with or without radiculopathy, we carried out a large explorative phase IV international, multicenter, prospective, randomized, double-blind, parallel group, placebo-controlled study (DANTE).
Methods
A total of 538 patients with or without a history of LBP and experiencing acute LPB of moderate to severe intensity [Numerical Rating Scale-Pain Intensity (NRS-PI) score > 5] were randomized 4:4:1:1 to DKP/TRAM 25/75 mg every 8 h (n = 211), tramadol (TRAM) 100 mg (n = 207), placebo-matched DKP/TRAM (n = 59), or placebo-matched TRAM (n = 61).
Results
The proportion of patients achieving the primary endpoint, defined as the time to first achieve NRS-PI score < 4 or pain intensity reduction ≥ 30% from drug intake up to 8 h after the first dose, was higher in the DKP/TRAM arm than in the placebo group, but the difference was not statistically significant (46.1% vs. 42.6%, respectively; hazard ratio 1.11; 95% confidence interval 0.775, 1.595; p = 0.566). DKP/TRAM achieved superiority over TRAM in total pain relief at 4, 6, and 8 h (p < 0.05). Conversely, in relation to the secondary endpoints, a significantly greater reduction in NRS-PI score was seen with DKP/TRAM versus placebo starting from 1 h, and this reduction remained numerically lower throughout 8 h. Summed pain intensity difference values were also significantly lower at 4, 6, and 8 h with DKP/TRAM compared to TRAM (p < 0.05). Overall, DKP/TRAM was well tolerated.
Conclusion
Although the primary endpoint was not met, secondary efficacy analyses suggest the superiority of DKP/TRAM over placebo and TRAM alone in terms of total pain relief. DKP/TRAM can be considered to be an effective and safe option for the treatment of moderate to severe acute LBP.
Dante Study Registration
EudraCT number: 2019–003656-37; ClinicalTrials.gov Identifier: NCT05170841.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
Dexketoprofen/tramadol 25/75 mg (DKP/TRAM) is a fixed-dose combination of a cyclooxygenase (COX) inhibitor and opioid receptor agonist |
We carried out a large explorative phase IV prospective, randomized, double-blind, parallel group, placebo-controlled study (DANTE) to investigate DKP/TRAM for the treatment of moderate to severe acute lower back pain (LBP) |
What was learned from the study? |
The primary endpoint (time to first achieve a Numerical Rating Scale-Pain Intensity [NRS-PI] score < 4 or a pain intensity reduction ≥ 30% from drug intake up to 8 h after the first dose) was not met |
Secondary efficacy analyses suggest the superiority of DKP/TRAM over placebo and TRAM in terms of total pain relief |
DKP/TRAM can be considered an effective and safe option for the treatment of moderate to severe acute LBP |
Introduction
Lower back pain (LBP) is a highly prevalent condition with an estimated lifetime prevalence of up to 80% [1]. LBP is a prominent cause of limitations in activity and employment, and is responsible for substantial social and economic burdens [2, 3]. Natural recovery from acute LBP depends on a number of factors, including gender, marital status, sports activities, and history of LBP [4]. While there are a number of treatment options for acute LBP, the majority still lack a high level of evidence [5,6,7]. A review of clinical practice guidelines for LBP reported that the most frequently recommended drugs for first-line treatment of acute LBP were non-steroidal anti-inflammatory drugs (NSAIDs) and that acetaminophen and skeletal muscle relaxants were not consistently recommended to treat acute LBP [8]. Notwithstanding, it should be highlighted that attention should be given to the prescribing of opioids to minimize the risk of opioid use disorder [9].
In general, achieving adequate pain control with pharmacological monotherapy is often sub-optimal, and combining drugs with diverse mechanisms of action is a valid strategy to provide greater pain relief and/or improved tolerability; notwithstanding, at present there is no solid evidence or clinical practice guidelines to support the routine use of combination therapy to manage LBP [10]. Indeed, recent meta-analyses found no evidence for benefit of NSAIDs administered alone in LBP [11, 12]. This may suggest that the combination of NSAIDs and a weak opioid might be a rationale means to achieve the desired benefit in LBP. In this regard, multimodal analgesia is currently regarded as a cornerstone of effective pain management [13, 14], and this is reflected in current guidance which now advocates the benefits of a multimodal and multidisciplinary approach to relieve pain [15].
Cyclooxygenase (COX) inhibitor/opioid receptor agonist combinations are considered to have great potential for multimodal management for pain since the combination can provide adequate analgesia while still providing a more desirable safety profile as the use of COX inhibitors has an opioid-sparing effect [16]. In daily practice, combination therapy with an opioid analgesic + acetaminophen or an NSAID is frequently used to manage LBP [10, 17]. This strategy is reflected in a recent Delphi survey in which it was agreed that combination therapy with an opioid and NSAID/paracetamol is useful in moderate to severe-acute refractory LBP [18].
Among the different fixed-dose combinations of a COX inhibitor + opioid receptor agonist, the dexketoprofen/tramadol 25/75 mg (DKP/TRAM) combination is being increasingly considered as a multimodal option due to its analgesic efficacy, fast onset of action, and prolonged duration, as reported in several models of pain [19,20,21,22,23]. Evidence of the analgesic efficacy of DKP/TRAM in LBP has been documented in observational studies, which have shown that the oral DKP/TRAM 25/75 mg fixed-dose combination is effective in patients with acute LBP associated with lumbar disc herniation [24], as well as in non-specific LBP [25]. However, these studies are limited by relatively small sample sizes and their single-center design [24, 25].
