
Abstract
In 1976 Ebola revealed itself to the world, marking the beginning of a series of localized outbreaks. However, it was the Ebola outbreak that began in 2013 that incited fear and anxiety around the globe. Since then, our comprehension of the virus has been steadily expanding. Ebola virus (EBOV), belonging to the Orthoebolavirus genus of the Filoviridae family, possesses a non-segmented, negative single-stranded RNA genome comprising seven genes that encode multiple proteins. These proteins collectively orchestrate the intricate process of infecting host cells. It is not possible to view each protein as monofunctional. Instead, they synergistically contribute to the pathogenicity of the virus. Understanding this multifaceted replication cycle is crucial for the development of effective antiviral strategies. Currently, two antibody-based therapeutics have received approval for treating Ebola virus disease (EVD). In 2022, the first evidence-based clinical practice guideline dedicated to specific therapies for EVD was published. Although notable progress has been made in recent years, deaths still occur. Consequently, there is an urgent need to enhance the therapeutic options available to improve the outcomes of the disease. Emerging therapeutics can target viral proteins as direct-acting antivirals or host factors as host-directed antivirals. They both have advantages and disadvantages. One way to bypass some disadvantages is to repurpose already approved drugs for non-EVD indications to treat EVD. This review offers detailed insight into the role of each viral protein in the replication cycle of the virus, as understanding how the virus interacts with host cells is critical to understanding how emerging therapeutics exert their activity. Using this knowledge, this review delves into the intricate mechanisms of action of current and emerging therapeutics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Ebola virus (EBOV) haunts humankind with a case fatality rate (CFR) of 60%, underscoring the imperative nature of studying and understanding how the virus interacts with host cells to effectively develop antiviral strategies. |
In 2022, a significant milestone in the field of Ebola virus disease (EVD) therapeutics was achieved with the publication of the first evidence-based clinical practice guideline dedicated to specific EBOV therapeutics for EVD. |
At present, two antibody-based therapeutics are approved for the treatment of EVD, but to further improve the outcomes of the disease it is crucial to focus on emerging therapeutics that may shift the grim course of the disease. |
Many different mechanisms of action are being investigated to halt viral infection, with potential molecules acting on the virus, as direct-acting antivirals, or acting on the host, as host-directed antivirals. |
Repurposing already approved drugs for non-EVD indications to treat EVD is a promising strategy to bypass some of the disadvantages associated with conventional drug discovery. |
Introduction
The twenty-first century has experienced a surge of severe infectious disease outbreaks, including the Ebola virus disease (EVD) epidemic in West Africa from 2013–2016 and the recent COVID-19 pandemic. Both outbreaks resulted in substantial morbidity and mortality while spreading across borders to infect people in multiple countries [1]. Technological advances have enabled swifter movement of people and pathogens over large distances, and increases in air travel have been associated with the risk of importing and exporting pathogens, with some capable of causing severe health crises [1,2,3,4].
According to The World Bank, the number of airline passengers almost tripled from 2000 to 2019, going from 1.67 billion in 2000 to 4.46 billion in 2019, excluding data from 2020 and forward because of travel limitations during the COVID-19 pandemic [5].
Since the volume of global air travel continues to increase annually, and passengers can introduce infections to new regions in short timeframes [2], as could be seen with the rapid global spread of the SARS-CoV-2 that prompted the COVID-19 pandemic [1], air travel poses a growing threat to global health security since it has the potential to cause swift and broad dissemination of emerging and re-emerging infectious diseases. Furthermore, it is possible for a traveler harboring an infection in one location on Earth to travel to virtually any other point of the planet in only 1–2 days [2].
EVD is a life-threatening disease caused by Ebola virus (EBOV). According to current terminology, EBOV is an Orthoebolavirus zairense species in the Orthoebolavirus genus [6]. EBOV has a pooled case fatality rate (CFR) of 60% in outbreaks from 2010 to 2020 [7, 8]. Considering the high CFR of EVD, it is no surprise that there is a rising concern for potential EVD transmission in distant locales via commercial air travel [9]. EVD outbreaks typically start from a single case, an index case, of probable zoonotic transmission, followed by human-to-human transmission [7].
Subsequently, the EVD outbreak in West Africa that occurred from 2013 to 2016 demonstrated how rapidly pathogens can spread to large urban centers following one spillover event (i.e., the spread of Ebola from animals to humans) [4]. Although Guinea, Sierra Leone, and Liberia bore the brunt of the outbreak, several cases were exported to nearby African countries and other countries around the globe as a result of the ease of international travel, inciting fear and anxiety around the world [2, 10, 11]. Later, the 2018–2019 EVD outbreak in the Democratic Republic of Congo (DRC) raised concern for EVD transmission via air traveling since cases were detected in Goma, a capital city with an international airport [9]. These outbreaks and numerous others serve as an incessant warning of the ongoing threat posed by EBOV to global public health and biosecurity [10].
To date, there are approved EBOV vaccines and antibody-based therapeutics. Nonetheless, the high fatality rate of EVD indicates that continuous research and development of antivirals is necessary to improve its current management and increase preparedness and vigilance for future emergencies [12]. With globalization, international travel, and foreign medical missions, a patient with EVD may present in any emergency department, and if misdiagnosed, serious repercussions for the patient and public health would ensue [13, 14].
This work aims to inform the reader about not only the state-of-the-art therapeutics for EVD but also emerging therapeutics. However, as a result of the available evidence, the focus of this work is directed only to the treatment of EVD, the disease caused by EBOV (O. zairense), and not other viruses of the Orthoebolavirus genus. This review has two main parts. Firstly, an introduction to the virus, the genome, proteins, and replication cycle is provided to give the reader a complete understanding of the virus while focusing on the different roles played by the EBOV proteins, as they serve as potential targets to halt viral infection. Secondly, approved and emerging EBOV therapeutics are highlighted, establishing a link between their mechanisms of action and the replication cycle of the virus, using detailed images to aid comprehension. Furthermore, this work compiles information about repurposing widely used licensed drugs for EVD.
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Ebola Virus: From a Virology Point of View
Classification
According to the current International Committee of Taxonomy of Viruses (ICTV) taxonomy release, the family Filoviridae is divided into eight genera, Cuevavirus, Dianlovirus, Orthoebolavirus (formerly Ebolavirus), Orthomarburgvirus (formerly Marburgvirus), Oblavirus, Striavirus, Tapjovirus, and Thamnovirus [15]. Frequently, viruses belonging to the Orthoebolavirus genus are referred to as ebolaviruses, and viruses belonging to the family Filoviridae are widely recognized as filoviruses [10].
The Orthoebolavirus genus has six species: O. zairense (formerly Zaire ebolavirus, often known as Ebola virus, abbreviated as EBOV), Orthoebolavirus sudanense (formerly Sudan ebolavirus, SUDV), Orthoebolavirus restonense (formerly Reston ebolavirus, RESTV), Orthoebolavirus bundibugyoense (formerly Bundibugyo ebolavirus, BDBV), Orthoebolavirus taiense (formerly Tai Forest ebolavirus, TAFV), and Orthoebolavirus bombaliense (formerly Bombali ebolavirus, BOMV) [6, 16, 17]. Both the genus and species names were officially changed in 2023 by the ICTV [18], and the scientific community predominantly adopts the former terminology. Virus names, i.e., commonly used names and abbreviations, are not official ICTV designations [19]. Therefore, this work uses the most routinely used ones. Figure 1 illustrates Orthoebolavirus genus taxonomy according to the ICTV, virus names, and corresponding abbreviations often used in the literature.

Schematic representation of the taxonomy of the genus Orthoebolavirus. Viral species are often referred as the commonly used Virus name and corresponding Abbreviation. Created with BioRender.com
Viruses of the genus Orthoebolavirus can cause life-threatening diseases. While EBOV, SUDV, and BDBV are highly lethal human pathogens, with EBOV being the leading cause of most outbreaks, TAFV has only one recorded non-lethal human disease, albeit severe. As far as is known, RESTV has only caused asymptomatic human infections, and no human cases of BOMV have been reported [12, 19, 20].
According to the World Health Organization (WHO) International Classification of Diseases Revision 11 (ICD-11) of 2018, the disease caused by any Orthoebolavirus is collectively referred to as Ebola disease (EBOD). Inside the EBOD subcategory, the disease caused by EBOV is defined as Ebola virus disease (EVD), i.e., EVD is defined as a disease only caused by EBOV (O. zairense) [21].
In the past, EVD was known as Ebola hemorrhagic fever on the basis of its clinical manifestations, which include coagulation abnormalities, bleeding, and shock. However, following the publication of “WHO Best Practices for the Naming of New Human Infectious Diseases” the term “hemorrhagic fever” is no longer used to refer to EVD because most Ebola-infected individuals do not develop overt hemorrhage, and it usually occurs in the terminal phase of fatal illness, when the individual is already in shock. Consequently, healthcare workers could misdiagnose EVD, or potentially infected individuals may not seek medical assistance on the basis of the absence of bleeding [21, 22].
Virion Morphology
Filoviruses derive their name from the Latin word filum, meaning “thread” [10], which corroborates with their predominantly filamentous appearance that is easily identifiable in electron micrographs, as depicted in Fig. 2. Depending on culture conditions and cell or tissue types, particles can assume different shapes, such as branched, toroid, U-shaped, and 6-shaped (“shepherd’s crook”) [23]. Ebolaviruses have also been observed to have spherical virion morphologies [6, 24].

Ebola virus morphology. A Digitally colorized electron microscopic image of an isolate of Ebola virus (CDC/Dr. Frederick Murphy). B Digitally colorized scanning electron microscopic (SEM) image of filamentous Ebola virus budding from a VERO cell (NIAID). C Transmission electron microscopic (TEM) image of a VERO cell infected with the Ebola virus (CDC/Dr. Stan Foster, B. Partin). D Digitally colorized SEM image of numerous filamentous Ebola virus particles (red) budding from a VERO E6 cell (blue) (NIAID). E SEM image of numerous filamentous Ebola virus particles budding from a VERO E6 cell (NIAID). All images were obtained from the Public Health Image Library. Created with BioRender.com
Filamentous particles are typically less than 1 µm long, but these particles can reach lengths greater than 20 µm. This extreme degree of length polymorphism occurs as a result of multiple copies of viral RNA being packaged to produce polyploid virus particles, in which a single virion contains more than one copy of the viral genome, each arranged end-to-end continuously or separated from one another by a short section of empty envelope. Particles were reported to have up to 22 genome copies [10, 23, 25].
Their diameters are more uniform, ranging from approximately 91 to 98 nm. Ebola virions possess a central core, the helically arranged nucleocapsid (NC), and an envelope of cellular origin [10, 23]. The lipid envelope is decorated with globular peplomers, i.e., viral proteins inserted in the lipoprotein bilayer envelope [23, 26]. Empty virions containing no viral genome have also been observed, possessing much smaller diameters and random lengths [25].
Genome Structure and Organization
Genomic Organization
Ebolaviruses genome comprises a single, non-segmented, linear strand of negative-sense RNA (ssRNA (−)) of approximately 19 kb. The genome contains seven genes, each of which encodes one of seven structural proteins: the nucleoprotein (NP), the viral protein 35 (VP35), VP40, the glycoprotein (GP), VP30, VP24, and the RNA-dependent RNA polymerase (RdRp) “Large” protein (L) [10, 27, 28]. Each gene contains a single open reading frame (ORF), except for the GP gene, which consists of three overlapping ORFs [6], further elaborated in the Sect. “Glycoprotein”.
Viruses from the Orthoebolavirus genus have the gene order 3′-NP-VP35-VP40-GP-VP30-VP24-L-5′ [23], as illustrated in Fig. 3. The genome itself is flanked by a conserved 3′ leader sequence and a 5′ trailer sequence [10]. They contain cis-acting signals for mRNA transcription and genome replication, including promoters. Each gene is flanked at its 3′ and 5′-end by highly conserved untranslated regions (UTRs), which contain transcription start and stop signals, respectively [23].

Schematic representation of an ebolavirus genome. The genome contains seven genes, the nucleoprotein (NP), the viral protein 35 (VP35), VP40, the glycoprotein (GP), VP30, VP24, and the RNA-dependent RNA polymerase (RdRp) “Large” protein (L). Each gene contains a single open reading frame (ORF), except for the GP gene, which consists of three overlapping ORFs. The ORFs are transcribed and subsequently translated into a nonstructural protein precursor of the secreted glycoprotein (pre-sGP), major glycoprotein (GP1,2) precursor (pre-GP), and secondary secreted glycoprotein precursor (pre-ssGP). After post-translational modifications, pre-sGP yields sGP and Δ-peptide, pre-GP yields GP1 and GP2, and pre-ssGP yields ssGP. The elements depicted in the image are not drawn to scale. The sizes and proportions of the objects are intentionally adjusted for visual clarity and ease of understanding. Created with BioRender.com
Genome Expression Products (EBOV Proteins and Their Functions)
EBOV particles consist of a central helical NC, in which the viral genome is encapsidated by the helically polymerized NP and is associated with four additional viral proteins: the polymerase cofactor (VP35), the polymerase (L), the transcriptional activator (VP30), and VP24 [29, 30]. The NC protects the viral RNA from degradation by endonucleases and host immune response [6]. This structure is embedded in the virion matrix composed of a layer of VP40 polymers surrounded by a cellular membrane-derived envelope containing GP1,2 peplomers, sGP-GP2, and, among other components, phosphatidylserine (PS) [10, 23]. Figure 4 provides an artistic rendering of an ebolavirus virion.

Artistic rendering of an ebolavirus virion. The viral particle has a central helical nucleocapsid (NC), in which the viral genome is encapsidated by the helically polymerized nucleoprotein (NP) and is associated with four additional viral proteins (VP): the polymerase cofactor (VP35), the polymerase (L), the transcriptional activator (VP30), and VP24. This structure is embedded in the virion matrix composed of a layer of VP40 polymers surrounded by a host membrane-derived envelope containing GP1,2 peplomers, sGP-GP2, and among other components, phosphatidylserine. The elements depicted in the image are not drawn to scale. The sizes and proportions of the objects are intentionally adjusted for visual clarity and ease of understanding. Created with BioRender.com
In the literature, the term NC is often used interchangeably with the term ribonucleoprotein (RNP) complex. Similar to other authors, herein, RNPs are considered to be functional complexes that are active in transcription and replication, composed of viral RNA, NP, VP35, and L, with or without the transcription initiation factor VP30. On the other hand, NCs are categorized as discrete condensed structures that are detected inside the viral particles composed of the genomic RNA, NP, VP35, L, VP30, and VP24. NCs are not active in transcription or replication [29].
EBOV virulence is partially attributed to the ability of the virus to evade innate antiviral responses and suppress dendritic cell maturation, which may blunt the development of effective adaptive immune responses [31]. Sections “Nucleoprotein” to “Polymerase” describe the known and principal functions of each viral protein, as well as their role in EBOV virulence, making them possible therapeutic targets. Please note that structural complexities are beyond the scope of this work.
Nucleoprotein (NP)
NP is the main determinant of the structure of ebolaviruses NCs owing to its homo-oligomerization resulting in the formation of helical structures [29]. During the assembly of the NC, the RNA genome is tightly coated with the NP, which protects it from degradation and recognition by the cellular immune response [32].
On the one hand, the NP plays a major role in the formation of cytoplasmic inclusion bodies (IBs), i.e., cytosolic membrane-less compartments, also known as viral factories, which are critical for viral RNA synthesis [33, 34]. The accumulation of this protein leads to the recruitment of the other NC proteins, i.e., VP35, L, VP30, and VP24, via direct or indirect protein–protein interactions. On the other hand, NP has a crucial function in the formation of ribonucleoprotein complexes, a consequence of NP encapsidation of positive-strand RNA genomes (antigenomic RNPs) and negative-strand RNA genomes (genomic RNPs) [29]. NP forms a complex with VP35, VP30, and L, the viral polymerase complex, which is essential for genome replication and transcription [35, 36].