To better understand the efficacy and safety of DKP/TRAM in the treatment of moderate to severe acute LBP with or without radiculopathy, we carried out a large, explorative, international, multicenter, prospective, randomized, double-blind trial, the DANTE study.
Methods
The design of the DANTE trial (EudraCT Number: 2019-003656-37) has been previously published in detail [26]. The salient features are reported herein. This study was performed in compliance with International Council for Harmonisation (ICH) Good Clinical Practices (GCP), including the archiving of essential documents, as well as the ethical principles of the Declaration of Helsinki of 1964 and its later amendments. The study protocol and protocol amendments, patient information leaflet, informed consent form (ICF), Summary of Product Characteristics (SmPC), and any other relevant documents according to National Regulations were reviewed and approved by an independent ethics committee (IECs) and the Health Authorities (HAs) of the participating countries. All local, national, and legal requirements for the conduct of a clinical study were followed. Prior to the patient’s enrollment into the study and before performing any study-related procedures, the Investigator or its authorized delegate obtained the patient’s written, dated, and signed informed consent to participate in the study and to allow the confidential disclosure, processing, and transferring of necessary documentation of the patient’s health and personal data to the contract research organization (CRO), Sponsor and its Affiliates, the competent HAs, and any other institutions, as legally required and in accordance with the local applicable privacy laws. All ethics committees involved in the study are listed in Electronic Supplementary Material (ESM) Table 1.
Study Design
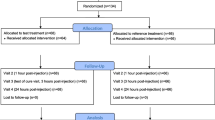
This was a phase IV, multicenter, randomized, double-blind, double-dummy parallel group, placebo, and active controlled study carried out from September 2020 to May 2022. The study was conducted at 36 sites in six European countries and recruited patients from hospitals (Emergency Department), private clinics, and site management organizations. The trial was divided into two phases: a single-dose phase (day 1, t0–t8h) followed by a multiple-dose phase beginning immediately (from t8h to 8 h after intake of the last dose at day 5). Study participation lasted up to 8 days, which included: (1) visit 1 (day 1), the screening phase, randomization, and first administration of study treatment; (2) day 1 to day 5, treatment and assessment period; (3) visit 2, end of the study visit (day 6 with an allowed window of ± 2 days). Randomization and blinding were carried out using an interactive web response system. Patients were randomized at 4:4:1:1 to one of four treatment groups: DKP/TRAM 25/75 mg; tramadol (TRAM) 100 mg; placebo-DKP/TRAM; or placebo-TRAM, with all capsules given every 8 h (Fig. 1). The immediate-release formulation of TRAM was used. TRAM 100 mg was administered as two capsules 50 mg each + one tablet placebo every 8 h. DKP/TRAM was given as one tablet 25/75 mg + two capsules placebo every 8 h. Patients receiving placebo were administered two capsules placebo + one tablet placebo.

Design of the DANTE study. Participants experiencing moderate to severe acute lower back pain were randomized 4:4:1:1 ratio to one of four treatment groups: DKP/TRAM 25/75 mg administered orally as a single film-coated tablet every 8 h; TRAM 100 mg administered as two capsules containing TRAM 50 mg every 8 h; placebo-DKP/TRAM; or placebo-TRAM [26]. Single asterisk (*) A total of 70 patients discontinued: DKP/TRAM, n = 28; TRAM, n = 25; Placebo-DKP/TRAM, n = 6; Placebo-TRAM, n = 11. Double asterisks (**) During the single-dose phase patients were randomized to receive placebo or DKP/TRAM or TRAM, and in the multiple-dose phase they were randomized to receive DKP/TRAM or TRAM. DKP Dexketoprofen, DKP/TRAM 25/75 mg dexketoprofen/tramadol fixed-dose combination, ERT eResearch Technology, ITT intention to treat, TRAM tramadol
The patients assigned to received the DKP/TRAM fixed combination or TRAM 100 mg during the single-dose phase continued to receive the same treatment during the multiple-dose phase; however, patients assigned to receive placebo during the single-dose phase received either the DKP/TRAM fixed combination or TRAM 100 mg during the multiple-dose phase. The double-dummy technique was used to ensure the double-blind condition of DKP/TRAM 25/75 mg versus TRAM 100 mg versus placebo. Paracetamol 500 mg orally for a maximum of 2 g per day was the recommended rescue medication, to be taken orally. Rescue medication could be taken at any time after the first dose if adequate pain relief was not achieved with the study treatment; however, patients were encouraged to wait for at least 60 min after dosing to allow time for the study treatment effect to take place.
Inclusion and Exclusion Criteria
Patients aged 18 to 65 years with or without a previous history of LBP experiencing new episodes of moderate to severe intensity [Numerical Rating Scale-Pain Intensity (NRS-PI) score > 5] with or without radiculopathy after a period of at least 2 months without any LBP were eligible for enrollment. The age range was chosen to avoid a population that can create a bias on the subjective evaluation of pain and also reduce the likelihood that the LBP was secondary to any other serious underlying conditions (e.g., cancer). The current acute LBP episode had to be within 48 h prior to screening. Exclusion criteria included acute LBP with radiation to limb and presence of neurologic signs according to the Quebec Task Force Classification [27]; spinal surgery within the preceding 6 months; known or suspected serious spinal pathology; and treatment with topical preparations/medications within 4 h prior to screening, with anesthetics and muscle relaxants within 8 h prior to screening, with short-acting analgesics within 4 h prior to screening, and with an opioid within 14 days prior to screening. No opioid-related eligibility criterion (e.g., history of long-term opioid use, opioid abuse, or opioid dependence) was used during recruitment.
Study Endpoints
The primary endpoint, assessed during the single-dose phase, was the time to first achieve an NRS-PI score < 4 or a pain intensity reduction ≥ 30% from drug intake up to 8 h after the first dose (t8h). The NRS-PI evaluates the severity of pain using a scale of 0–10, with 0 indicating no pain and 10 indicating the worst pain imaginable [28]. Key secondary endpoints included total pain relief (TOTPAR), percentage of maximum TOTPAR (%max TOTPAR), Roland Morris Disability Questionnaire (RMQ), and summed pain intensity difference (SPID) at different timepoints during the single- and multiple-dose phases. In the single-dose phase, key secondary endpoints were percentage of maximum SPID (%max SPID) at 4, 6, and 8 h after the first dose, patient global evaluation (PGE) of the study medication at 8 h after the first dose, time to rescue medication, and percentage of patients who required rescue medication within the first 4, 6, or 8 h post-dose. The exploratory efficacy endpoint was the time to first achieve an NRS-PI score < 4 and a pain intensity reduction ≥ 30% from drug intake at 6-, or 8-h post-dose.
Safety Assessments
Safety endpoints considered the incidence, severity, grade, and causality of adverse events (AEs) as well as clinically significant changes in laboratory evaluations. AEs were considered to be related to the study treatment unless they met the definition of either “unlikely related” (i.e., a causal relationship cannot be definitively ruled out, but other drugs or underlying disease provide plausible explanations and/or the temporal relation to the administration of the drug makes a causal relation improbable) or “not related” (i.e., any of the following are present: existence of a clear alternative explanation, and/or unreasonable temporal relationship between drug and event, and/or non-plausibility).
Data Management
Data collected during the study were recorded in an electronic case report form (eCRF). The investigator (or designee) was responsible for entering study data into the eCRF in accordance to the eCRF user guidelines, and ensured the accuracy, the completeness, and the consistency of the data entered in the eCRF. On the eCRF, patients were identified by a patient number, which was assigned at the screening visit. The patient number was a number composed of numeric values. During the conduct of the clinical part of the study, the eCRF was to be always available and up-to-date, in order to reflect the latest observations on the respective patient. The investigator or any designee was responsible for entering study data into the eCRF.
Data management of the eCRFs was performed by the contract research organization appointed by the study sponsor. All data were verified in a timely manner for missing information, inconsistencies, and for any necessary medical clarifications. Queries arising from the edit checks (either programmed or manual) were sent to the investigator for response. Once all data queries had been resolved, and comments/changes arising from the blind Data Review Meeting incorporated, the study data were declared to be “clean”, and the study database was locked ready for analysis. Once the database was locked and the data blind review report was approved, unblinding was performed.
Statistical Analysis
For the primary efficacy analysis, the intention-to-treat (ITT) population was considered. The original study power was set at 90%, but given the COVID-19 pandemic a sample size re-estimation was proposed in order to achieve a desired power level of 80%. At this level, a sample size of 510 patients was required to detect the difference between DKP/TRAM and placebo and to demonstrate the non-inferiority of DKP/TRAM versus TRAM for the time to first achieve an NRS-PI score < 4 or a pain intensity reduction ≥ 30% from drug intake up to 8 h after the first dose. A modified ITT approach was also used which maintained the 4:4:1:1 ratio of patients across the treatment arms considering the first cohort of 510 patients randomized to the four treatment arms. The primary efficacy endpoint was analyzed for the superiority of DKP/TRAM 25/75 mg versus placebo on the ITT population using a Cox proportional hazard (CPH) model with treatment, baseline pain intensity categories, and baseline radiculopathy categories as covariates. If more than one consecutive data were missed, the last-observation-carried-forward (LOCF) method was applied. A p value < 0.05 was considered to indicate significance. Non-inferiority of DKP/TRAM versus TRAM was tested with a one-sided significance level of 2.5%. For time-to-event variables, the non-inferiority margin was 0.80, based on the hazard ratio (HR), while for continuous variables the non-inferiority margin was 20% (0.20), based on the least squares (LS) mean of treatment difference. For binary variables, the non-inferiority margin was 0.80, based on the odds ratio (OR). Lastly, for non-parametric comparisons, the Wilcoxon rank-sum test was used.
Results
Baseline Characteristics
The study design of DANTE is shown in Fig. 1. A total of 544 patients with acute LBP were screened, of whom 538 were randomized to the treatment groups as follows: DKP/TRAM (n = 211); TRAM (n = 207); placebo-DKP/TRAM (n = 59); placebo-TRAM (n = 61). Baseline characteristics are shown in Table 1. The mean (± standard deviation [SD]) age of the entire cohort was 42.9 ± 12.5 years and the proportion of males and females was 52.6% and 47.4%, respectively. Overall, about one-half of patients had radiculopathy, and more than one-third of them had radiation to a distal or proximal extremity. The mean NSR-PI score was 7.0 ± 1.3 for all groups, and one-third of patients had severe pain. Of the 538 patients enrolled, 468 (87.0%) completed the study treatment, and the remaining 70 (13.0%) discontinued the study after the first dose. The main reason for discontinuation was AEs (n = 55, 78.6%), which are described in detail below.