In a paper published in 2020, it was suggested that NP recruits CAD (carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase), a trifunctional multi-domain enzyme involved in the biosynthesis of pyrimidines, into IBs to provide pyrimidines for EBOV genome replication and transcription, and thus hijacks a host cellular pathway to facilitate EBOV genome replication and transcription [32]. Another research article demonstrated that NP recruits NXF1, a host nuclear RNA export factor, into IBs to facilitate viral protein expression. More specifically, the authors propose that upon recruitment of NXF1 into IBs, it forms complexes with viral mRNAs and promotes its export from these sites. This is extremely relevant, as viral mRNAs need to be transported outside IBs to reach free cytosolic ribosomes for translation [37].
Additionally, NP interacts with host chaperones, including heat shock protein (HSP) 70 and 90 [12, 30]. The stability of NP was shown to depend on HSP70, and disruption of this stability negatively impacts viral RNA synthesis [38]. HSPs are stress proteins known to cooperate with numerous viruses to promote their replication. A review on the topic can be found elsewhere [39].
In summary, it has been shown that NP is vital for viral genome replication and transcription, RNA encapsidation, NC formation, and capsid assembly, and these functions are completed, in part, via NP interactions with other viral proteins and host proteins [6, 23, 29, 30]. Therefore, it has been hypothesized that developing a therapeutic candidate targeting NP could hinder NC formation and NP–host protein interactions, thus halting virus transcription and assembly [6].
Viral Polymerase Complex Protein 35 (VP35)
VP35 is a structural component of the NC and an essential cofactor for the viral polymerase [31]. It is also a component of the RNP complex [23, 29]. Inside IBs, the maturation of thin-walled helices (RNPs) into thick-walled helices (corresponding to NCs) can only be observed in the presence of VP35 [29].
VP35 plays a vital role in immune evasion by antagonizing the innate host immune response [6]. Several in vitro studies have demonstrated that EBOV VP35 inhibits both the production of type I interferon (IFN) and dendritic cell maturation by interfering with host retinoic acid-inducible gene I (RIG-I)-like receptor (RLR) signaling [31]. The RLRs, such as RIG-I and MDA-5, are cytosolic pattern recognition receptors (PRRs) that detect pathogen-associated molecular patterns (PAMPs), such as viral dsRNA [40]. VP35 is capable of binding to viral dsRNA to prevent recognition by RIG-I and MDA-5, which consequently inhibits the induction of IFN production and dendritic cell maturation [31, 41, 42]. This binding to dsRNA is dependent on the Interferon Inhibitory Domain (IID) of VP35, which acts as an RNA-binding domain. IID is also crucial for the polymerase cofactor function of VP35, although this function is independent of IID binding to dsRNA [6, 43].
The importance of the virulence of VP35 has been shown in vivo by Woolsey and colleagues, where a mutant VP35 (mVP35) EBOV inoculated in non-human primates did not cause lethal disease and elicited adaptative immune responses that protected the animals against a wild-type EBOV challenge. The mVP35 did not inhibit the RIG-1 pathway [31].
VP35 also interacts with NP. NP has a hydrophobic pocket that either binds to another NP or to VP35. This interaction regulates viral RNA synthesis since binding to VP35 regulates the oligomerization of NP and results in the release of RNA from NP–RNA complexes, which are essential to viral replication [6, 30, 36].
Haasnoot et al. documented the activity of VP35 as a suppressor of RNA silencing. RNA silencing or interference (iRNA) serves as an innate antiviral defense triggered by viral dsRNA produced during infection. By inhibiting iRNA pathways, VP35 promotes EBOV replication [44].
According to the findings of Zhu and colleagues, VP35 hijacks the host PKA-CREB1 pathway by binding to AKIP1, which, in turn, activates PKA and CREB1, a transcription factor. During EBOV infection, CREB1 is recruited into RNP complexes inside IBs and is employed for viral replication, as it increases the interaction between L and VP35. Additionally, several coagulation-related genes are upregulated by the VP35-dependent CREB1 activation, which may contribute to the hemorrhage associated with EVD [45].
Furthermore, it has been shown that TRIM6 is an important host factor for EBOV replication since it ubiquitinates the IID of VP35, which promotes viral replication. It is suggested that VP35 hijacks TRIM6 to enhance replication [23, 46].
Additionally, VP35 prevents stress granule assembly [23]. Stress granules are cytoplasmic aggregates of translationally silenced mRNAs that assemble in response to environmental stress and play an important role in antiviral innate immunity [47].
Taking all the above into consideration, VP35 is a multifunctional protein essential for various EBOV replication steps and immune evasion. Thus, targeting VP35 might enable the host to mount a robust immune response and inhibit viral replication [6].
Matrix Protein 40 (VP40)
VP40 is the major matrix protein of EBOV that surrounds the NCs [6, 30, 48]. It has an important role in the maintenance of the structural stability of viral particles and aggregation at the cell membrane for virion budding and egress [27, 49]. It drives particle budding by manipulating host cell membranes that are embedded with the viral GP [30]. VP40 is essential and sufficient for viral assembly and egress, as proven by the fact that independent expression of VP40 leads to the production of virus-like particles (VLPs) [50,51,52].
Furthermore, VP40 contains late-budding motifs that are pivotal for budding since they mediate the recruitment of host proteins required for virus–cell separation [23, 51]. It allows VP40 to interact with the host endosomal sorting complex required for transport (ESCRT) machinery, which promotes the budding and release of new virions [53]. The recruitment of NEDD4, a host E3 ubiquitin ligase associated with the ESCRT complex, is one of the mandatory interactions [51]. Findings from a recent study suggest that P300, a host acetyltransferase, acetylates NEDD4, which promotes the NEDD4–VP40 interaction, with subsequent VP40 ubiquitination, which alters VP40 subcellular localization since ubiquitination can be regarded as a signal to drive VP40 migration to the plasma membrane (PM). The localization of VP40 at the PM is a requirement for efficient EBOV egress [52]. In addition to ubiquitination, other post-translational modifications (PTMs), such as phosphorylation and SUMOylation, are implicated in efficient VP40-driven EBOV release. The host c-Abl1 tyrosine kinase (TK) phosphorylates VP40, which is reported to increase EBOV release [30, 52].
This viral protein is classified as a transformer protein owing to its ability to have multiple conformational states—dimers, linear hexamers, and circular octamers. Dimers bind to cellular membranes, hexamers polymerize to matrix filaments, and octamers bind to ssRNA [6, 23, 48].
VP40 dimers at the infected cell plasma membrane recruit NCs since the matrix protein directly binds NP and VP35. Interacting with PS, VP40 reorganizes into linear hexamers, resulting in virion matrix filaments that engulf NCs. Lastly, octameric conformations play an important role in virion replication since they do not have a PM localization, but instead bind to RNA, likely negatively regulating transcription [23, 48, 54].
From an evolutionary perspective, when a virus synthesizes enough elements for packaging, i.e., genome and proteins, the virus should shut down further synthesis and promote assembly and budding of the viral particles, to ensure viral amplification [33]. VP40 is a negative regulator of genome transcription and replication [23, 29]. In a recent publication, Wu et al. proposed a two-stage interaction between VP40 and NP that results in a switch from RNA transcription and replication to virion assembly and budding [33].
Additionally, in a recent article, VP40 was shown to interact with PS-enriched regions at the PM inner leaflet as a dimer, enhancing the clustering of PS. Moreover, VP40 membrane binding and oligomerization are dependent on PS in the PM and, thus, the enrichment of PS within regions of the PM provides additional PS molecules available to recruit VP40 to platforms of viral budding. Thus, PS clustering by VP40 is a critical step in viral budding [50]. PS is also actively involved in viral entry and will be discussed in detail in Sect. “Attachment”.
VP40 also has a role in immune evasion via iRNA suppression and bystander lymphocyte apoptosis. The latter is achieved because VP40 induces the formation of extracellular vesicles (EVs) that are capable of inducing apoptosis [54, 55]. Pleet et al. demonstrated these phenomena, where VP40-containing EVs induced apoptosis of T cells and monocytes, which may be, in part, responsible for the decimation of immune cells during EBOV infection [56]. The recent review by Pleet et al. summarizes the current state of knowledge regarding extracellular vesicles in EBOV infection [55].
Considering the aforementioned, VP40 is a promising target for EBOV therapy, as it is involved in the budding process and replication, transcription, and immune evasion [6, 12].
Glycoprotein (GP)
The Ebolavirus GP gene contains three ORFs. The 0 frame is transcribed and subsequently translated to a nonstructural protein precursor of the secreted glycoprotein (pre-sGP). The expression of the mRNA corresponding to the − 1 frame yields the major glycoprotein (GP1,2) precursor (pre-GP), and the mRNA corresponding to the + 1 frame yields the secondary secreted glycoprotein precursor (pre-ssGP). The expression ratio of these pre-proteins (pre-sGP/pre-GP/pre-ssGP) has the following range: 61–72%:19–28%:1–5% [23].
Pre-sGP is synthesized in the endoplasmic reticulum (ER) and is cleaved post-translationally by furin-like endoproteases into sGP and Δ-peptide [6, 23]. sGP is not structural and is secreted from infected cells through the Golgi apparatus, thereby leading to high sGP concentrations in the blood of patients with EDV. As a result of its resemblance to GP1, since sGP carries the N-terminal 295 amino acids of GP1,2, sGP is partially cross-reactive with several anti-GP1 antibodies, including anti-GP1,2, suggesting an antibody decoy activity of circulating sGP in infected individuals [23, 57]. Furuyama et al. provided some insight into EBOV sGP pathogenicity in vivo. By infecting BALB/c mice with wild-type EBOV, and subsequently injecting them with EBOV sGP, they observed higher viral titers in the liver compared to the group that did not receive sGP. Curiously, the group that received RESTV sGP did not reveal a higher viral titer in the liver. Lastly, the observed viral titers in the group that received EBOV sGP resembled the titers from the group that was inoculated with PMA, a compound that activates the MAPK signaling pathway previously known to affect the robustness of EBOV replication. The authors hypothesize that EBOV sGP activates the MAPK signaling pathway, which contributes to higher viral replication [58]. All the possible known roles of sGP are discussed elsewhere [59].
The Δ-peptide is a conserved product of post-translational processing of the abundant pre-sGP [23, 60]. It is a secreted nonstructural protein that assembles as a pentameric chloride-selective viroporin, i.e., a nonstructural virus-encoded protein that permeabilizes cellular or viral membranes [23, 61]. Viroporins are known to adapt host cells for effective viral spread. He et al. speculated that the viroporin activity of the EBOV Δ-peptide could give it enterotoxin activity, which could be associated with the gastrointestinal symptoms experienced by patients with EVD [61]. The actual function of the peptide during in vivo infection remains to be determined [23], but recently, Melnik et al. used BALB/c mice to demonstrate that the Δ-peptide is indeed a potent enterotoxin that induces diarrhea via direct cellular damage and regulation of genes that encode proteins involved in fluid secretion. Since gastrointestinal symptoms are prominent clinical features of EVD, and diarrhea is a significant predictor of fatal outcomes, this discovery suggests that Δ-peptide might contribute to EBOV-induced gastrointestinal pathology [60].
Pre-GP contains a signal peptide that targets the protein to the ER and a type I transmembrane domain (TM) that tethers the protein to the ER membrane. In the ER, it is glycosylated in the mucin-like subdomain [23]. ER α-glucosidases facilitate proper folding and maturation of glycoproteins by ensuring their accurate N-glycosylation [30]. The glycosylated pre-GP dimerizes via a disulfide bond and is posteriorly cleaved by furin-like endoproteases into GP1 and GP2 subunits that remain attached through a disulfide bond. Before transport to the PM, these heterodimers trimerize to form the mature GP1,2 peplomer that is incorporated into the viral envelopes. GP1,2 is a trimeric type I transmembrane and class I fusion protein consisting of disulfide-linked GP1–GP2 heterodimers that assume the shape of a chalice with the three GP1 subunits angling outward but bound together at their bases where they connect to the GP2 trimer [23].
GP1 is the virion surface-exposed GP1,2 moiety that mediates cell-surface attachment. GP1 is structured into a base, head, glycan cap, and mucin-like subdomains. The receptor-binding site (RBS) is situated in the head, but it is masked by the glycan cap. The RBS binds to the endolysosomal Niemann-Pick C1 (NPC1) receptor, described in detail in Sect. “Fusion: The Endolysosomal Escape”, and is critical for viral entry. The mucin-like subdomain is located upward and outward from these caps, and while it does not directly impact viral entry, it shields GP1,2 from host antibodies and can stimulate host dendritic cells via MAPK and NF-κβ pathways [6, 23]. GP2 has a fusion loop that wraps around the outside of the GP2 trimer and contributes to the fusion of viral and host cell membranes after NPC1 binding due to conformational changes that lead to the insertion of the fusion loop into the endolysosomal membrane. It also has a TM that tethers GP1,2 to the virion membrane. Additionally, it has an immunosuppressive motif [6, 23].
Proteolytic cleavage of EBOV GP1,2 near the TM domain of GP2 by ADAM metallopeptidase domain 17 (ADAM17, formerly TACE) produces a soluble, trimeric GP1,2 variant with a truncated GP2, referred to as shed glycoprotein (GP1,2Δ). Because the structure is highly similar to that of GP1,2, it is hypothesized to be an anti-GP1,2 antibody decoy [23, 59]. GP1,2Δ activates non-infected dendritic cells and macrophages to secrete cytokines that increase endothelial vascular permeability [6, 23].
A poorly understood situation is when GP1 is replaced by sGP, producing a functional sGP-GP2 protein. sGP monomers connect to GP2 monomers via a disulfide bridge, and the resulting heterodimer is anchored to the ER and, later, to the PM via the GP2 TM domain and may be incorporated into the envelope of budding virions. As a result of significant similarities between sGP and GP1, sGP-GP2 is hypothesized to function as an anti-GP1,2 antibody decoy directly on the virion, thereby protecting GP1,2 peplomers from host recognition [23].
Pre-ssGP undergoes maturation in the ER, becomes N-glycosylated, and is subsequently secreted from infected cells as a disulfide-linked homodimer (ssGP). The function remains unclear, but its similarity to sGP and sGP-GP2, ssGP could also serve as an antibody decoy [23].
Nanbo et al. demonstrated that GP1,2 promotes the incorporation of host XK-related protein (Xkr) 8 into viral particles, which enhances viral entry by apoptotic mimicry [62]. This mechanism of infection is further detailed in Sect. “Attachment”.
Overall, since GP1,2 is the viral protein on the viral envelope and is responsible for viral attachment and entry into host cells, agents targeting various GP gene products are promising candidates for therapeutics, and specifically targeting GP1,2 could be an excellent method for neutralizing the virus. In fact, it is the most extensively studied antiviral target [6, 12, 63].
Minor Nucleoprotein (VP30)
VP30 is a structural component of the NC and is indispensable for RNA transcription initiation [6, 29, 54]. It is also a component of the RNP complex and IBs [23, 29].
The viral polymerase complex executes both replication and transcription and is composed of the polymerase L, VP35, and NP. Transcription is also dependent on the transcriptional initiation factor VP30 [29, 35]. The transcription-activating function of VP30 depends on an RNA hairpin structure that is formed at the transcription start signal of the NP gene [23, 27, 64, 65].
The transcriptional support activity of VP30 is switched off upon phosphorylation, enhancing its binding to NP while inhibiting its interaction with RNA and VP35, thereby inhibiting transcription and favoring replication [29, 35, 66]. Meanwhile, genome transcription requires VP30 in its unphosphorylated form [35]. Dephosphorylation weakens the binding between VP30 and NP, and VP30 recruits the polymerase complex via the binding of VP35 and initiates transcription [66]. Thus, the phosphorylation of VP30 regulates the balance between transcription and replication [29].
In addition, VP30 is speculated to promote L transcription reinitiation during the sequential transcription of all the EBOV genes and to regulate cotranscriptional GP gene editing since VP30 acts as a trans-acting factor to cis-acting sequences that regulate the cotranscriptional editing of the GP gene [23, 65].
Phosphorylation of VP30 is mediated by two cellular kinases, serine-arginine protein kinase 1 (SRPK1) and SRPK2, and dephosphorylation is catalyzed by phosphatase PP2A and host protein phosphatase 1 (PP1) [29, 66, 67]. Kruse and colleagues have shown that NP recruits the host PP2A.B56 phosphatase [35]. Takamatsu et al. demonstrated that VP30 recruits SRPK1 into IBs [66]. VP30 is predominantly dephosphorylated inside IBs and phosphorylated in released virions [67].
In addition to its RNA-binding role in transcription, VP30 also interferes with cellular RNA silencing [23, 54].
The above discussion suggests that VP30 plays an active role in viral transcription and replication; hence, agents targeting this protein are promising candidates for antiviral therapy [6].
Minor Viral Protein (VP24)
VP24 is a component of the NC and IBs [6, 23, 29]. The presence of VP24 is essential for the condensation of RNP complexes to NCs through direct interaction with NP [23, 29]. High levels of VP24 function as a negative regulator of genome transcription and replication, suggesting that increasing levels of VP24 in IBs block viral RNA synthesis in favor of NC maturation, thereby facilitating NC transport to budding sites and packaging into virions [23, 29].
VP24 is also known as the minor viral matrix protein as it can be associated with cellular membranes [23]. However, a recent publication observed that EBOV VP24 does not associate with lipids, so its presence on the viral matrix layer likely does not depend on direct lipid interactions [68].
Along with VP35, VP24 is an important virulence factor of EBOV. It counters the innate host cell antiviral response by binding to nuclear transporter karyopherin-α (KPNA1, KPNA5, and KPNA6), which transports STAT proteins to the nucleus to activate IFN-stimulated genes (ISGs). By doing so, VP24 reduces transcriptional activation of ISGs, preventing the establishment of an antiviral state in the host cells [23, 69, 70]. Additionally, VP24 inhibits IFN responses by blocking p38 phosphorylation which inhibits the p38 MAPK pathway, and by blocking the activation of NF-κβ [6].
Vidal et al. observed that EBOV VP24 interacts with emerin, lamin A/C, and lamin B, which compromises the integrity of the nuclear envelope. This nuclear membrane disruption induces a laminopathy-like cellular phenotype characterized by nuclear morphological abnormalities with activation of a DNA damage response. This activity of VP24 most likely contributes to EBOV pathogenesis [71].
Since VP24 is essential for NC formation and counters the host immune response, targeting this viral protein may hinder viral replication [6, 12].
Polymerase (L)
The viral polymerase is a component of the RNP complex, IBs, and NC [23, 29]. It is the largest EBOV protein with four distinct domains, namely (a) RNA-dependent RNA polymerase with transcription/replication and polyadenylation activity, and polyribonucleotidyl transferase activity (PRNTase), (b) a connector domain with an organizational role, (c) methyltransferase activity (MTase), and (d) a small C-terminal domain (CTD) [6, 23, 72].
With these four domains, the L protein binds to the genome and antigenomes, which possess a single L entry site (promoter) at the 3′ ends of the encapsidated genomes and antigenomes, respectively. After binding to the promoter region, L either replicates the genome or antigenomes over their entire lengths (replication) or scans the genome, but not antigenomes, for gene start and stop signals to transcribe individual genes (transcription). Transcription includes viral mRNA guanosine (G) capping, N-7 methylation of the G cap and 2′-O-methylation, cap-independent methylation, and polyadenylation. EBOV L protein also mediates cotranscriptional mRNA editing [23, 73].
The 5′ cap is an evolutionarily conserved modification of eukaryotic mRNA and plays a major role in protein translation and innate immunity. Obviously, viruses have evolved to produce capped mRNA for efficient protein synthesis and evasion of innate immune responses. Specifically, the methylation of the N-7 of the G cap and the methylation of the 2′ group in the hydroxyl position of the first ribose are key factors for immune evasion. These methylations avoid binding of the viral mRNA to the PRRs RIG-I and MDA5, which trigger IFN responses to inhibit viral infections. Since EBOV replicates in the cytosol, and the cellular machinery for cap synthesis has a nuclear location, the virus has its own cap machinery. The EBOV L protein has a domain with PRNTase activity and another with MTase activity that synthetizes the cap structure. In the presence of S-adenosylmethionine (SAM), the MTase catalyzes the transfer of a methyl group from SAM to the N-7 of the G cap, generating S-adenosyl homocysteine (SAH), and afterward, in the presence of SAM, it catalyzes the transfer of a methyl group to the 2′ group in the hydroxyl position in the first ribose, generating the final cap structure that resembles a self mRNA that swiftly avoids innate immune responses [23, 72].
In essence, the viral polymerase plays an essential role in genome transcription, replication, and immune evasion, thus representing another target for antiviral drug development [12].
Replication Cycle
Overview
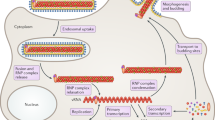
An overview of the EBOV replication cycle can be found in Fig. 5. This figure accompanies the discussion in Sects. “Genome Structure and Organization” and “Replication Cycle” and highlights the key steps in the replication cycle as well as the functions of the viral proteins.

Artistic rendering of the EBOV replication cycle. 1 The virus attaches to host cells through carbohydrate-binding receptors on the cell surface that bind to heavily glycosylated GP1,2 or to phosphatidylserine (PS) receptors on the cell surface that bind to PS in the viral envelope. 2 The virus is then internalized via macropinocytosis and is transported through the progressively acidic endocytic pathway. 3 In endolysosomes, after GP1,2 is cleaved by host cathepsins, GP1 interacts with NPC1 and GP2 promotes fusion. 4 Once inside the cytosol, the nucleocapsids relax and the genome is transcribed, producing viral mRNAs. 5 Free ribosomes translate viral mRNA into viral proteins. Increasing levels of viral proteins lead to the formation of inclusion bodies (IBs). 6 Secondary transcription takes place inside IBs. 7 The viral genome is replicated inside IBs. 8 Once enough viral proteins and genomic RNA are replicated, the nucleocapsids are assembled and then transported to the plasma membrane. 9 Budding of the virus from the plasma membrane releases the virion. Created with BioRender.com
Viral Entry
The virus replication cycle begins with virion attachment to a host cell. Although ebolaviruses are thought to preferentially infect dendritic cells and macrophages in early infection, they have broad cell and tissue tropism, except for lymphocytes [10]. Interestingly, a recent publication demonstrated a previously unknown susceptibility to EBOV in human adipocytes, which may contribute to viral pathogenesis owing to the pro-inflammatory nature of these cells [74].
Attachment
Several factors have been reported as EBOV receptors or co-receptors [49]. Attachment to the host cell is mediated by at least two distinct mechanisms: (a) carbohydrate-binding receptors on the cell surface interacting with the heavily glycosylated GP1,2 peplomer, and (b) phosphatidylserine receptors interacting with phosphatidylserine in the viral envelope [10, 23]. PS receptors typically bind to apoptotic cell membrane PS and orchestrate the uptake and clearance of apoptotic debris. Like many enveloped viruses, Ebola also contains exposed PS and can exploit these receptors for cell entry [75].
The glycans on GP1,2 bind directly to a broad range of C-type lectins (CLECs) because this family of lectins contains carbohydrate recognition domains (CRDs) that bind to the glycan cap, such as asialoglycoprotein receptor 1 (ASGR1), CD209 (formerly DC-SIGN), C-type lectin domain family 4 member M (CLEC4M, formerly DC-SIGNR), C-type lectin domain containing 10A (CLEC10A, formerly MGL), C-type lectin domain family 4 member G (CLEC4G, formerly LSECTin), and mannose-binding lectin 2 (MBL2) [23, 49].
It can also be mediated by hepatitis A virus cellular receptor 1 (HAVCR1, formerly TIM-1) and T cell immunoglobulin and mucin domain containing 4 (TIMD4, formerly TIM-4), which bind phosphatidylserine at the outer leaflet of the viral envelope [23].
PS plays a central role in EBOV entry as is involved in a phenomenon that the virus exploits, known as apoptotic mimicry. During apoptotic mimicry, PS is transferred from the inner leaflet of the PM, where it is the most abundant anionic phospholipid, to the outer leaflet of the PM, which causes PS to become a component of the outer viral envelope during infection. Subsequently, the exposed PS is recognized by target cell receptors, such as the aforementioned ones, resulting in EBOV internalization [50, 75]. The transfer of PS to the outer leaflet is mediated by XK-related protein 8 (Xkr8), a host scramblase responsible for the exposure of PS in the PM of apoptotic cells [62, 75]. Xkr8 is transported to budding sites together with GP and, once incorporated in nascent viral particles, is thought to be activated by caspases, which leads to the externalization of PS in the envelope of EBOV particles. This is particularly noteworthy since if the asymmetrical distribution of PS in the PM occurred prior to budding, the infected cell might not be able to escape phagocytosis, as PM in the outer leaflet functions as a signaling mechanism for phagocytic engulfment [62].
In addition, other cell-surface factors, such as the TAM (TYRO3, AXL, MERTK) family receptor tyrosine kinases, have been shown to facilitate ebolavirus attachment [23, 30, 62, 76].
Importantly, the broad distribution and redundancy of these attachment factors likely help to explain the broad cell and tissue tropism exhibited by ebolaviruses since none of these receptors are indispensable for EBOV attachment. Instead, they appear to work in a complementary manner to adsorb virions at the target cell surface, i.e., if a particular factor is absent, another will take over this function [6, 10, 23, 77].
Uptake: The Virus Enters the Cell
Upon binding to the receptor, EBOV enters host cells via three mechanisms: (a) macropinocytosis, (b) clathrin-mediated endocytosis, and (c) caveolin-mediated endocytosis. [6]. The primary uptake mechanism is macropinocytosis [6, 10, 23].
EBOV associates with sphingomyelin (SM)-enriched sites at the PM. SM associates with cholesterol to form lipid rafts, microdomains rich in cholesterol, and sphingolipids that recruit cellular receptors [78, 79]. Acid sphingomyelinase (ASM) is a lysosomal hydrolase that catalyzes the degradation of SM to phosphorylcholine and ceramide. ASM is present in lysosomes, but since these compartments are constantly recycled to the PM, it can also be found on the cell surface, where it promotes the formation of ceramide in the outer leaflet of the cell membrane [80]. EBOV was shown to recruit ASM to the cell surface. Ceramide in the outer membrane leaflet promotes the formation of a macropinosome. Therefore, EBOV requires SM and ASM for infection [78].
Moreover, other host factors are needed for macropinocytosis internalization of the virus, such as AMP-activated protein kinase (AMPK) and p38 mitogen-activated protein kinase (MAPK) [12, 81].
Fusion: The Endolysosomal Escape
The internalized virion is then trafficked through the progressively acidic endocytic pathway. In the low pH environment of the endolysosome, the GP1 subunit is cleaved by host low-pH-dependent cathepsins B and L, thereby releasing the mucin-like subdomains and glycan caps and exposing the receptor-binding site. Once exposed, the GP1 receptor-binding site engages the NPC1 receptor, an intracellular cholesterol transporter that is ubiquitously expressed and located on the interior membrane of late endosomes and lysosomes [10, 23, 82, 83]. Cleavage of the glycan cap was shown to induce changes in GP1, which in turn enabled flexibility in GP2, which is essential for fusion. It also ensures that GP2 is not prematurely triggered prior to the arrival of the virion in a late endosome that contains the necessary factors for viral fusion [84].
Host N-acetylglucosamine-1-phosphate transferase (GlcNAc-1-phosphotransferase) is a Golgi-resident enzyme that is required for the correct intracellular localization of lysosomal proteins, such as cathepsin B, which is mandatory for proteolytic cleavage of EBOV-GP1,2. Naturally, this host factor is crucial for EBOV entry and infection. It has been demonstrated that fibroblasts from patients with mucolipidosis II, a lysosomal storage disease associated with loss of functional GlcNAc-1-phosphotransferase, are refractory to EBOV [83].
NPC1 is critical for ebolavirus entry, and cells lacking this molecule are resistant to infection [10]. Herbert et al. demonstrated that wild-type (WT) mice (NPC1+/+) had high viral loads of EBOV, while transgenic mice (NPC1−/−) were entirely free of viral replication and thus protected from EBOV infection. Curiously, heterozygous mice (NPC+/−) revealed a survival advantage compared to WT mice [85]. In humans, loss of NPC1 function is associated with Niemann-Pick type C disease (NPC), a rare autosomal recessive lysosomal storage disease. Thus, dysfunctional NPC1 receptors lead to the accumulation of cholesterol and other lipids in lysosomes, with abnormal accumulation within various tissues that dictate the symptomatology of the disease. Life expectancy in patients with NPC disease ranges from a few days to several decades [83, 86, 87].
The GP1–NPC1 interaction results in conformational changes in the GP1,2, resulting in the unwinding of the fusion loop within the GP2, which contains a fusion peptide that penetrates the endolysosomal membrane. Subsequently, folding of GP2 into a hairpin structure pulls the endolysosomal membrane into proximity of the viral envelope, thereby triggering membrane–membrane fusion and release of the viral NC into the cytosol of the host cell, where primary transcription commences thereafter [10, 23, 88]. Igarashi et al. provided structural insight into the interaction between GP1 and NPC1 [89].
The binding to NPC1 is independent of its cholesterol-transporting activity. However, cholesterol plays a significant role in enhancing viral fusion. The TM of GP2 is anchored in the viral envelope, which has cholesterol since the envelope originated from the host cell. Lee et al. demonstrated that cholesterol interacts with the TM of EBOV-GP2 and promotes membrane fusion, facilitating viral entry [88].
In addition to NPC1, EBOV entry into host cells also requires the activity of endolysosomal two-pore channels (TPCs), namely TPC1 and TPC2. TPCs are Ca2+-permeable ion channels triggered by nicotinic acid adenine dinucleotide phosphate (NAADP) to release Ca2+ from endosomes and lysosomes [84, 90,91,92]. TPCs are also activated by the endolysosomal lipid PI(2,3)P2 [90, 92]. Following internalization via endocytosis, the Ca2+ concentration decreases because of TPCs. Inhibiting TPC function prevents EBOV infection, which implicates endolysosomal Ca2+ in EBOV entry, although the exact mechanism of action of how ebolaviruses require TPCs remains undetermined [23, 77, 84]. Das et al. found that acidic pH, endosomal Ca2+, and NPC1 binding synergistically induce conformational changes in GP2 essential for membrane fusion. They observed that maximal GP-mediated lipid mixing occurred over a range of 0.1–0.5 mM Ca2+, whereas excess Ca2+ (> 1 mM) limited membrane fusion, offering some insight into how Ca2+ might modulate EBOV entry [84].
As previously mentioned, HAVCR1 (formerly TIM-1) is an attachment factor for EBOV. However, a study found that this receptor is also located inside endolysosomes and interacts with NPC1. This interaction proved to be mandatory for EBOV fusion [93].
As a result of the endolysosomal localization of these triggering factors, EBOV entry is dependent on internalization in host cells and trafficking through endocytic pathways. To do so, EBOV requires the activity of cellular trafficking factors, but exactly how EBOV is trafficked and delivered to NPC1 remains unclear [82, 94].
Recent research elucidated that for EBOV to be trafficked to NPC1 compartments, it requires the HOPS tethering complex and its regulator, UVRAG. These host factors are both involved in vesicular trafficking, strengthening the notion that EBOV requires a host trafficking pathway for efficient infection [82]. EBOV particles were shown to activate the PI3K-Akt-Rac1 pathway to facilitate vesicular trafficking and fusion [76]. Another research pinpointed the role of PI(3,5)P2, a critical regulator of endosomal membrane homeostasis and progression of cargo through the endolysosomal trafficking system, to promote efficient delivery to NPC1 [94].