Efficacy
The primary endpoint of the study was not met. Specifically, although the proportion of patients with NRS-PI score < 4 or pain intensity reduction ≥ 30% was numerically higher in the DKP/TRAM arm compared with the placebo arm (46.1% vs. 42.6%, respectively), the HR was 1.11 and not statistically significant [95% confidence interval (CI) 0.775, 1.595; p = 0.566] using a CPH model with treatment, baseline pain intensity categories, and baseline radiculopathy categories as factors. In relation to the secondary endpoints, the mean time to reach an NRS-PI score < 4 or pain intensity reduction ≥ 30% from drug intake up to 8 h after the first dose was 105 (range 15–480) min in the DKP/TRAM group compared to 120 (range 15–360) min in the placebo groups (ESM Table 2).
Changes in TOTPAR at 4, 6, and 8 h are shown in Fig. 2. During the single-dose phase, DKP/TRAM achieved superiority over TRAM in TOTPAR at 4, 6, and 8 h in the modified ITT population (4h: LS mean difference 0.78; 95% CI 0.163, 1.402; p = 0.013; 6h: LS mean difference 1.33; 95% CI 0.380, 2.280; p = 0.006; 8h: LS mean difference 1.59; 95% CI 0.336, 2.840; p = 0.013). The DKP/TRAM combination also achieved superiority over placebo in TOTPAR at 6 and 8 h (6h: LS mean difference 1.28; 95% CI 0.118, 2.450; p = 0.031; 8h: LS mean difference 1.73; 95% CI 0.194, 3.270; p = 0.027). The LS mean %max TOTPAR at 4, 6, and 8 h was significantly higher with DKP/TRAM compared to TRAM [per protocol (PP) population at 4 h: 30.10 vs. 24.55; p = 0.005; at 6 h: 32.16 vs. 25.61; p = 0.001; at 8 h: 33.32 vs. 27.15; p = 0.002]. There was a significantly higher percentage of patients achieving at least 50% of maximum TOTPAR at 4, 6, and 8 h after the first dose in the DKP/TRAM arm compared with the TRAM arm (modified ITT population, at 4 h: 20.6% vs 8.8%; p < 0,001; at 6 h: 22.5% vs 9.8%; p < 0,001; at 8h 23.5% vs 11.3% p = 0.001) (Fig. 3). The percentage of achieving changes in SPID in the PP population are shown in Fig. 4; significantly lower values were seen at 4, 6, and 8 h versus placebo. Changes in SPID are shown in Fig. 2b. In the single-dose phase, SPID values were significantly lower at 4, 6, and 8 h in the DKP/TRAM group compared with the TRAM group. The LS mean of SPID was significantly lower in the DKP/TRAM compared with TRAM arm at 4, 6, and 8 h after dosing in the PP population (t4h: LS mean difference − 1.55; 95% CI − 2.637, − 0.454; p = 0.003; t6h: LS mean difference − 2.41; 95% CI − 4.156, − 0.671; p = 0.003; t8h: LS mean difference − 2.95; 95% CI − 5.247, − 0.653; p = 0.006).

Changes in total pain relief (TOTPAR) at 4, 6, and 8 h after the first dose (T4h, T6h, T8h) in the modified intent-to-treat population. Asterisk (*) indicates significant difference at p < 0.05 vs. placebo; obelisk (†) indicates significant difference at p < 0.05 vs. TRAM. DKP/TRAM Dexketoprofen/tramadol fixed-dose combination, TRAM tramadol

Percentage of patients achieving at least 50% of maximum total pain relief (TOTPAR) in the modified intent-to-treat population. Asterisk (*) indicates significant difference at p < 0.05 vs. placebo at all timepoints; obelisk (†) indicates significant difference at p < 0.05 vs. TRAM at all timepoints. DKP/TRAM Dexketoprofen/tramadol fixed-dose combination, TRAM tramadol, T4h, T6h, T8h 4, 6, and 8 h after the first dose

Changes in summed pain intensity difference (SPID) at 4, 6, and 8 h after the first dose(T4h, T6h, T8h) per protocol population. Obelisk (†) indicates significant difference at p < 0.05 vs. TRAM. DKP/TRAM Dexketoprofen/tramadol, TRAM Tramadol
The time to first achieve an NRS-PI score < 4 or pain intensity reduction ≥ 30% from drug intake until 8 h after the first dose is shown in ESM Tables 3 and 4, respectively. The related HR for DKP/TRAM versus placebo was 1.10 (p = 0.576) in the ITT population and 1.17 (p = 0.397) in the PP population. A significantly greater reduction in NRS-PI was seen with DKP/TRAM versus placebo starting from 1 h, which remained numerically lower, but not statistically significant, throughout 8 h (Fig. 5). The analysis of the PGE in the single-dose phase is presented in ESM Table 5.