For the viral genome to be released inside the cytosol, the nucleocapsid needs to dissociate from the matrix layer composed of VP40, and the nucleocapsid needs to dissociate from the viral RNA. These processes are referred to as viral uncoating. Winter et al. demonstrated that the low pH in endolysosomes promotes the disassembly of the matrix layer composed of VP40 prior to GP-mediated fusion. This research elucidated that the protons passively equilibrate between the endolysosomes and the viral envelope. Hence, the VP40 interactions with the negatively charged lipids on the viral envelope are weakened through neutralization, and the matrix layer disassembles, which is a requirement for membrane fusion [95].
Primary Transcription and Translation
After entry into the cell, nucleocapsids relax as a result of the dissociation of VP24, which allows the viral RNP complex to become transcriptionally active, performing primary transcription, a critical step for EBOV since the viral genome is negative sense and cannot be directly translated into proteins. Therefore, positive sense viral mRNAs are transcribed from the viral genome. The replication cycle takes place solely in the cytosol [10, 23, 29, 77].
For primary transcription to take place, VP30 is dephosphorylated by host phosphatases and associates with VP35 by RNA interaction and clamps the RNA template together with L and VP35, initiating the primary transcription of viral mRNAs [66]. The viral polymerase L binds to a conserved promoter in the 3′ end of the NP-encapsidated genome and moves successively toward the 5′ end, initiating transcription at each gene transcriptional start and stop site, respectively. Newly transcribed viral mRNAs are capped and polyadenylated by L, but unlike the viral genome, they are not encapsidated by NP [10, 23].
There is a gradient for mRNA concentrations since mRNAs produced from the utmost 3′ gene (NP) are synthesized in high abundance, whereas mRNAs produced from the utmost 5′ gene (L) are synthesized in the lowest concentrations. This gradient is likely because the polymerase complex enters the genome at the 3′ end and then moves along the template until it recognizes a gene end signal, which contains a short stretch of uridines. Here, the polymerase stutters, leading to the addition of a poly(A) tail to the nascent mRNA strand. Concomitantly, the polymerase occasionally falls off the template, so the most 5′-located genes are less likely to be transcribed by the polymerase. This results in a gradient of mRNA concentrations, with transcripts for NP being the most abundant and transcripts for L being the least abundant [10, 23].
Except for GP, which is translated at ER-bound ribosomes, all the other viral proteins are translated at free ribosomes in the cytosol [29]. EBOV requires the host translation initiation factor eIF4A, a component of the eIF4F translation initiation complex, for the translation of viral mRNAs into viral proteins. This factor binds to the mRNA cap structure and enables the recruitment of ribosomes to the 5′-UTR regions of mRNA [30, 96, 97].
Increasing amounts of viral proteins, especially NP, in the cytosol, lead to the formation of IBs, sites of secondary transcription, genome replication, and NC assembly [29]. Bodmer et al. recently demonstrated that IBs are liquid organelles [98].
Replication
As transcription and translation continue, increasing levels of VPs are thought to trigger the switch to replication. As previously elucidated, phosphorylation of VP30 causes its dissociation from the RNP complex and shifts RNA synthesis toward replication [10]. Replication of the genome occurs in IBs [29].
NP-encapsidated genomes (genomic RNPs) serve as a template for viral RNA synthesis, which requires viral polymerase L and VP35. The polymerase complex binds to a replication promoter in the leader sequence of the genome and commences the synthesis of full-length complementary positive-sense genomes, referred to as the antigenome, which is encapsidated by NP as the antigenome chain elongates, thus forming RNP complexes (antigenomic RNPs) [6, 10, 23, 29].
Using the antigenome promoter, these antigenomic RNPs serve as templates for synthesizing progeny genomic RNPs. Synthesized genomic RNPs accumulate in cytoplasmic perinuclear inclusion bodies [6, 10, 23].
Assembly and Budding
Assembly and Transport of Nucleocapsids
Still in the IBs, genomic RNPs, which appear as thin-walled helices mature into nucleocapsids, which appear as thick-walled helices, by the recruitment of VP24. VP35 also plays a role in RNP maturation. High concentrations of VP24 inside IBs are believed to inhibit viral transcription and replication, thus favoring NC assembly [29]. As previously stated, VP40 also plays a role in transcription and replication inhibition [23, 29, 33].
Since NCs are formed in IBs, which have perinuclear localization, they need to be trafficked to budding sites at the plasma membrane for the final phase of the replication cycle to take place. The recruitment of host factors is of the utmost importance since the size of the NCs restrings their diffusion to the budding site, and the virus does not encode transport factors. This transport is mediated by an actin-dependent and microtubule-independent mechanism [23, 29, 34]. Outside the IBs, actin tails that are WAVE1, Rac1, and Arp2/3 complex-dependent are formed at one end of the NC in the cytosol, which drives their transport to the plasma membrane, where budding takes place [29]. Meanwhile, VP40 transport to budding sites is not associated with NC transport from IBs to the PM, and the GP is transported to the PM through the classical secretory pathway [6, 29, 34].
Furthermore, VP40 contains late-budding motifs that interact with components of ESCRT, such as TSG101, a protein that escorts proteins from the cytosol to the cell membrane. Without these host proteins, the assembly and budding of virions would not be possible [12, 23, 77].
Budding from the Host Cell
Budding of ebolaviruses occurs mainly at filopodia, i.e., long and thin membrane protrusions containing characteristic parallel actin filaments enriched with a VP40 layer and GP1,2 trimers [23, 29, 99]. Filopodia accumulate GP1,2 in a tubulin-dependent manner [23]. Once located near the PM, the NCs associate with VP40 [34]. The movement of cargo along actin filaments within the filopodia is mediated by myosin 10, and VP40 was shown to use this mechanism for transport inside filopodia along with NCs [29]. Subsequently, several host factors are recruited, such as IQAP1, that enable the budding of the virus [34].
Consequently, budding viruses contain part of the PM, and the membrane incorporates GP1,2, which is crucial for attachment and viral entry [23]. The PS is flipped from the inner leaflet of the PM by cellular scramblases, such as Xkr8, that are transported inside GP-containing vesicles [23, 62].
Ebola Virus: From a Therapeutic Point of View
Treatment: State of the Art
2022 as a Turning Point in EVD: The Makings of a Guideline
The year 2022 was a breakthrough for EVD therapeutics. The first evidence-based clinical practice guideline on specific EVD therapies was published. This guideline, from the WHO, incorporates the latest high-quality evidence and provides new recommendations on EBOV-specific therapeutics for EVD. As a result of the evidence available, this guideline is directed only to the treatment of EVD, the disease caused by EBOV, i.e., O. zairense [8, 100].
Some countries and regions around the world have society and government-sponsored guidelines, such as Canada, the USA, some European countries, and Japan. These guidelines, for the most part, are not updated and focus on supportive care. They can be consulted and are discussed elsewhere [101].
The WHO published an international guideline on 19 August 2022 with the most up-to-date recommendations for the clinical management of people with EVD. This guideline, entitled “Therapeutics for Ebola Virus Disease”, contains new recommendations regarding the use of therapeutics for EVD and is written to accompany the “Optimized Supportive Care for EVD standard operating procedures”, also by the WHO [8, 102].
In 2019, the limited evidence for therapeutics for EVD was augmented by the publication of the Pamoja Tulinde Maisha (PALM) trial, “Together Save Lives” in the Kiswahili language, a randomized controlled trial (RCT), which compared Zmapp with three investigational agents: remdesivir, REGN-EB3, and mAb114. This RCT demonstrated superior efficacy to two EVD therapeutics and, thus, the WHO proposed developing a new guideline [8, 100, 103].
The WHO formulated an international guideline panel, the WHO Guideline Development Group for Therapeutics for Ebola Virus Disease, to develop a clinical practice guideline based on all available evidence from RCTs [8, 100].
Gao et al., funded by the WHO, did a systematic review and network meta-analysis of RCTs to evaluate the efficacy and safety of therapies for patients with EVD. They identified 7840 records through database searching, of which two RCTs with a total of 753 patients met the inclusion criteria [100]. The PREVAIL II trial was reported by Davey et al. in 2016, and the PALM trial was reported by Mulango et al. in 2019 [8, 103, 104]. The two trials investigated four therapeutics, ZMapp, remdesivir, mAb114, and REGN-EB3 [8].
ZMapp is a cocktail of three monoclonal antibodies (mAbs), c2G4, c4G7, and c13C6, against the EBOV-GP. c13C6 binds to the glycan cap in GP1 and is non-neutralizing. It mediates effector functions through antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and antibody-dependent complement deposition (ADCD). c2G4 and c4G7 are neutralizing antibodies that have overlapping binding sites on the GP1–GP2 interface, thereby preventing the insertion of the fusion loop into the endolysosomal membrane [8, 105]. Remdesivir, commercial name Veklury™ (Gilead Sciences, Inc), is a prodrug that is activated intracellularly to GS-443902, an adenosine triphosphate nucleoside analogue that competes with ATP for RNA incorporation, thus inhibiting the RdRp of EBOV [106]. REGN-EB3 is a cocktail of three mAbs that target EBOV-GP. mAb114 is a single mAb that targets EBOV-GP [8]. REGN-EB3 and mAb114 are explored in detail in Sect. “Approved Ebola-Specific Therapies”.
PREVAIL II was an RCT done in 2015, during the 2013–2016 outbreak in West Africa, with two treatment arms, where one received the current standard of care, and the other received ZMapp plus the current standard of care, in patients with EVD that were diagnosed by a positive RT-qPCR for EBOV. The CFR of the control group was 37% and the CFR of the Zmapp group was 22%. The result did not reach the prespecified statistical threshold for efficacy, even though the estimated effect of using Zmapp appeared to be beneficial [8, 104, 107].
The PALM clinical trial was an RCT done during the DRC outbreak in 2018–2019 that compared Zmapp with three newer investigational drugs [3, 8, 103, 107, 108]. All groups received standard of care. Patients were assigned in a 1:1:1:1 ratio to receive Zmapp, remdesivir, mAb114, or REGN-EB3. Zmapp was chosen as the control on the basis of data from the aforementioned PREVAIL II trial. REGN-EB3 was added later, so it was compared to a Zmapp subgroup, i.e., patients who received Zmapp on or after the time the REGN-EB3 group was added. Curiously, on August 9, 2019, the Data Safety Monitoring Board (DSMB) recommended terminating random assignment to the Zmapp and remdesivir groups. The REGN-EB3 arm had crossed an interim boundary for efficacy, and the analysis of mortality showed a clear separation between the mAb144 and REGN-EB3 groups and the ZMapp and remdesivir groups. The overall CFR for mAb114 was 35.1%, compared to 49.7% in the ZMapp group, and 33.5% for the REGN-EB3 group, compared to 51.3% in the ZMapp subgroup. The remdesivir group had an overall CFR of 53.1%, compared to the 49.7% of the ZMapp group [8, 103, 107, 108].
These values are even further apart when considering the CFRs of patients with high viral loads at baseline (Ct ≤ 22) and patients with low viral loads at baseline (Ct > 22). For patients with Ct ≤ 22, the ZMapp group had a CFR of 84.5%, the remdesivir group had 85.3%, and the mAb114 group had 69.9%. The REGN-EB3 group accounted for 63.6%, and the ZMapp subgroup accounted for 86.2%. Considering the patients with Ct > 22, the ZMapp group had a CFR of 24.5%, the remdesivir group had a CFR of 29%, and the mAb114 group had a CFR of 9.9%. The REGN-EB3 group had a CFR of 11.2% and the ZMapp subgroup had a CFR of 25.8% [103].
Following the DSMB recommendation, the study continued to enroll only in the mAb114 and REGN-EB3 groups in an extension phase of the trial [107, 108]. In summary, REGN-EB3 and mAb114 demonstrated efficacy as compared to the control arm, ZMapp [107].
Partially based on the PALM RCT results, in 2020, the US Food and Drug Administration (FDA) approved both REGN-EB3 and mAb114 for the treatment of patients with EVD, and in 2022, the WHO published the first evidence-based clinical practice guideline focusing on specific therapies for EVD [8, 107, 108]. The recommendations from the guideline are addressed in Sect. “Therapeutics for Ebola Virus Disease”.
Following the PALM trial results, the two therapeutic mAbs were used for the treatment of EVD in the subsequent EBOV outbreaks under Expanded Access Programs (EAP), which corresponds to the emergency use of unapproved, investigational products outside of a clinical trial [3, 108]. These uses were not RCTs, but observational data suggested that the use of EBOV-specific therapeutics reduced the CFR [108]. In fact, in an outbreak in DRC, from June 1 to November 18, 2020, both medicines were employed to evaluate their behavior with a different strain of EBOV from the one that circulated during the PALM trial. The CFRs, though observational, were 6% (2/32) for the patients who received one of the mAbs, and 54% (53/98) for the patients who did not receive an EBOV-specific treatment [109].
Guidelines: Recommendations
Therapeutics for Ebola Virus Disease
There is a strong recommendation for treatment with either mAb114 or REGN-EB3 for patients with RT-qPCR-confirmed EVD and neonates of unconfirmed EVD status, 7 days or younger, born to mothers with confirmed EVD. Note that these two medicines should not be given together, as they should be viewed as alternatives. The choice depends on availability. Furthermore, this recommendation is exclusive to EVD, the disease caused by O. zairense. The use of these medicines does not replace optimized supportive care, and they should be administered as soon as possible after diagnosis [8].
There are conditional recommendations against treatment with remdesivir or Zmapp for patients with RT-qPCR-confirmed EVD because of considerable uncertainty of mortality reduction compared with the standard of care. Nevertheless, since remdesivir has a different mechanism of action from the other medicines, all neutralizing monoclonal antibodies, there may be a rationale to include this therapeutic in future trials of combination therapy, especially for patients at higher risk of mortality [8].
Optimized Supportive Care for Ebola Virus Disease: Clinical Management Standard Operating Procedures
As previously stated, the “Therapeutics for Ebola Virus Disease” guideline is to be accompanied by the “Optimized Supportive Care for Ebola Virus Disease”, because, despite the PALM study finding REGN-EB3 and mAb114 to be effective against EBOV, some treated patients still succumbed with high viral load. During the 2013–2016 West African outbreak, it was demonstrated that supportive care could potentially contribute to help support vital functions and increase survival [102, 107]. The level of supportive care provided by Ebola Treatment Units (ETUs) within the same outbreak and across outbreaks varies significantly, so this evidence-based guideline was developed to serve as the basis for optimized supportive care (oSoC), which should be followed to ensure the best possible chance of survival and enable reliable comparison of investigational therapeutic interventions as part of an RCT [102].
oSoC includes volume resuscitation, symptom control, laboratory and bedside monitoring of glucose, electrolyte levels, and organ dysfunction, and prompt detection and management of co-infections [102].
Approved Ebola-Specific Therapies
REGN-EB3 (Atoltivimab, Maftivimab, and Odesivimab-ebgn; Inmazeb®)
REGN-EB3, trade name Inmazeb® (Regeneron Pharmaceuticals, Inc), is a co-formulated cocktail, i.e., a combination of three fully human IgG1 monoclonal antibodies—atoltivimab (REGN3470), maftivimab (REGN3479), and odesivimab (REGN3471), in a 1:1:1 ratio, for the treatment of EBOV infection [107, 108, 110,111,112]. The three antibodies bind to non-overlapping epitopes in EBOV-GP1,2 in a so-called multi-antibody approach, which may increase efficacy and decrease the likelihood of escape mutants, which would require the simultaneous selection of escape mutations in the GP1,2 to each component of the drug [57, 107, 112]. Additionally, Inmazeb® recruits immune effectors for the destruction of both viral particles and infected cells by eliciting ADCC, ADCP, and ADCD. Synergically, this antibody cocktail halts viral entry [8, 110]. Nevertheless, the EBOV genome can change over time, so it is recommended that the drug susceptibility patterns for circulating strains be ascertained when deciding whether to use this drug [111, 112].
The three antibodies in the REGN-EB3 cocktail were obtained from genetically engineered mice previously immunized with DNA constructs encoding EBOV-GP1,2 or recombinant purified GP1,2 and were selected on the basis of their ability to bind EBOV-GP1,2 simultaneously and on their complementary combination of functional properties [8, 57, 108, 110]. Odesivimab targets the GP1 head and sGP, which indicates that it binds within the first 295 amino acids of the common region of EBOV-GP and EBOV-sGP [8, 57, 110]. It is poorly neutralizing but mediates effector function through FcγRIIIa to trigger ADCC function. Maftivimab targets the conserved GP2 fusion loop and is potently neutralizing. Atoltivimab targets the GP1 glycan cap, is partially neutralizing, and mediates Fc effector functions, promoting the killing of EBOV-infected cells [8, 57, 107].
Rayaprolu et al. provide direct evidence that targeting non-overlapping epitopes of EBOV is an effective strategy to protect against the rapid emergence of EBOV escape mutants and therefore mitigate the risk of drug-induced viral resistance, a phenomenon reported with monotherapy treatment for SARS-CoV-2 infection [57]. Notwithstanding these findings, ZMapp, also a cocktail of three monoclonal antibodies, has characterized escape mutants. Notably, mutations in amino acid residues 273 and 508 of EBOV GP1,2 abrogate binding to all the mAbs of the cocktail [113]. Nevertheless, it is pertinent to note that although ZMapp is composed of three mabs, two of them bind to the same epitope on the GP1,2 [105].
Inmazeb® is the first FDA-approved therapeutic against EDV [57, 63, 110]. It received its first approval on October 14, 2020 in the USA, following the results of the PALM trial. Inmazeb® is indicated for the treatment of infection caused by EBOV in adult and pediatric patients, including neonates born to a mother who is RT-qPCR positive for EBOV infection. Its efficacy has not been established for other species of the Orthoebolavirus genus [110,111,112]. Meanwhile, this treatment is not yet approved in the EU, but on May 25, 2018, it received orphan designation (EU/3/18/2027) by the European Commission (EC) for the treatment of EVD [114, 115]. To the date of writing, the orphan designation in the EU remains active [116].
Ansuvimab (Ansuvimab-zykl; mAb114; Ebanga™)
Ansuvimab, trade name Ebanga™ (Ridgeback Biotherapeutics, LP), is a human monoclonal IgG1 antibody produced by recombinant DNA technology in Chinese hamster ovary (CHO) cells, that binds to the conserved LEIKKPDGS epitope located in the RBS of the GP1 subunit of EBOV-GP1,2 to block its interaction with NPC1, thereby blocking EBOV endolysosomal escape and halting viral entry [8, 107, 108, 117, 118]. Additionally, ansuvimab elicits ADCC [117]. Owing to the vital role of RBS in the infectivity of EBOV, ansuvimab binding to this conserved domain could mitigate the risk of escape mutants while preserving the high neutralizing activity of the antibody, as alterations in RBS can result in a decline in viral fitness [63, 107]. Nevertheless, the EBOV genome can change over time, which could alter the clinical benefit of the antiviral drug. Therefore, the drug susceptibility patterns for circulating strains should be ascertained [63, 117,118,119].
It received its first approval on December 21, 2020 in the USA, following the results of the PALM trial. Ebanga™ is indicated for the treatment of infection caused by EBOV in adult and pediatric patients, including neonates born to a mother who is RT-qPCR positive for EBOV infection. Its efficacy has not been established for other species of the Orthoebolavirus genus [117,118,119].
Ansuvimab was initially selected after isolation and screening of a panel of memory B cells isolated from the blood of a human survivor of the 1995 Ebola outbreak in Kikwit, 11 years after infection, which demonstrated potent neutralization of EBOV [107, 117, 118, 120].
At the date of writing, Ebanga™ is the most recent FDA-approved drug for treating EVD [63]. Meanwhile, this treatment is not yet approved in the EU, nor does it have an orphan designation [114].
Ebanga™ and Inmazeb®: A Perfect Combination?
The potential synergistic effects of combining these two drugs, a total of four different monoclonal antibodies, for the treatment of EDV have not been investigated either in a clinical or laboratory trial. Notwithstanding the non-studied safety implications of using them simultaneously, their mechanism of action differs, as all mAbs bind to distinct epitopes, so the potential to combine Ebanga™ and Inmazeb® might be a possibility [63]. Moreover, since they target distinct epitopes, there is some insurance against the emergence of a virus that is resistant to both treatments [108].
Nevertheless, as previously stated, the WHO does not recommend the concomitant use of the two medicines, and the choice of which to use depends, essentially, on availability [8].
Emerging Therapeutics
Despite the improvements in EVD therapy over the previous few years, deaths in the PALM study were nevertheless accounted for by 35.1% (61/174) and 33.5% (52/155) of people receiving the mAb114 and REGN-EB3 therapies, respectively. As described in Sect. “2022 as a Turning Point in EVD: The Makings of a Guideline”, in the subset of participants who had a high viral load at baseline (Ct ≤ 22), the mortality was greater than the subset of people with a lower viral load (Ct > 22) [107]. Despite receiving either Inmazeb® or Ebanga™, 69.9% and 63.6% of patients with Ct ≤ 22 succumbed to the disease, respectively [103]. Moreover, both approaches are specific to EBOV, and outbreaks caused by other species of the Orthoebolavirus genus also have considerable mortality [121].
Therefore, there exists a need to improve EVD outcomes by improving the arsenal of treatments against EBOV. Possible approaches include improving the existing ones, such as combining mAbs or optimizing potency, dose, and Fc effector function. It is, however, conceivable that results might be improved by combining Inmazeb® or Ebanga™, or other effective mAbs, with a second EBOV-specific therapy, with a different mechanism of action, such as small molecules that inhibit viral steps in the replicative cycle of the virus [108].
Besides, no small-molecule compounds are licensed for EVD, and these provide some advantages compared to mAbs, such as being generally easy and cheap to produce, transport, and store, which makes them interesting and promising candidates, especially for treating patients in remote locations [122].
Furthermore, some EVD survivors have evidence of viral persistence. The molecular weight of monoclonal antibodies limits their penetration into immune sanctuaries where EBOV persists. Therefore, combining them with promising antiviral small molecules may help to clear the virus completely, avoiding potential transmission and the occurrence of sequelae [107].
The following sections describe the emerging pharmacological approaches that aim to halt EBOV infection. Some of the molecules described are only used in research settings and are not approved for clinical use in humans. Several of them were identified through in silico screening studies, and their antiviral effects remain to be validated with biological experiments. Others have been tested in vitro and/or in vivo (non-human). Lastly, a number of molecules have been approved for human use for non-EVD indications and are now being studied for possible drug repurposing to treat EBOV infection. Importantly, none of these molecules have been tested in clinical trials for EVD, so their efficacy remains to be proven. They represent future perspectives on the treatment of EVD. The section is dedicated to exploring the mechanisms of action (MoA) of these molecules, using the replication cycle of the virus as a visual aid to facilitate understanding.
Host-Directed Antivirals
As previously discussed in Sect. “Replication Cycle”, EBOV hijacks and exploits host pathways and cellular proteins to facilitate various aspects of its replication cycle [30, 123, 124]. Targeting essential host factors in the viral replication cycle is a key strategy to perturb viral infection. Moreover, host factors impose a higher barrier to mutations compared to viral proteins, especially from RNA viruses such as EBOV, with a low fidelity RdRp without proofreading activity, making them attractive candidates for antiviral intervention, as they are less likely to become ineffective because of viral mutations, i.e., they do not select drug-resistant strains. On the other hand, several viruses usurp the same host factors and, therefore, targeting these host functions could lead to the development of broad-spectrum antivirals [12, 81, 123, 125,126,127,128]. Nevertheless, to develop host-targeting antivirals, it is necessary to understand the host–virus molecular mechanisms [66, 129].
Nonetheless, targeting host factors is not free of drawbacks. The host factor that is being targeted needs to be essential for the virus to replicate but not mandatory for host cell functions, risking toxicity and undesired off-target effects. If not finely controlled, inhibition of host factors has the potential to disrupt vital cellular processes since the impairment of numerous host processes is detrimental to the cell [35, 77, 123, 124, 127,128,129]. Regardless, in the context of acute EBOV infection, these treatments are required for a relatively short timeframe, which potentially mitigates toxicity and side effects [77]. Table 1 summarizes the benefits and drawbacks of host-directed antivirals.
Excellent up-to-date reviews on host-directed EBOV antivirals are available in the literature, with a multitude of mechanisms of action. Lo et al. and Liu et al. compiled information about S-adenosyl-homocysteine hydrolase inhibitors, kinase and phosphatase inhibitors, protein folding and maturation inhibitors, proteolytic processing inhibitors, NPC1 inhibitors, ion channel inhibitors, drugs that perturb cholesterol and calcium regulation in endosomes and sphingosine depletion, immune response enhancers, antioxidants, and inhibitors of host translational factors. These diverse MoAs culminate in host-targeting inhibitors that perturb viral entry, replication, egress, or immune evasion [12, 30]. Salata et al. provided an extensive review of MoAs of entry inhibitors that target host factors [81]. Liu and colleagues also compiled potential combination treatments for EVD [12].
As previously discussed, NPC1 is a key entry receptor for EBOV. Inhibition of this host factor induces an NPC phenotype in cells, a phenotype characterized to be resistant to EBOV infection [130]. Morales-Tenorio et al. reviewed pharmacological strategies targeting NPC1 [131].
As explored in Sect. “Attachment”, for apoptotic mimicry to be feasible, PS needs to be exposed on the outer leaflet of the EBOV envelope. To do so, the virus GP recruits a host scramblase, Xkr8, that is incorporated into viral particles. Moreover, Xkr8 is activated by caspases. Nanbo et al. verified that upon cell treatment with Z-VAD-FMK, a pan-caspase inhibitor, the externalization of PS in the envelope and internalization of viral-like particles were suppressed [62].
As previously discussed in Sect. “Nucleoprotein”, NP recruits the host factor CAD into IBs to provide increased amounts of pyrimidines for EBOV genome replication and transcription. Even though the authors did not test any compound in their study, they acknowledged that CAD inhibitors, such as the antinucleoside N-phosphonacetyl-l-aspartate (PALA), which inhibits the aspartate transcarbamylase activity of CAD and proved effective in vitro against various viruses, might be promising for EBOV, although this remains to be investigated [32].
As referenced in Sect. “Viral Polymerase Complex Protein 35”, VP35 sequesters the PKA-CREB1 pathway, which enhances viral replication and virus-induced coagulopathy. Zhu and colleagues demonstrated that a CREB1 inhibitor, 666-15, showed promising efficacy in suppressing viral replication. A PKA inhibitor, H89, also proved to have promising efficacy, albeit to a lesser extent [45].
Selected molecules and their respective MoAs are illustrated in Fig. 6. This figure accompanies the text in Sects. “Host-Directed Antivirals” and “Repurposing Widely Used Licensed Drugs Against Ebolavirus Infection: Host-Directed Antivirals”.

Artistic rendering of the mechanisms of action of emerging approaches that target host factors to treat Ebola infection. Blue text indicates already approved drugs for non-EVD indications. A 666-15 is a CREB1 inhibitor and H89 is a PKA inhibitor. B N-Phosphonacetyl-l-aspartate (PALA) inhibits the aspartate transcarbamylase activity of CAD. C Z-VAD-FMK is a pan-caspase inhibitor that inhibits the caspases needed for Xkr8 activation. D S-Adenosyl-homocysteine hydrolase (SAAH) inhibitors inhibit the hydrolase of SAH, resulting in the accumulation of SAH which acts as a negative feedback regulator of mRNA cap methylation. E, F PP1 inhibitors and PP2.B56 inhibitors decrease viral transcription by accumulation of the inactive phosphorylated form of VP30. G Inhibiting SRPK1/SRPK2 downregulates replication by accumulation of the active dephosphorylated form of the transcription factor VP30. H α-Glucosidase inhibitors prevent proper folding and maturation of GP by disrupting accurate N-glycosylation. I PF-429242 inhibits GlcNAc-1-phosphotransferase activity by inhibition of S1P, a protease that cleaves the precursor of GlcNAc-1-phosphotransferase. J Inhibition of host cathepsins disrupts GP interaction with NPC1. K Inhibiting NPC1 prevents the interaction with GP, which inhibits fusion. L Blocking the interaction between HAVCR1 and NPC1 prevents membrane fusion. M Inhibition of TPCs leads to the accumulation of endolysosomal Ca2+, which inhibits fusion. N Fendiline lowers PS levels in the plasma membrane by inhibiting ASM. O Statins lower the cholesterol available to bind to the transmembrane domain of GP2, perturbing efficient fusion. P Benproperine inhibits the actin-related protein 2/3 complex, which is crucial for actin polymerization and NC transport. Q TSG101 inhibitors prevent proper escort of viral proteins to the plasma membrane, thus interfering with budding. R Inhibition of VP40 phosphorylation decreases budding. S Geldanamycin, radicicol, and 17AAG inhibit HSP90. VER-155008 inhibits HSP70, and EGCG inhibits HSPA5 (a member of the HSP70 family). T Compound C inhibits AMPK, and the other molecules inhibit p38 MAPK. They inhibit macropinocytosis. U Fluoxetine inhibits the endosome-residing ASM, leading to cholesterol accumulation and impaired EBOV fusion. Created with BioRender.com
Some of the medicines in these reviews are already approved for non-EVD indications. This phenomenon, called drug repurposing, has emerged as a novel concept to combat pathogens [30, 132] and is discussed in Sect. “Repurposing Widely Used Licensed Drugs Against Ebolavirus Infection: Host-Directed Antivirals”. Herein, drug repurposing is defined as using approved drugs by the FDA, European Medicines Agency (EMA), and other regulatory authorities for a non-EVD indication to treat EVD. Some molecules that were developed and studied for other indications but are not yet approved are also included in this section.
Repurposing Widely Used Licensed Drugs Against Ebolavirus Infection: Host-Directed Antivirals
EVD is a rare disease caused by EBOV, a Biosafety Level 4 (BSL-4) pathogen, which imposes significant obstacles to the development of countermeasures for the virus. Additionally, conventional drug discovery takes years and is astonishingly expensive [12, 132, 133]. These factors combined hamper the development of highly needed therapeutics for EVD [81].
Repurposing already approved drugs to be anti-EBOV agents has emerged as a novel concept to combat Ebola, as it provides faster development, especially because the absorption, distribution, metabolism, and excretion (ADME), toxicology properties, and safety profile of approved drugs are already assessed beforehand [128, 132, 134].
Dhama and colleagues reviewed some of the drugs that have been suggested to be repurposable for EVD, which have been screened with diverse methodologies [134].
Besides the various molecules described in the aforementioned review, recent articles are being published that illuminate new potential targets. As described in Sect. “Matrix Protein 40”, VP40 induces the clustering of PS and promotes viral budding. PS also fulfills an essential function in viral entry. As per the investigation carried out by Husby et al., the drug fendiline lowers PS content in the PM and reduces PS clustering, which perturbs viral budding and entry. Using fendiline for EBOV is an example of repurposing, as this drug was approved by the FDA in the 1970s as a non-selective calcium channel blocker to treat coronary heart disease. Herein, the MoA was identified to be calcium-independent and associated with the inhibition of ASM to alter sphingomyelin levels in the PM, which are necessary to maintain proper PS levels in the PM. Additionally, fendiline treatment significantly reduced VP40 oligomerization [50].