Summary of Numerical Rating Scale-Pain Intensity (NRS-PI) score by timepoints (single-dose phase) in the modified intent-to-treat population. There were no statistically significant differences between groups at any timepoint. DKP/TRAM Dexketoprofen/tramadol
All 510 patients in the single-dose phase continued to the multiple-dose phase, and none were lost. During the multiple-dose phase, DKP/TRAM achieved superiority over TRAM in TOTPAR at 24, 48, and 72 h in the modified ITT population (24 h: LS mean difference 3.62; 95% CI 0.083, 7.165; p = 0.045; 48 h: LS mean difference 7.62; 95% CI 0.263, 14.973; p = 0.042; 72 h: LS mean difference 11.99; 95% CI 0.721, 23.257; p = 0.037). The LS mean %max TOTPAR at 24, 48, 72, and 96 h was significantly higher in the DKP/TRAM arm than in the TRAM arm in the PP population (at 24 h: 47.75 vs. 42.97; p = 0.007; at 48 h: 52.61 vs. 47.68; p = 0.007; at 72 h: 56.28 vs. 51.17; p = 0.007; at 96 h: 59.36 vs. 54.42; p = 0.010) (Fig. 6). The percentage of patients achieving at least 50% of maximum TOTPAR at 24, 48, 72, and 96 h after the first dose was higher in the DKP/TRAM arm compared with the TRAM group; it was also significantly higher in the DKP/TRAM arm versus the TRAM arm at 48 and 72 h in the PP population (at 24 h: 46.4% vs. 42.6%; p = 0.170; at 48 h: 60% vs. 46.5%; p = 0.001; at 72 h: 62.6% vs. 53.5%; p = 0.019; at 96 h: 65.1% vs. 60.4%; p = 0.146).

Percentage of maximum total pain relief (TOTPAR) at 24, 48, 72, and 96 h of the multiple-dose phase (T24h, T48h, T72h, T96h) estimated from the analysis of covariance arm in the per protocol population. Asterisk (*) indicates a signficant difference at p < 0.05 vs. TRAM at all timepoints. DKP/TRAM Dexketoprofen/tramadol, TRAM Tramadol
There was no significant difference in the LS means of SPID between the DKP/TRAM and the TRAM arms at any timepoint during the multiple-dose phase (24, 48, 72, and 96 h).
At 96 h of the multiple-dose phase, there was a reduction in mean RMQ total scores from 64.1 at baseline to 29.6 at 104 h in the DKP/TRAM arm and from 65.2 at baseline to 35.2 at 104 h in the TRAM arm. The mean percentage change in RMQ score from baseline to t104 h was − 53.5% and − 46.1% in the DKP/TRAM and TRAM arms, respectively. There was no significant difference in LS mean score at 104 h between the DKP/TRAM and the TRAM arms (p = 0.067). Data on PGE at 96 h of the multiple-dose phase and RMQ total score are presented in ESM Tables 6 and 7, respectively.
Use of Rescue Medication
Time to rescue medication ranged from 79 to 450 min in the DKP/TRAM arm and from 91 to 365 min in the TRAM arm. No significant difference in the time to first use of rescue medication was reported between the DKP/TRAM and TRAM arms. In the single-dose phase, 11 (5.2%) patients in the DKP/TRAM arm, 14 (6.8%) in the TRAM arm, and nine (7.5%) in the placebo arm received rescue medication. Of these, nine (4.3%) patients in DKP/TRAM arm, 11 (5.3%) in the TRAM arm, and five (4.2%) in the placebo arm took rescue medication once. The frequency of taking rescue medication twice or ≥ 3 times was < 2% in all treatment arms.
Safety
Overall, the DKP/TRAM fixed-dose combination was well tolerated in patients with moderate to severe acute LBP after a single-dose (first 8 h) and during the multiple-dose phase (from 8 h up to day 5). No clinically significant hematological abnormalities were reported in the study and there were no major changes in vital signs.
In the single-dose phase (Table 2), a total of 70 (13.0%) patients had at least one treatment-emergent adverse event (TEAE), with a rate of 13.3%, 15%, and 9.2% in the DKP/TRAM, TRAM, and placebo groups, respectively. There were no statistically significant differences in TEAEs between the DKP/TRAM and TRAM groups in either the single- or multiple-dose phases, or between DKP/TRAM and placebo in the single-dose phase. The majority of TEAEs were mild or moderate in intensity. One severe TEAE (urinary calculus) was reported in the DKP/TRAM arm and was considered to be serious and unrelated to treatment. Treatment-related TEAEs were reported in 63 (11.7%) patients, with an incidence of 11.8%, 13.0%, 9.2% in the DKP/TRAM, TRAM, and placebo arms, respectively. In the single-dose phase, > 1% of the patients in the DKP/TRAM, TRAM, and placebo arms had the following TEAEs: dizziness (5.7%, 5.8%, and 1.7%, respectively) and nausea (3.8%, 2.9%, and 4.2%, respectively). In the multiple-dose phase, > 1% of the patients in the DKP/TRAM and in the TRAM arms had the following TEAEs: nausea (6.7%, 9.3%, respectively), dizziness (6.3%, 6%, respectively), vomiting (4.8%, 8.2%, respectively), somnolence (4.4%, 2.6%, respectively), headache (3%, 1.9%, respectively), and constipation (1.9%, 1.1%, respectively).
TEAEs leading to discontinuation in the single-dose phase are shown in Table 2. Treatment discontinuations due to TEAEs were reported in 17 (3.2%) patients, with a comparable incidence between groups. In the multiple-dose phase, 141 (26.2%) patients reported TEAEs, with a comparable incidence between the DKP/TRAM (25.2%) and TRAM (27.2%) arms (Table 2). The majority of the TEAEs were mild to moderate in intensity. Three (0.6%) patients had severe TEAEs. Treatment-related TEAEs were reported in 127 (23.6%) patients, with a comparable incidence between the DKP/TRAM and the TRAM arms (22.6% and 24.6%, respectively). Treatment discontinuations due to TEAEs were reported in 38 (7.1%) patients with an incidence of 7.5% versus 6.7% in the TRAM and DKP/TRAM groups, respectively (ESM Table 8).
Discussion
Dexketoprofen/tramadol 25/75 mg is an oral fixed-dose combination that acts through a multimodal approach to moderate-to-severe acute pain since it has central analgesic action along with a peripheral analgesic effect and anti-inflammatory activity [14]. DANTE is the first phase IV trial investigating the effects of DKP/TRAM on LBP. The primary efficacy objective was to evaluate the analgesic efficacy of DKP/TRAM compared to placebo in patients with moderate to severe acute LBP for the first 8 h following the initial dose. A composite primary endpoint was selected to address this research question, namely, time to first achieve an NRS-PI score < 4 or a pain intensity reduction ≥ 30% from drug intake up to 8 h after the first dose (t8h); this endpoint was not met. Nevertheless, some considerations should be taken into account when interpreting the study results.
First, the primary endpoint was quite ambitious, aiming at demonstrating a faster reduction of pain intensity beyond a certain threshold. In fact, this endpoint was a dichotomous variable, which did not punctually quantify pain reduction following the administration of either DKP/TRAM or placebo.
Second, DKP/TRAM showed a positive trend over placebo in terms of first achieving an NRS-PI score < 4 or a pain intensity reduction ≥ 30% from drug intake up to 8 h after the first dose, but the difference did not reach statistical significance. This was mainly related to the overall lower rates of patients achieving the primary endpoint compared to those estimated. In addition, the high level of pain at baseline (mean NRS-PI score of 7) may have influenced this result. It should also be noted that the use of rescue medication was low in all treatment arms, which might have contributed to the overall low number of events in either arm.
Third, when it comes to continuous mode of pain intensity assessment, as defined by the secondary efficacy endpoints, the effectiveness of the fixed-dose combination clearly emerged. The discrete measurement of pain intensity chosen for the primary efficacy analysis did not allow capture of the analgesic effect as a whole, flattening the differences in pain reduction. This finding may be helpful for the design of future studies in the setting of LBP in which each point of NRS-PI reduction may translate into clinically meaningful advantages. The safety results indicated that the DKP/TRAM fixed combination is safe and well-tolerated in patients with moderate to severe acute LBP after both single and repeated doses. The majority of the TEAEs during the study were mild or moderate in intensity; there were only two serious adverse events, both of which were unrelated to the study medication. Overall, the discontinuation rate due to TEAEs was low (3.2%) in both the single-dose and multiple-dose phases, confirming that the DKP/TRAM fixed combination was well-tolerated. The majority of patients had no clinically significant abnormalities in laboratory parameters. Moreover, the spectrum and frequency of adverse events was similar between the different treatment arms.
In the clinical setting of acute LBP, the DKP/TRAM fixed combination was previously evaluated in a smaller observational study [24]. Compared to diclofenac/thiocolchicoside, DKP/TRAM provided significantly greater and sustained analgesia at days 3 and 7, with a higher proportion of responders. The present trial extends those results in a larger number of patients and with a randomized design. In the present study, the results with TOTPAR, but not SPID, were significantly greater with DKP/TRAM compared to TRAM at 24, 48, and 72 h.
Even if a wide range of treatment approaches are available, there is no consensus on the most effective pharmacological therapy for LBP at present. However, multimodal analgesia is highlighted by current guidelines as a valid strategy [5,6,7, 15]. While it is acknowledged that the primary endpoint of the DANTE study was not met, the results of key secondary efficacy endpoints, such as TOTPAR and SPID, clearly demonstrated that the DKP/TRAM fixed combination is useful and effective to treat acute LBP. It is conceivable that the primary efficacy outcome was not met since it was a composite endpoint based on challenging, time-dependent, and dichotomous variables. In this regard, however, the compelling results in terms of secondary endpoints comparing continuous/categorical variables clearly demonstrate statistical significance and that the DKP/TRAM fixed combination can be considered to be an effective and safe option in the acute moderate to severe LBP treatment armamentarium. In particular, it should be noted that DKP/TRAM achieved superiority over TRAM in TOTPAR at 4, 6, and 8 h after the initial dose, which is an encouraging result. Moreover, the combination DKP/TRAM spares the use of opioids and is well tolerated.
Among the limitations of the DANTE study are the use of a composite primary endpoint, and the lack of a comparator arm receiving a COX inhibitor/opioid combination. In addition, full return to activity was not evaluated, no pain phenotyping was performed, a potential neuropathic component was not assessed, and patients with or without radiculopathy were enrolled. Moreover, sleep was not evaluated, which can also be a surrogate indicator of pain. On the other hand, its main strengths are the heterogeneity of patients included, which is reflective of routine practice, use of multiple assessments for analgesic efficacy, and the large size of the cohort, which to our knowledge is the largest randomized trial to date investigating a COX inhibitor/opioid combination as treatment for acute LBP compared to prior studies [24, 25, 29, 30]. The safety profile was also good overall.