Kummer and colleagues explored the endolysosomal host–pathogen interface as a suitable target for antiviral treatment. They demonstrated that itraconazole, an already-approved antifungal, and fluoxetine, an already-approved antidepressant, disrupt the endolysosomal cholesterol balance by inducing the sequestration of cholesterol within the endolysosomal system. Itraconazole inhibits NPC1, and fluoxetine inhibits the endosome-residing ASM. Moreover, the massive cholesterol accumulation by the two drugs resulted in decreased EBOV infection rates [121]. Inevitably, these drugs can only be used in humans as potential treatments for EVD if their daily dose is tolerated and does not show toxicity [135]. In this article, the concentrations required for the anti-EBOV activity were lower than the plasma concentrations required for the antifungal and antidepressant activity of itraconazole and fluoxetine, respectively, which makes them plausible candidates for drug repurposing [121]. Curiously, research that screened databases of compounds for repurposing also identified itraconazole as a lead compound to inhibit EBOV [122].
As illustrated in Fig. 6, blocking TPCs is a strategy to halt viral entry. Tetrandrine is a potent TPC inhibitor that prevents EBOV from escaping the endosomal network, as demonstrated by Sakurai and colleagues [90]. Naringenin also blocks TPCs, albeit with a lower affinity. Nevertheless, these molecules are not selective TPC antagonists, and having multiple targets makes them prone to undesired off-target effects. The research conducted by Penny et al. identified multiple FDA-approved drugs that potentially target TPC, such as dopamine receptor antagonists that are used as antipsychotics (fluphenazine, trifluoperazine, prochlorperazine, and thioridazine) and selective estrogen receptor modulators (SERMs) used for the treatment of breast cancer and osteoporosis (clomiphene, tamoxifen, toremifene, raloxifene, bazedoxifene) [92]. By blocking TPCs, these molecules promote the accumulation of endolysosomal Ca2+, which inhibits GP-mediated virus entry [84], as explained in Sect. “Fusion: The Endolysosomal Escape”. Previously, multiple drug repurposing screenings showed that SERMs were active against EBOV, and experimental data pointed out that SERMs reduced levels of cellular sphingosine, which consequently led to the accumulation of endolysosomal Ca2+, but the exact MoA remained elusive. Penny et al. revealed their anti-EBOV MoA as a TPC blocker [92].
During the 2013–2016 EBOV outbreak, statins were suggested as an adjunct therapy for EVD owing to their pleiotropic MoA, especially because they possess the ability to improve endothelial integrity, which is lost during EVD [88, 136]. Statins are cholesterol-lowering drugs that inhibit HMG-CoA reductase. Moreover, in a screening of FDA-approved drugs for repurposing for EVD, simvastatin was a hit molecule [88]. Curiously, in the research conducted by Penny and colleagues, simvastatin was also one of the hits for TPC inhibitors [92], which corroborates the pleiotropic MoA exhibited by statins. As previously discussed in “Fusion: The Endolysosomal Escape”, cholesterol in the viral envelope interacts with the TM of GP2 to enhance fusion. The study conducted by Lee et al. illustrated that VLPs produced under treatment with lovastatin were defective in infecting the next set of cells. This was due to the VLPs having less cholesterol available to bind to the TM of GP2 and influencing their structure for optimal fusion [88]. Shrivastava-Ranjan et al. previously demonstrated similar results, although the mechanism behind the anti-EBOV activity of lovastatin was not completely elucidated [136].
Fascinatingly, Wu et al. reported the results of a retrospective cohort study about the risk of viral infections in statin users in 2022. The study compared statin users (n = 20,202) and non-statin users (n = 20,202) from a population with hyperlipidemia. Statin treatment was associated with a significantly lower risk of viral infection in all age groups older than 18 years in both men and women, and this risk was reduced as the duration of treatment increased [137]. While this study only observed people with hyperlipidemia, and the mechanism by which statins might be associated with a lower risk of viral infections was not explored, the data obtained are certainly interesting when considering the potential of repurposing statins for EBOV infection.
Lastly, in a recent research endeavor, two large repurposing compound libraries were screened for their activity against EBOV. It should be noted that the compounds in those libraries have been previously studied for certain indications, but not all of them are approved for human use. Importantly, eight novel EBOV inhibitors were identified. Three of the hit molecules inhibit host factors. Itraconazole, already discussed above, was one of them. Z-FA-FMK inhibits cathepsins B and L. Doramapimod directly inhibits p38 MAPK, which is involved in interferon type I response. Three of the hits have less clear MoAs. Evans blue is thought to inhibit EBOV by altering cytosolic Ca2+ concentrations. UNC1999 might counteract some pro-viral manipulation of host factor pathways during EBOV infection. Benproperine inhibits the actin-related protein 2/3 complex, which is crucial for actin polymerization and subsequent NC transport. Lastly, two hits without elucidated MoA were identified. Retapamulin, a topical antibiotic, and MMV1782214, originally developed for HIV [122].
Direct-Acting Antivirals
The development of antiviral drugs that directly target viral components has been the gold standard of antiviral drug development [123, 128, 138]. Hence, most approved antiviral drugs target viral proteins [124]. A prime example is Ebanga™ and Inmazeb®, the only approved drugs for EVD that, indeed, target a viral protein [8]. Direct-acting antivirals (DAAs) include monoclonal antibodies and small molecules that inhibit viral proteins to suppress the replication cycle. Importantly, they are selective for viral proteins, which ideally minimizes the impact on host cells and off-target effects. Nevertheless, a major challenge of DAAs is the emergence of drug-resistant variants. As covered throughout this work, RNA viruses are more prone to mutations, and some of them might lead to the selection of variants that escape DAAs [77, 128]. As previously mentioned in Sect. “REGN-EB3”, this phenomenon has been observed for Zmapp, and according to the actual recommendations, when using Ebanga™ and Inmazeb®, the drug susceptibility patterns for circulating strains should be ascertained exactly because of this occurrence [110, 113, 117]. Table 1 summarizes the benefits and drawbacks of direct-acting antivirals.
In a similar manner to emerging host-directed antivirals for EVD, Lo et al. and Liu et al. provide current and relevant insights about emerging DAAs to treat EVD [12, 30]. These DAAs inhibit EBOV proteins, viral protein interactions, or interactions between viral proteins and host factors. They can be sorted into VP35 inhibitors, VP40 inhibitors, GP inhibitors, VP30 inhibitors, VP24 inhibitors, L inhibitors, and NP inhibitors [12, 30, 77]. They mainly target the entry process, the viral RNA synthesis machinery, or morphogenesis and budding [77].
As research is being conducted, new molecules are being identified as potential emerging DAAs against EBOV. According to the findings of Yi and colleagues, the natural compound EEI-10, also known as berbamine, inhibits EBOV replication in vitro and in vivo. This inhibition was attributed to its capacity to bind to GP after being cleaved by host cathepsins, thus preventing GP binding to NPC1 [139]. Although this research suggests that employing berbamine for EBOV is drug repurposing, berbamine is not yet approved for human use, as no results are returned upon searching for this compound on the FDA or EMA websites and on drug databases [140,141,142]. This compound is used in traditional Chinese medicine for treating various diseases, such as cancer [143]. In the present work, berbamine is considered to be a potential direct-acting antiviral, albeit not a repurposing agent.
In light of recent research findings, 42 natural-product-derived compounds were identified as VP40 inhibitors with anti-EBOV activity and desirable ADME and toxicity profiles. These compounds were virtually screened from Chinese and African sources, 1 from the Northern African Natural Products Database (NANPDB), 2 from the AfroDb, and 39 from the Traditional Chinese Medicine (TCM) database [144].
As elucidated earlier, blocking the interaction between NPC1 and EBOV-GP is a strategy to inhibit viral entry. To do so, molecules that directly bind to and inhibit NPC1 can be used, as described in Sect. “Host-Directed Antivirals”, or molecules that act at the GP level. Some molecules bind to a hydrophobic pocket at the interface between GP1 and GP2. This binding compromises the interaction between GP1 and NPC1, thus perturbing viral fusion. Benzodiazepine derivative 7, compound 35, and compound 38 have this MoA [131]. Other molecules are described in Sect. “Repurposing Widely Used Licensed Drugs Against Ebolavirus Infection: Direct-Acting Antivirals”.
Ma et al. screened compounds that bind to a hydrophobic pocket on VP30. This pocket is where NP binds to VP30. By competitively inhibiting the binding of NP to VP30, the author and colleagues demonstrated impaired EBOV transcription and replication. Two leads were identified, Embelin and Kobe2602. Interestingly, the binding pockets of these two compounds do not overlap, so they postulate the synergistic use of both compounds [65].
As previously described, VP35 is a virulence factor that binds to dsRNA and prevents engagement by the host sensor RIG-I, which prevents its activation and inhibits IFN production. Therefore, blocking the VP35–dsRNA interaction is a potential drug target. Corona et al. screened a library of natural compounds and found that cynarin inhibits the interferon inhibitory domain of VP35, which is essential for VP35-dsRNA. Cynarin was shown to inhibit EBOV replication by acting directly on VP35 and subverting its IFN antagonism [42].
A recently published article demonstrated in vivo protection of EBOV by rintatolimod, a mismatched dsRNA that acts as a PAMP for Toll-like receptor 3 (TLR3), a PRR. The authors also revealed that this drug is a competitive antagonist for the IDD domain of VP35. By blocking this domain, it prevents viral dsRNA sequestration by VP35, allowing RIG-I to detect it [43]. Curiously, rintatolimod has an orphan designation in the EU [145].
Antonius et al. screened four potential natural compounds that inhibit EBOV-NP, hesperidin, cucurbitacin, ginsenoside RH2, and ginsenoside RO [146].
Repurposing Widely Used Licensed Drugs Against Ebolavirus Infection: Direct-Acting Antivirals
Dhama and colleagues also review some of the potential drugs that have been suggested to be DAAs repurposable for EVD [134].
Zhao et al. performed drug repurposing on FDA-approved (n = 1766) and experimental drugs (n = 259) to identify those with potential anti-EBOV activities. The study focused on two targets, the MTase domain of L and VP24. Zhao and colleagues reported that indinavir, an HIV protease inhibitor, may be effective in reducing the virulence of the virus owing to its high affinity for VP24. Several approved antiviral drugs (e.g., maraviroc, abacavir, telbivudine, and cidofovir) may inhibit the viral polymerase owing to their affinity for the MTase domain [132].
Yuan et al. demonstrated that suramin blocks nucleoside triphosphate (NTP) entry into the RdRp domain of L, thus hindering its polymerization activity [147]. Although suramin is not approved by the FDA or EMA, it is used to treat human African trypanosomiasis in sub-Saharan countries and is present in the List of Essential Medicines by the WHO [140, 141, 148, 149].
In the above-mentioned research orchestrated by Broni and colleagues, 23 approved drugs were predicted to have anti-EBOV activity because they directly inhibit VP40, which can be further explored so that they may be repurposed for EVD treatment [144].
As previously discussed, binding to the hydrophobic pocket at the GP1–GP2 interface is a MoA for destabilizing the interaction with NPC1. Some approved drugs have been shown to bind to this pocket, such as toremifene, benztropine, sertraline, paroxetine, bepridil, imipramine, clomipramine, and thioridazine [131].
Conclusion, Gaps, and Future Perspectives
Currently, two antibody-based therapeutics have received approval for treating EVD and in 2022 the first evidence-based clinical practice guideline dedicated to specific therapies for EVD was published. Ebanga™ or Inmazeb® are strongly recommended for patients with RT-qPCR-confirmed EVD alongside optimized supportive care [8].
Despite the improvements in EVD therapy over the years, deaths still occur. The PALM trial had an overall CFR of 35.1% for Inmazeb ® and 33.5% for Ebanga™. Consequently, there is an urgent need to improve the arsenal of treatments against EBOV [103, 107].
No small-molecule compounds are licensed for EVD, and such molecules provide some advantages compared to mAbs, such as being generally easy and cheap to produce, transport, and store. Additionally, EVD survivors have evidence of viral persistence. The molecular weight of mAbs limits their penetration into immune sanctuaries where EBOV persists [107, 122].
This review highlights many different mechanisms of action that are being investigated to halt viral infection, with potential molecules acting on the virus, as direct-acting antivirals, or acting on the host, as host-directed antivirals. Repurposing already approved drugs for non-EVD indications to treat EVD is also a promising strategy to bypass some of the disadvantages associated with conventional drug discovery.
Despite the advancements showcased, several gaps and limitations persist in this area of research. Some of the emerging therapeutics have been identified through in silico screening studies, and their antiviral effects remain to be validated through biological experiments. Others have been tested in vitro or/and in vivo (non-human) but have never been tested in clinical trials for EVD, which happens during outbreaks. Consequently, their efficacy remains to be demonstrated, and their approval by regulatory authorities depends on these results. Collaboration among multidisciplinary teams, including researchers, clinicians, patients, governments, and industry partners, will be crucial in accelerating the translation of these innovations from bench to bedside.
In summary, by addressing the identified gaps and leveraging collaborative efforts, the evolution of these emerging pharmacological approaches has the potential to reshape the landscape of therapeutic interventions for EVD, ultimately improving the outcomes of the disease.
Data availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References
Baker RE, Mahmud AS, Miller IF, et al. Infectious disease in an era of global change. Nat Rev Microbiol. 2022;20:193–205. https://doi.org/10.1038/s41579-021-00639-z.
Findlater A, Bogoch II. Human mobility and the global spread of infectious diseases: a focus on air travel. Trends Parasitol. 2018;34:772–83. https://doi.org/10.1016/j.pt.2018.07.004.
Amaral R, Torre C, Rocha J, Sepodes B. Ebola outbreaks: a stress test of the preparedness of medicines regulatory systems for public health crises. Drug Discov Today. 2021;26:2608–18. https://doi.org/10.1016/j.drudis.2021.06.019.
Kraemer MUG, Golding N, Bisanzio D, et al. Utilizing general human movement models to predict the spread of emerging infectious diseases in resource poor settings. Sci Rep. 2019;9:5151. https://doi.org/10.1038/s41598-019-41192-3.
The World Bank. Air transport, passengers carried. https://data.worldbank.org/indicator/IS.AIR.PSGR?end=2020&start=1970&type=shaded&view=chart. Accessed 4 Mar 2023.
Jain S, Martynova E, Rizvanov A, Khaiboullina S, Baranwal M. Structural and functional aspects of Ebola virus proteins. Pathogens. 2021;10:1330. https://doi.org/10.3390/pathogens10101330.
Jacob ST, Crozier I, Fischer WA, et al. Ebola virus disease. Nat Rev Dis Prim. 2020;6:13. https://doi.org/10.1038/s41572-020-0147-3.
World Health Organization. Therapeutics for Ebola virus disease. https://www.who.int/publications/i/item/9789240055742. Accessed 5 May 2023.
Tuite AR, Watts AG, Khan K, Bogoch II. Ebola virus outbreak in North Kivu and Ituri provinces, Democratic Republic of Congo, and the potential for further transmission through commercial air travel. J Travel Med. 2019. https://doi.org/10.1093/jtm/taz063.
Marzi A, Banadyga L. Ebola virus (Filoviridae). In: Encyclopedia of virology. Amsterdam: Elsevier; 2021. p. 232–44.
Malvy D, McElroy AK, Clerck H, Günther S, Griensven J. Ebola virus disease. Lancet. 2019;393:936–48. https://doi.org/10.1016/S0140-6736(18)33132-5.
Liu CH, Hu YT, Wong SH, Lin LT. Therapeutic strategies against Ebola virus infection. Viruses. 2022;14:579. https://doi.org/10.3390/v14030579.
Richardson KJ. Ebola virus disease. Adv Emerg Nurs J. 2015;37:102–15. https://doi.org/10.1097/TME.0000000000000063.