Conclusion
The DANTE trial found that the DKP/TRAM fixed combination did not meet the primary endpoint of the study, but it was superior over placebo and TRAM in terms of total pain relief and significantly superior in terms of pain reduction over placebo at early timepoints. The combination also achieved superiority over TRAM in TOTPAR at early timepoints and was well tolerated with no safety concerns, while sparing the use of opioids. Overall, the results stress the validity of multimodal analgesia and the DKP/TRAM combination to treat acute LBP.
References
Rubin DI. Epidemiology and risk factors for spine pain. Neurol Clin. 2007;25(2):353–71.
Grabovac I, Dorner TE. Association between low back pain and various everyday performances: activities of daily living, ability to work and sexual function. Wien Klin Wochenschr. 2019;131(21–22):541–9.
Hartvigsen J, Hancock MJ, Kongsted A, et al. What low back pain is and why we need to pay attention. Lancet. 2018;391(10137):2356–67.
Perrot S, Allaert FA, Concas V, Laroche F. “When will I recover?” A national survey on patients’ and physicians’ expectations concerning the recovery time for acute back pain. Eur Spine J. 2009;18(3):419–29.
O’Connell NE, Cook CE, Wand BM, Ward SP. Clinical guidelines for low back pain: a critical review of consensus and inconsistencies across three major guidelines. Best Pract Res Clin Rheumatol. 2016;30(6):968–80.
Oliveira CB, Maher CG, Pinto RZ, et al. Clinical practice guidelines for the management of non-specific low back pain in primary care: an updated overview. Eur Spine J. 2018;27(11):2791–803.
Qaseem A, Wilt TJ, McLean RM, et al. Noninvasive treatments for acute, subacute, and chronic low back pain: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2017;166(7):514–30.
Price MR, Cupler ZA, Hawk C, et al. Systematic review of guideline-recommended medications prescribed for treatment of low back pain. Chiropr Man Therap. 2022;30(1):26.
Amaechi O, Huffman MM, Featherstone K. Pharmacologic therapy for acute pain. Am Fam Physician. 2021;104(1):63–72.
Mathieson S, Kasch R, Maher CG, et al. Combination drug therapy for the management of low back pain and sciatica: systematic review and meta-analysis. J Pain. 2019;20(1):1–15.
Cashin AG, Wand BM, O’Connell NE, et al. Pharmacological treatments for low back pain in adults: an overview of Cochrane Reviews. Cochrane Database Syst Rev. 2023;4(4):CD013815.
Wewege MA, Bagg MK, Jones MD, et al. Comparative effectiveness and safety of analgesic medicines for adults with acute non-specific low back pain: systematic review and network meta-analysis. BMJ. 2023;380:e072962.
Hanna M, Montero A, Perrot S, Varrassi G. Tramadol/dexketoprofen analgesic efficacy compared with tramadol/paracetamol in moderate to severe postoperative acute pain: subgroup analysis of a randomized, double-blind, parallel group Trial-DAVID Study. Pain Ther. 2021;10(1):485–503.
Varrassi G, Hanna M, Macheras G, et al. Multimodal analgesia in moderate-to-severe pain: a role for a new fixed combination of dexketoprofen and tramadol. Curr Med Res Opin. 2017;33(6):1165–73.
Allegri M, Montella S, Salici F, et al. Mechanisms of low back pain: a guide for diagnosis and therapy. F1000Res. 2016;5. https://doi.org/10.12688/f1000research.8105.2.
Varrassi G, Yeam CT, Rekatsina M, et al. The expanding role of the COX inhibitor/opioid receptor agonist combination in the management of pain. Drugs. 2020;80(14):1443–53.
Williams CM, Maher CG, Hancock MJ, et al. Low back pain and best practice care: a survey of general practice physicians. Arch Intern Med. 2010;170(3):271–7.
Varrassi G, Moretti B, Pace MC, et al. Common clinical practice for low back pain treatment: a modified Delphi study. Pain Ther. 2021;10(1):589–604.
Gay-Escoda C, Hanna M, Montero A, et al. Tramadol/dexketoprofen (TRAM/DKP) compared with tramadol/paracetamol in moderate to severe acute pain: results of a randomised, double-blind, placebo and active-controlled, parallel group trial in the impacted third molar extraction pain model (DAVID study). BMJ Open. 2019;9(2): e023715.
McQuay HJ, Moore RA, Berta A, et al. Randomized clinical trial of dexketoprofen/tramadol 25 mg/75 mg in moderate-to-severe pain after total hip arthroplasty. Br J Anaesth. 2016;116(2):269–76.
Montero Matamala A, Bertolotti M, Contini MP, et al. Tramadol hydrochloride 75 mg/dexketoprofen 25 mg oral fixed-dose combination in moderate-to-severe acute pain: sustained analgesic effect over a 56-h period in the postoperative setting. Drugs Today (Barc). 2017;53(6):339–47.
Moore RA, Gay-Escoda C, Figueiredo R, et al. Dexketoprofen/tramadol: randomised double-blind trial and confirmation of empirical theory of combination analgesics in acute pain. J Headache Pain. 2015;16:541.
Moore RA, McQuay HJ, Tomaszewski J, et al. Dexketoprofen/tramadol 25 mg/75 mg: randomised double-blind trial in moderate-to-severe acute pain after abdominal hysterectomy. BMC Anesthesiol. 2016;17(1):159.
Meloncelli S, Divizia M, Germani G. Efficacy and tolerability of orally administered tramadol/dexketoprofen fixed-dose combination compared to diclofenac/thiocolchicoside in acute low back pain: experience from an Italian, single-centre, observational study. Curr Med Res Opin. 2020;36(10):1687–93.
Perna A, Ricciardi L, Barone G, et al. Medical management of acute non-specific low back pain: comparison of different medical treatments, one center’s retrospective analysis. J Biol Regul Homeost Agents. 2018;32(6 Suppl. 1):121–9.
Varrassi G, Hanna M, Coaccioli S, et al. DANTE Study: The first randomized, double-blind, placebo and active-controlled, parallel arm group study evaluating the analgesic efficacy and safety of Dexketoprofen trometAmol aNd Tramadol hydrochloride oral fixEd dose combination on moderate to severe acute pain in patients with acute low back pain-rationale and design. Pain Ther. 2022;11(3):1055–70.
Atlas SJ, Deyo RA, Patrick DL, et al. The Quebec Task Force classification for Spinal Disorders and the severity, treatment, and outcomes of sciatica and lumbar spinal stenosis. Spine (Phila Pa 1976). 1996;21(24):2885–92.
Breivik H, Borchgrevink PC, Allen SM, et al. Assessment of pain. Br J Anaesth. 2008;101(1):17–24.
Babej-Dolle R, Freytag S, Eckmeyer J, et al. Parenteral dipyrone versus diclofenac and placebo in patients with acute lumbago or sciatic pain: randomized observer-blind multicenter study. Int J Clin Pharmacol Ther. 1994;32(4):204–9.
Lasko B, Levitt RJ, Rainsford KD, et al. Extended-release tramadol/paracetamol in moderate-to-severe pain: a randomized, placebo-controlled study in patients with acute low back pain. Curr Med Res Opin. 2012;28(5):847–57.
Acknowledgements
The study was sponsored by the Menarini Group. We also thank the participants of the study.
Medical Writing, Editorial, and Other Assistance
Medical writing and editing assistance, including preparation of a draft manuscript under the direction and guidance of the authors, incorporation of author feedback, and manuscript submission, were provided by Content Ed Net and funded by Menarini, with the helpful support of Patrick Moore, PhD.
Data Availability
The data are available at https://www.clinicaltrialsregister.eu/ctr-search/trial/2019-003656-37/results.
Funding
Medical writing assistance was funded by the Menarini group, which also funded the journal’s rapid service fee.
Author information
Authors and Affiliations
Contributions
Giustino Varrassi, Magdi Hanna, Serge Perrot, and Stefano Coaccioli, as members of the Steering Committee of the study, have made substantial contributions to the conception and design of the study, acquisition, analysis, and interpretation of data, and drafting and revising of the article. Paolo Fabrizzi and Simone Baldini were part of the Sponsor’s team of the study and revised the manuscript. Ivan Kruljac and Carlos Brotons participated as authors as main investigators involved in the study. All authors take the responsibility for the integrity of the work as a whole and have given their approval for this version to be published.
Corresponding author
Ethics declarations
Conflicts of Interest
Giustino Varrassi, Magdi Hanna, and Serge Perrot have received honoraria and consultancy fees from Menarini. Giustino Varrassi is a member of the Editorial Board of Pain & Therapy, but was not involved in the selection of peer reviewers for the manuscript nor in any of the subsequent editorial decisions. Stefano Coaccioli has received consultancy fees from Menarini. Ivan Kruljac and Carlos Brotons have no conflicts of interest to report. Paolo Fabrizzi and Simone Baldini are employed by A. Menarini Industrie Farmaceutiche Riunite, Florence, Italy.
Ethics/Ethical Approval
This study was performed in compliance with International Council for Harmonisation (ICH) Good Clinical Practices (GCP), including the archiving of essential documents, as well as the ethical principles of the Declaration of Helsinki of 1964 and its later amendments. The study protocol and protocol amendments, patient information leaflet, informed consent form (ICF), Summary of Product Characteristics (SmPC), and any other relevant documents according to National Regulations were reviewed and approved by an independent ethics committee (IECs) and the Health Authorities (HAs) of the participating countries. All local, national, and legal requirements for the conduct of a clinical study were followed. Prior to the patient’s enrollment into the study and before performing any study-related procedures, the Investigator or its authorized delegate obtained the patient’s written, dated, and signed informed consent to participate in the study and to allow the confidential disclosure, processing, and transferring of necessary documentation of the patient’s health and personal data to the contract research organization (CRO), Sponsor and its Affiliates, the competent HAs, and any other institutions, as legally required and in accordance with the local applicable privacy laws.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Varrassi, G., Hanna, M., Coaccioli, S. et al. Dexketoprofen Trometamol and Tramadol Hydrochloride Fixed-Dose Combination in Moderate to Severe Acute Low Back Pain: A Phase IV, Randomized, Parallel Group, Placebo, Active-Controlled Study (DANTE). Pain Ther (2024). https://doi.org/10.1007/s40122-024-00623-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40122-024-00623-4