Chavez S, Koyfman A, Gottlieb M, et al. Ebola virus disease: a review for the emergency medicine clinician. Am J Emerg Med. 2023;70:30–40. https://doi.org/10.1016/j.ajem.2023.04.037.
ICTV. Current ICTV taxonomy release. https://ictv.global/taxonomy. Accessed 25 Jun 2023.
Kuhn JH, Biedenkopf N, Bukreyev A, et al. Rename two genera (Mononegavirales: Filoviridae). 2022. https://ictv.global/files/proposals/approved?fid=11668#block-teamplus-page-title. Accessed 25 Jun 2023
Kuhn JH, Postler TS, Biedenkopf N, et al. Rename all species in the family to comply with the ICTV-mandated binomial format (Mononegavirales: Filoviridae). 2021. https://ictv.global/files/proposals/approved?fid=11668#block-teamplus-page-title. Accessed 25 Jun 2023
ICTV. Approved proposals. https://ictv.global/files/proposals/approved?fid=11668#block-teamplus-page-title. Accessed 25 Jun 2023.
Kuhn JH, Amarasinghe GK, Basler CF, et al. ICTV virus taxonomy profile: filoviridae. J Gen Virol. 2019;100:911–2. https://doi.org/10.1099/jgv.0.001252.
Bell BP, Damon IK, Jernigan DB, et al. Overview, control strategies, and lessons learned in the CDC response to the 2014–2016 Ebola epidemic. MMWR Suppl. 2016;65:4–11. https://doi.org/10.15585/mmwr.su6503a2.
Kuhn JH, Adachi T, Adhikari NKJ, et al. New filovirus disease classification and nomenclature. Nat Rev Microbiol. 2019;17:261–3. https://doi.org/10.1038/s41579-019-0187-4.
Patel P, Shah S. Ebola virus—StatPearls—NCBI bookshelf. https://www.ncbi.nlm.nih.gov/books/NBK560579/#_NBK560579_pubdet_. Accessed 2 Apr 2023.
Howley PM, Knipe DM, Whelan S. Fields virology: emerging viruses. 7th ed. USA: LWW; 2020.
Bharat TAM, Noda T, Riches JD, et al. Structural dissection of Ebola virus and its assembly determinants using cryo-electron tomography. Proc Natl Acad Sci USA. 2012;109:4275–80. https://doi.org/10.1073/pnas.1120453109.
Beniac DR, Melito PL, Devarennes SL, et al. The organisation of Ebola virus reveals a capacity for extensive, modular polyploidy. PLoS One. 2012;7:e29608. https://doi.org/10.1371/journal.pone.0029608.
Burrell CJ, Howard CR, Murphy FA. Virion structure and composition. In: Fenner and White’s medical virology. Amsterdam: Elsevier; 2017. p. 27–37.
Dutta P, Halder AK, Basu S, Kundu M. A survey on Ebola genome and current trends in computational research on the Ebola virus. Brief Funct Genomics. 2018;17:374–80. https://doi.org/10.1093/bfgp/elx020.
Furuyama W, Sakaguchi M, Yamada K, Nanbo A. Development of an imaging system for visualization of Ebola virus glycoprotein throughout the viral lifecycle. Front Microbiol. 2022;13:3910. https://doi.org/10.3389/fmicb.2022.1026644.
Dolnik O, Becker S. Assembly and transport of filovirus nucleocapsids. PLOS Pathog. 2022;18:e1010616. https://doi.org/10.1371/journal.ppat.1010616.
Lo MK, Spengler JR, Erickson BR, Spiropoulou CF. Developing therapeutics for Ebola virus disease: a multifaceted approach. In: Neglected tropical diseases: drug discovery and development. New Jersey: Wiley; 2019. p. 49–91.
Woolsey C, Menicucci AR, Cross RW, et al. A VP35 mutant Ebola virus lacks virulence but can elicit protective immunity to wild-type virus challenge. Cell Rep. 2019;28:3032. https://doi.org/10.1016/j.celrep.2019.08.047.
Brandt J, Wendt L, Bodmer BS, Mettenleiter TC, Hoenen T. The cellular protein CAD is recruited into Ebola virus inclusion bodies by the nucleoprotein NP to facilitate genome replication and transcription. Cells. 2020;9:1126. https://doi.org/10.3390/cells9051126.
Wu L, Jin D, Wang D, et al. The two-stage interaction of Ebola virus VP40 with nucleoprotein results in a switch from viral RNA synthesis to virion assembly/budding. Protein Cell. 2022;13:120–40. https://doi.org/10.1007/s13238-020-00764-0.
Diot C, Cosentino G, Rameix-Welti MA. Ribonucleoprotein transport in negative strand RNA viruses. Biol Cell. 2023;115:10. https://doi.org/10.1111/boc.202200059.
Kruse T, Biedenkopf N, Hertz EPT, et al. The Ebola virus nucleoprotein recruits the host PP2A-B56 phosphatase to activate transcriptional support activity of VP30. Mol Cell. 2018;69:136–145.e6. https://doi.org/10.1016/j.molcel.2017.11.034.
Easton V, McPhillie M, Garcia-Dorival I, et al. Identification of a small molecule inhibitor of Ebola virus genome replication and transcription using in silico screening. Antiviral Res. 2018;156:46–54. https://doi.org/10.1016/j.antiviral.2018.06.003.
Wendt L, Brandt J, Bodmer BS, et al. The Ebola virus nucleoprotein recruits the nuclear RNA export factor NXF1 into inclusion bodies to facilitate viral protein expression. Cells. 2020;9:187. https://doi.org/10.3390/cells9010187.
García-Dorival I, Wu W, Armstrong SD, et al. Elucidation of the cellular interactome of Ebola virus nucleoprotein and identification of therapeutic targets. J Proteome Res. 2016;15:4290–303. https://doi.org/10.1021/acs.jproteome.6b00337.
Wan Q, Song D, Li H, Liang He M. Stress proteins: the biological functions in virus infection, present and challenges for target-based antiviral drug development. Signal Transduct Target Ther. 2020;5:1–40. https://doi.org/10.1038/s41392-020-00233-4.
Kell AM, Gale M. RIG-I in RNA virus recognition. Virology. 2015;479–480:110–21. https://doi.org/10.1016/j.virol.2015.02.017.
Pattabhi S, Knoll ML, Gale M, Loo YM. DHX15 is a coreceptor for RLR signaling that promotes antiviral defense against RNA virus infection. J Interf Cytokine Res. 2019;39:331. https://doi.org/10.1089/jir.2018.0163.
Corona A, Fanunza E, Salata C, et al. Cynarin blocks Ebola virus replication by counteracting VP35 inhibition of interferon-beta production. Antiviral Res. 2022;198: 105251. https://doi.org/10.1016/j.antiviral.2022.105251.
Corona A, Strayer D, Distinto S, et al. Ebola virus disease: in vivo protection provided by the PAMP restricted TLR3 agonist rintatolimod and its mechanism of action. Antiviral Res. 2023;212:105554. https://doi.org/10.1016/j.antiviral.2023.105554.
Haasnoot J, De Vries W, Geutjes EJ, Prins M, De Haan P, Berkhout B. The Ebola virus VP35 protein is a suppressor of RNA silencing. PLOS Pathog. 2007;3:e86. https://doi.org/10.1371/journal.ppat.0030086.
Zhu L, Gao T, Huang Y, et al. Ebola virus VP35 hijacks the PKA-CREB1 pathway for replication and pathogenesis by AKIP1 association. Nat Commun. 2022;13:2256. https://doi.org/10.1038/s41467-022-29948-4.
Bharaj P, Atkins C, Luthra P, et al. The host E3-ubiquitin ligase TRIM6 ubiquitinates the Ebola virus VP35 protein and promotes virus replication. J Virol. 2017. https://doi.org/10.1128/jvi.00833-17.
Le Sage V, Cinti A, McCarthy S, et al. Ebola virus VP35 blocks stress granule assembly. Virology. 2017;502:73–83. https://doi.org/10.1016/j.virol.2016.12.012.
Le H, Spearman P, Waggoner SN, Singh K. Ebola virus protein VP40 stimulates IL-12- and IL-18-dependent activation of human natural killer cells. JCI Insight. 2022;7:16. https://doi.org/10.1172/jci.insight.158902.
Yu DS, Weng TH, Wu XX, et al. The lifecycle of the Ebola virus in host cells. Oncotarget. 2017;8:55750–9. https://doi.org/10.18632/oncotarget.18498.
Husby ML, Amiar S, Prugar LI, et al. Phosphatidylserine clustering by the Ebola virus matrix protein is a critical step in viral budding. EMBO Rep. 2022;23:e51709. https://doi.org/10.15252/embr.202051709.
Loughran HM, Han Z, Wrobel JE, et al. Quinoxaline-based inhibitors of Ebola and Marburg VP40 egress. Bioorg Med Chem Lett. 2016;26:3429–35. https://doi.org/10.1016/j.bmcl.2016.06.053.
Zhang L, Zhou S, Chen M, et al. P300-mediated NEDD4 acetylation drives ebolavirus VP40 egress by enhancing NEDD4 ligase activity. PLOS Pathog. 2021;17:e1009616. https://doi.org/10.1371/journal.ppat.1009616.
Rojas M, Monsalve DM, Pacheco Y, et al. Ebola virus disease: an emerging and re-emerging viral threat. J Autoimmun. 2020;106:102375. https://doi.org/10.1016/j.jaut.2019.102375.
Cantoni D, Rossman JS. Ebolaviruses: new roles for old proteins. PLoS Negl Trop Dis. 2018;12:e0006349. https://doi.org/10.1371/journal.pntd.0006349.
Pleet ML, DeMarino C, Stonier SW, et al. Extracellular vesicles and Ebola virus: a new mechanism of immune evasion. Viruses. 2019;11:410. https://doi.org/10.3390/v11050410.
Pleet ML, Erickson J, Demarino C, et al. Ebola virus VP40 modulates cell cycle and biogenesis of extracellular vesicles. J Infect Dis. 2018;218:S365–87. https://doi.org/10.1093/infdis/jiy472.
Rayaprolu V, Fulton BO, Rafique A, et al. Structure of the Inmazeb cocktail and resistance to Ebola virus escape. Cell Host Microbe. 2023;31:260–272.e7. https://doi.org/10.1016/j.chom.2023.01.002.
Furuyama W, Shifflett K, Feldmann H, Marzi A. The Ebola virus soluble glycoprotein contributes to viral pathogenesis by activating the MAP kinase signaling pathway. PLOS Pathog. 2021;17:e1009937. https://doi.org/10.1371/journal.ppat.1009937.
Zhu W, Banadyga L, Emeterio K, Wong G, Qiu X. The roles of Ebola virus soluble glycoprotein in replication, pathogenesis, and countermeasure development. Viruses. 2019;11:999. https://doi.org/10.3390/v11110999.
Melnik LI, Guha S, Ghimire J, et al. Ebola virus delta peptide is an enterotoxin. Cell Rep. 2022;38:110172. https://doi.org/10.1016/j.celrep.2021.110172.
He J, Melnik LI, Komin A, et al. Ebola virus delta peptide is a viroporin. J Virol. 2017. https://doi.org/10.1128/jvi.00438-17.
Nanbo A, Maruyama J, Imai M, et al. Ebola virus requires a host scramblase for externalization of phosphatidylserine on the surface of viral particles. PLoS Pathog. 2018;14:e1006848. https://doi.org/10.1371/journal.ppat.1006848.
Taki E, Ghanavati R, Navidifar T, Dashtbin S, Heidary M, Moghadamnia M. Ebanga™: the most recent FDA-approved drug for treating Ebola. Front Pharmacol. 2023. https://doi.org/10.3389/fphar.2023.1083429.
Basler CF, Krogan NJ, Leung DW, Amarasinghe GK. Virus and host interactions critical for filoviral RNA synthesis as therapeutic targets. Antiviral Res. 2019;162:90. https://doi.org/10.1016/j.antiviral.2018.12.006.
Ma Yh, Hong X, Wu F, et al. Inhibiting the transcription and replication of Ebola viruses by disrupting the nucleoprotein and VP30 protein interaction with small molecules. Acta Pharmacol Sin. 2023;44:1487–99. https://doi.org/10.1038/s41401-023-01055-0.
Takamatsu Y, Krähling V, Kolesnikova L, et al. Serine–arginine protein kinase 1 regulates ebola virus transcription. MBio. 2020. https://doi.org/10.1128/mbio.02565-19.
Takamatsu Y, Yoshikawa T, Kurosu T, et al. Role of VP30 phosphorylation in Ebola virus nucleocapsid assembly and transport. J Virol. 2022. https://doi.org/10.1128/jvi.01083-22.
Su Y, Stahelin RV. The minor matrix protein VP24 from Ebola virus lacks direct lipid-binding properties. Viruses. 2020;12:869. https://doi.org/10.3390/v12080869.
Yamaoka S, Ebihara H. Pathogenicity and virulence of ebolaviruses with species- and variant-specificity. Virulence. 2021;12:885–901. https://doi.org/10.1080/21505594.2021.1898169.
Ramanathan P, Tigabu B, Santos RI, et al. Ebolavirus species-specific interferon antagonism mediated by VP24. Viruses. 2023;15:1075. https://doi.org/10.3390/v15051075.
Vidal S, Sánchez-Aparicio M, Seoane R, et al. Expression of the Ebola virus VP24 protein compromises the integrity of the nuclear envelope and induces a laminopathy-like cellular phenotype. MBio. 2021;12:972–93. https://doi.org/10.1128/mbio.00972-21.
Ramanathan A, Robb GB, Chan SH. mRNA capping: biological functions and applications. Nucleic Acids Res. 2016;44:7511–26. https://doi.org/10.1093/nar/gkw551.
Drazkowska K, Tomecki R, Warminski M, et al. 2′-O-Methylation of the second transcribed nucleotide within the mRNA 5′ cap impacts the protein production level in a cell-specific manner and contributes to RNA immune evasion. Nucleic Acids Res. 2022;50:9051–71. https://doi.org/10.1093/nar/gkac722.
Gourronc FA, Rebagliati MR, Kramer-Riesberg B, et al. Adipocytes are susceptible to Ebola virus infection. Virology. 2022;573:12–22. https://doi.org/10.1016/j.virol.2022.05.007.
Acciani MD, Lay Mendoza MF, Havranek KE, et al. Ebola virus requires phosphatidylserine scrambling activity for efficient budding and optimal infectivity. J Virol. 2021. https://doi.org/10.1128/jvi.01165-21.
Saeed MF, Kolokoltsov AA, Freiberg AN, Holbrook MR, Davey RA. Phosphoinositide-3 Kinase-Akt pathway controls cellular entry of Ebola virus. PLOS Pathog. 2008;4:e1000141. https://doi.org/10.1371/journal.ppat.1000141.
Hoenen T, Groseth A, Feldmann H. Therapeutic strategies to target the Ebola virus life cycle. Nat Rev Microbiol. 2019;17:593–606. https://doi.org/10.1038/s41579-019-0233-2.
Miller ME, Adhikary S, Kolokoltsov AA, Davey RA. Ebolavirus requires acid sphingomyelinase activity and plasma membrane sphingomyelin for infection. J Virol. 2012;86:7473–83. https://doi.org/10.1128/jvi.00136-12.
Jin C, Che B, Guo Z, et al. Single virus tracking of Ebola virus entry through lipid rafts in living host cells. Biosaf Heal. 2020;2:25–31. https://doi.org/10.1016/j.bsheal.2019.12.009.
Carpinteiro A, Edwards MJ, Hoffmann M, et al. Pharmacological inhibition of acid sphingomyelinase prevents uptake of SARS-CoV-2 by epithelial cells. Cell Reports Med. 2020. https://doi.org/10.1016/j.xcrm.2020.100142.
Salata C, Calistri A, Alvisi G, Celestino M, Parolin C, Palù G. Ebola virus entry: from molecular characterization to drug discovery. Viruses. 2019;11:274. https://doi.org/10.3390/v11030274.
Bo Y, Qiu S, Mulloy RP, Côté M. Filoviruses use the HOPS complex and UVRAG to traffic to Niemann-Pick C1 compartments during viral entry. J Virol. 2020. https://doi.org/10.1128/jvi.01002-20.
Flint M, Chatterjee P, Lin DL, et al. A genome-wide CRISPR screen identifies N-acetylglucosamine-1-phosphate transferase as a potential antiviral target for Ebola virus. Nat Commun. 2019;10:285. https://doi.org/10.1038/s41467-018-08135-4.
Das DK, Bulow U, Diehl WE, et al. Conformational changes in the Ebola virus membrane fusion machine induced by pH, Ca2+, and receptor binding. PLoS Biol. 2020;18(2):e3000626. https://doi.org/10.1371/journal.pbio.3000626.
Herbert AS, Davidson C, Kuehne AI, et al. Niemann-pick C1 is essential for ebolavirus replication and pathogenesis in vivo. MBio. 2015. https://doi.org/10.1128/mbio.00565-15.
National Organization for Rare Disorders—Niemann Pick disease type C—symptoms, causes, treatment. https://rarediseases.org/rare-diseases/niemann-pick-disease-type-c/. Accessed 8 Jun 2023.
Geberhiwot T, Moro A, Dardis A, et al. Consensus clinical management guidelines for Niemann-Pick disease type C. Orphanet J Rare Dis. 2018. https://doi.org/10.1186/s13023-018-0785-7.
Lee J, Kreutzberger AJB, Odongo L, et al. Ebola virus glycoprotein interacts with cholesterol to enhance membrane fusion and cell entry. Nat Struct Mol Biol. 2021;28:181–9. https://doi.org/10.1038/s41594-020-00548-4.
Igarashi M, Hirokawa T, Takadate Y, Takada A. Structural insights into the interaction of filovirus glycoproteins with the endosomal receptor Niemann-Pick C1: a computational study. Viruses. 2021;13:913. https://doi.org/10.3390/v13050913.
Sakurai Y, Kolokoltsov AA, Chen CC, et al. Two pore channels control Ebolavirus host cell entry and are drug targets for disease treatment. Science. 2015;347:995–8. https://doi.org/10.1126/science.1258758.
Gunaratne GS, Marchant JS. The ins and outs of virus trafficking through acidic Ca2+ stores. Cell Calcium. 2022;102:102528. https://doi.org/10.1016/j.ceca.2022.102528.
Penny CJ, Vassileva K, Jha A, et al. Mining of Ebola virus entry inhibitors identifies approved drugs as two-pore channel pore blockers. Biochim Biophys Acta Mol Cell Res. 2019;1866:1151–61. https://doi.org/10.1016/j.bbamcr.2018.10.022.
Kuroda M, Fujikura D, Nanbo A, et al. Interaction between TIM-1 and NPC1 is important for cellular entry of Ebola virus. J Virol. 2015;89:6481–93. https://doi.org/10.1128/jvi.03156-14/.
Qiu S, Leung A, Bo Y, et al. Ebola virus requires phosphatidylinositol (3,5) bisphosphate production for efficient viral entry. Virology. 2018;513:17–28. https://doi.org/10.1016/j.virol.2017.09.028.
Winter SL, Golani G, Lolicato F, et al. The Ebola virus VP40 matrix layer undergoes endosomal disassembly essential for membrane fusion. EMBO J. 2023. https://doi.org/10.15252/embj.2023113578.
Biedenkopf N, Lange-Grünweller K, Schulte FW, et al. The natural compound silvestrol is a potent inhibitor of Ebola virus replication. Antiviral Res. 2017;137:76–81. https://doi.org/10.1016/j.antiviral.2016.11.011.
Montero H, Pérez-Gil G, Sampieri CL. Eukaryotic initiation factor 4A (eIF4A) during viral infections. Virus Genes. 2019;55:267–73. https://doi.org/10.1007/s11262-019-01641-7.
Bodmer BS, Vallbracht M, Ushakov DS, Wendt L, Chlanda P, Hoenen T. Ebola virus inclusion bodies are liquid organelles whose formation is facilitated by nucleoprotein oligomerization. Emerg Microbes Infect. 2023;12:2223727. https://doi.org/10.1080/22221751.2023.2223727.
Mattila PK, Lappalainen P. Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol. 2008;9:446–54. https://doi.org/10.1038/nrm2406.
Gao Y, Zhao Y, Guyatt G, et al. Effects of therapies for Ebola virus disease: a systematic review and network meta-analysis. Lancet Microbe. 2022;3:e683–692. https://doi.org/10.1016/S2666-5247(22)00123-9.
UpToDate. Society guideline links: infection control—up to date. https://www.uptodate.com/contents/society-guideline-links-infection-control?search=ebola&topicRef=113258&source=see_link. Accessed 30 Apr 2023.
World Health Organization. Optimized supportive care for Ebola virus disease: clinical management standard operating procedures. https://apps.who.int/iris/bitstream/handle/10665/325000/9789241515894-eng.pdf. Accessed 8 May 2023.
Mulangu S, Dodd LE, Davey RT, et al. A randomized, controlled trial of Ebola virus disease therapeutics. N Engl J Med. 2019;381:2293–303. https://doi.org/10.1056/NEJMoa1910993.
The PREVAIL II Writing Group for the Multi-National PREVAIL II Study Team. A randomized, controlled trial of ZMapp for Ebola virus infection. N Engl J Med. 2016;375:1448–56. https://doi.org/10.1056/NEJMoa1604330.
Tran EEH, Nelson EA, Bonagiri P, et al. Mapping of Ebolavirus neutralization by monoclonal antibodies in the ZMapp cocktail using cryo-electron tomography and studies of cellular entry. J Virol. 2016;90:7618–27. https://doi.org/10.1128/jvi.00406-16.
Bakheit AH, Darwish H, Darwish IA, Al-Ghusn AI. Remdesivir. Profiles Drug Subst Excip Relat Methodol. 2023;48:71–108. https://doi.org/10.1016/bs.podrm.2022.11.003.
TshianiMbaya O, Mukumbayi P, Mulangu S. Review: insights on current FDA-approved monoclonal antibodies against Ebola virus infection. Front Immunol. 2021. https://doi.org/10.3389/fimmu.2021.721328.
Crozier I, Britson KA, Wolfe DN, et al. The evolution of medical countermeasures for Ebola virus disease: lessons learned and next steps. Vaccines. 2022;10:1213. https://doi.org/10.3390/vaccines10081213.
Mulangu S, Mbala-Kingebeni P, Mbaya OT. Antibody use during an outbreak of Ebola virus disease in the democratic Republic of Congo, 2020. N Engl J Med. 2022;386:1188–91. https://doi.org/10.1056/NEJMc2113505.
Markham A. REGN-EB3: first approval. Drugs. 2021;81:175–8. https://doi.org/10.1007/s40265-020-01452-3.
FDA. Inmazeb® leaflet. www.fda.gov/medwatch. Accessed 23 May 2023.
Inmazeb: The first FDA-approved treatment for Zaire ebolavirus. https://www.inmazeb.com/. Accessed 23 May 2023.
Di Paola N, Sanchez-Lockhart M, Zeng X, Kuhn JH, Palacios G. Viral genomics in Ebola virus research. Nat Rev Microbiol. 2020;18:365–78. https://doi.org/10.1038/s41579-020-0354-7.
European Medicines Agency. Ebola. https://www.ema.europa.eu/en/human-regulatory/overview/public-health-threats/ebola#ebola-outbreak-in-2018-19-section. Accessed 23 May 2023.
EMA. Public summary of opinion on orphan designation Three human monoclonal antibodies against the EBOV glycoprotein for the treatment of Ebola virus disease. https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu-3-18-2027. Accessed 26 May 2023.
EMA. EU/3/18/2027: Orphan designation for the treatment of Ebola virus disease. https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu-3-18-2027. Accessed 28 May 2023.
Ansuvimab LA. First approval. Drugs. 2021;81:595–8. https://doi.org/10.1007/s40265-021-01483-4.
Ebanga™. https://www.ebanga.com/. Accessed 21 May 2023.
FDA. Ebanga™ leaflet. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761172s000lbl.pdf. Accessed 21 May 2023
Black L, Wittman M, Wagner J. Treating Zaire ebolavirus with Ansuvimab-zykl. Contag Infect Dis Today. 2021;6. https://www.contagionlive.com/view/treating-zaire-ebolavirus-with-ansuvimab-zykl.
Kummer S, Lander A, Goretzko J, Kirchoff N, Rescher U, Schloer S. Pharmacologically induced endolysosomal cholesterol imbalance through clinically licensed drugs itraconazole and fluoxetine impairs Ebola virus infection in vitro. Emerg Microbes Infect. 2022;11:195–207. https://doi.org/10.1080/22221751.2021.2020598.
Vanmechelen B, Stroobants J, Chiu W, et al. Identification of novel Ebola virus inhibitors using biologically contained virus. Antiviral Res. 2022;200:105294. https://doi.org/10.1016/j.antiviral.2022.105294.
Kumar N, Sharma S, Kumar R, et al. Host-directed antiviral therapy. Clin Microbiol Rev. 2020. https://doi.org/10.1128/cmr.00168-19.
Chitalia VC, Munawar AH. A painful lesson from the COVID-19 pandemic: the need for broad-spectrum, host-directed antivirals. J Transl Med. 2020;18:390. https://doi.org/10.1186/s12967-020-02476-9.
Peck KM, Lauring AS. Complexities of viral mutation rates. J Virol. 2018. https://doi.org/10.1128/jvi.01031-17.
Ma J, Zhang X, Soloveva V, et al. Enhancing the antiviral potency of ER α-glucosidase inhibitor IHVR-19029 against hemorrhagic fever viruses in vitro and in vivo. Antiviral Res. 2018;150:112–22. https://doi.org/10.1016/j.antiviral.2017.12.008.
Mahajan S, Choudhary S, Kumar P, Tomar S. Antiviral strategies targeting host factors and mechanisms obliging +ssRNA viral pathogens. Bioorg Med Chem. 2021;46:116356. https://doi.org/10.1016/j.bmc.2021.116356.
Roa-Linares VC, Escudero-Flórez M, Vicente-Manzanares M, Gallego-Gómez JC. Host cell targets for unconventional antivirals against RNA viruses. Viruses. 2023;15:776. https://doi.org/10.3390/v15030776.
Badia R, Garcia-Vidal E, Ballana E. Viral-host dependency factors as therapeutic targets to overcome antiviral drug-resistance: a focus on innate immune modulation. Front Virol. 2022. https://doi.org/10.3389/fviro.2022.935933.
Basu A, Mills DM, Mitchell D, et al. Novel small molecule entry inhibitors of Ebola virus. J Infect Dis. 2015;212:S425–34. https://doi.org/10.1093/infdis/jiv223.
Morales-Tenorio M, Ginex T, Cuesta-Geijo MA, et al. Potential pharmacological strategies targeting the Niemann-Pick C1 receptor and Ebola virus glycoprotein interaction. Eur J Med Chem. 2021;223:113654. https://doi.org/10.1016/j.ejmech.2021.113654.
Zhao Z, Martin C, Fan R, Bourne PE, Xie L. Drug repurposing to target Ebola virus replication and virulence using structural systems pharmacology. BMC Bioinform. 2016. https://doi.org/10.1186/s12859-016-0941-9.
Rao N, Poojari T, Poojary C, Sande R, Sawant S. Drug repurposing: a shortcut to new biological entities. Pharm Chem J. 2022;56:1203–14. https://doi.org/10.1007/s11094-022-02778-w.
Dhama K, Karthik K, Khandia R, et al. Advances in designing and developing vaccines, drugs, and therapies to counter Ebola virus. Front Immunol. 2018. https://doi.org/10.3389/fimmu.2018.01803.
Sun W, He S, Martínez-Romero, et al. Synergistic drug combination effectively blocks Ebola virus infection. Antiviral Res. 2017;137:165–72. https://doi.org/10.1016/j.antiviral.2016.11.017.
Shrivastava-Ranjan P, Flint M, Bergeron É, et al. Statins suppress Ebola virus infectivity by interfering with glycoprotein processing. MBio. 2018. https://doi.org/10.1128/mbio.00660-18.
Wu BR, Chen DH, Liao WC, et al. Statin therapy and the risk of viral infection: a retrospective population-based cohort study. J Clin Med. 2022;11:5626. https://doi.org/10.3390/jcm11195626.
Ji X, Li Z. Medicinal chemistry strategies toward host targeting antiviral agents. Med Res Rev. 2020. https://doi.org/10.1002/med.21664.
Yi D, Li Q, Wang H, et al. Repurposing of berbamine hydrochloride to inhibit Ebola virus by targeting viral glycoprotein. Acta Pharm Sin B. 2022;12:4378–89. https://doi.org/10.1016/j.apsb.2022.05.023.
FDA. Drug Approvals and Databases. https://www.fda.gov/drugs/development-approval-process-drugs/drug-approvals-and-databases. Accessed 13 Jun 2023.
European Medicines Agency. Medicines. https://www.ema.europa.eu/en/medicines. Accessed 13 Jun 2023.
DrugBank Online. Database for drug and drug target info. https://go.drugbank.com/. Accessed 25 Jun 2023.
Zhao W, Bai B, Hong Z, Zhang X, Zhou B. Berbamine (BBM), a natural STAT3 inhibitor, synergistically enhances the antigrowth and proapoptotic effects of sorafenib on hepatocellular carcinoma cells. ACS Omega. 2020;5:24838–47. https://doi.org/10.1021/acsomega.0c03527.
Broni E, Ashley C, Adams J, et al. Cheminformatics-based study identifies potential Ebola VP40 inhibitors. Int J Mol Sci. 2023;24:6298. https://doi.org/10.3390/ijms24076298.
European Medicines Agency. EU/3/15/1480. https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu3151480. Accessed 18 Jun 2023.
Antonius Y, Ongko J, Sukweenadhi J, Putra SED. Identification of potential Ebola virus nucleoprotein (EBOV NP) inhibitor derivate from various traditional medicinal plants in Indonesia: in silico study. Media Pharm Indonesia. 2021;3:217–26. https://doi.org/10.24123/mpi.v3i4.4716.
Yuan B, Peng Q, Cheng J, et al. Structure of the Ebola virus polymerase complex. Nature. 2022;610:394–401. https://doi.org/10.1038/s41586-022-05271-2.
African sleeping sickness. https://www.bayer.com/en/pharma/african-sleeping-sickness. Accessed 18 Jun 2023.
WHO—Electronic essential medicines list. https://list.essentialmeds.org/recommendations/819. Accessed 18 Jun 2023.
Funding
The journal’s rapid service fee was funded by the authors. No funding was received for the writing of this review.
Author information
Authors and Affiliations
Contributions
Francisca Almeida-Pinto initiated the conceptualization, devised the structure, conducted the literature review, and authored all versions of the manuscript. Rui Pinto provided essential scientific insights as a pharmacology expert, approved the manuscript’s structure, and contributed to refining the manuscript. João Rocha provided expertise as a member of the Committee for Orphan Medicinal Products (COMP) of the European Medicines Agency (EMA), offering critical insights and guidance, significantly enriching the manuscript. All authors read and approved the final manuscript. The figures and table were created by Francisca Almeida-Pinto.
Corresponding author
Ethics declarations
Conflict of Interest
Francisca Almeida-Pinto, João Rocha, and Rui Pinto declare that they have no competing interests.
Ethical Approval
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Almeida-Pinto, F., Pinto, R. & Rocha, J. Navigating the Complex Landscape of Ebola Infection Treatment: A Review of Emerging Pharmacological Approaches. Infect Dis Ther 13, 21–55 (2024). https://doi.org/10.1007/s40121-023-00913-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-023-00913-y