
Abstract
Introduction
Antiviral drugs have shown limited effectiveness in treating patients with coronavirus disease 2019 (COVID-19). We aimed to assess the effects of a favipiravir and hydroxychloroquine combination on treating moderate-to-severe COVID-19 patients.
Methods
An investigator-initiated, multicenter, open-label, randomized trial at nine hospitals. Eligible patients were adults with moderate-to-severe COVID-19 defined as oxygen saturation (SaO2) of ≤ 94% while breathing ambient air or significant clinical symptoms with chest x-ray changes requiring hospital admission. Randomization was in a 1:1 ratio to receive standard care (control group) or standard care plus favipiravir and hydroxychloroquine. The primary outcome was time to clinical improvement of two points (from the status at randomization) on a seven-category ordinal scale or live discharge from the hospital within 14 days. Analyses were done in an intention-to-treat population.
Results
From May 2020 to Jan 2021, 254 patients were enrolled; 129 were assigned to standard of care and 125 to the treatment. The mean age was 52 (± 13) years, and 103 (41%) were women. At randomization, six patients were on invasive mechanical ventilation, 229 (90.15%) were requiring supplemental oxygen only (with or without non-invasive ventilation), and 19 (7.48%) were receiving neither. The time to clinical improvement was not significantly different between the groups: median of 9 days in the treatment group and 7 days in the control group (HR: 0.845; 95% CI 0.617–1.157; p-value = 0.29). The 28-day mortality was not significantly different between the groups (7.63% treatment) vs. (10.32% control); p-value = 0.45. The most prevalent adverse events were headache, elevation in ALT, and the prolonged QTc interval in the treatment group.
Conclusion
The combination of favipiravir and hydroxychloroquine did not result in a statistically significant clinical benefit in patients with moderate-to-severe COVID-19.
Clinical Trial Registration
ClinicalTrials.gov (NCT04392973).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Antiviral drugs have shown limited effectiveness in treating COVID-19 patients |
The study assessed if the combination of antiviral therapy (favipiravir and hydroxychloroquine) will improve the clinical outcome of hospitalized patients with moderate-to-severe COVID-19 |
What was learned from the study? |
The combination of favipiravir and hydroxychloroquine did not result in a statistically significant clinical benefit in patients with moderate-to-severe COVID-19 |
The routine clinical use or further studies to evaluate this combination are discouraged for this patient population |
Introduction
As of 15 April 2021, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) had spread globally, with 135 million reported cases and > 2.9 million deaths globally since the start of the pandemic [1]. Although > 80% of infections are self-limiting, the rest may lead to severe symptoms and require hospital admission [2]. COVID-19 is associated with a high in-hospital mortality rate (19–35%) after Intensive Care Unit (ICU) admission [3, 4]. The treatment for patients with SARS-CoV-2 pneumonia is mainly supportive. Dexamethasone is the only treatment that has been shown to reduce mortality [5]. A more recent trial in critically ill adult patients with COVID-19 receiving organ support in intensive care, the IL-6 receptor antagonists (tocilizumab and sarilumab), showed improved outcomes, including survival [6]. Favipiravir is an RNA-dependent RNA polymerase (RdRp) inhibitor with activity against the influenza virus. In addition to its anti-influenza virus activity, favipiravir is capable of blocking the replication of flavi-, alpha-, filo-, bunya-, arena-, noro-, and other RNA viruses and can theoretically be active against SARS-CoV-2 [7, 8]. Favipiravir was compared earlier in the COVID-19 pandemic to other antivirals and showed promising results [9]. A study in mild-to-moderate COVID-19 patients showed significant improvement in the time to clinical cure when using favipiravir [10]. Using a combination of various potential therapies for COVID-19 is not an uncommon approach, such as lopinavir/ritonavir NCT04499677, tocilizumab NCT04310228, chloroquine NCT04319900, and hydroxychloroquine NCT04303299, among others, which all have not been completed yet. The selected combination was based on limited clinical and in vitro evidence regarding the efficacy of favipiravir and hydroxychloroquine in SARS-CoV-2. The two medications were listed in many guidelines as treatment options, with ongoing trials assessing their efficacy and safety. Theoretically, combining the agents might result in a synergistic effect due to different target sites. We report the results of a randomized, controlled trial to assess the efficacy of combining hydroxychloroquine and favipiravir plus standard of care in patients with moderate-to-severe COVID-19.
Methods
Study Design
This was an investigator-initiated, multicenter, open-label, randomized trial. Adult patients aged 18 years and older and admitted to the hospital from 21 May 2020 for COVID-19, virologically confirmed using RT-PCR, were included. Patients were recruited from nine hospitals in Saudi Arabia (King Abdulaziz Hospital Al Ahsa, AlMadina General Hospital, Al-Qatif Central Hospital, Imam Abdulrahman Al Faisal Hospital Dammam, King Abdulaziz Medical City Jeddah, King Abdulaziz Hospital Makkah, King Abdulaziz Medical City Riyadh, Imam Abdulrahman Alfaisal Hospital Riyadh, and Al-Iman General Hospital Riyadh). Ethical approval was obtained from the Institutional Review Board (IRB) at the Ministry of National Guard-Health Affairs (MNGHA; study no. RC20/174/R) and Ministry of Health (MOH; log no. 20-83E). The trial was overseen by an independent data and safety monitoring board (DSMB). The trial was done according to the Declaration of Helsinki principles and the International Conference on Harmonization-Good Clinical Practice guidelines. The trial protocol and the statistical analysis plan were submitted for publication before the interim analysis [11].
Patients
Eligible patients were at least 18 years of age, male or non-pregnant females, and diagnosed with COVID-19 by RT-PCR-confirmed SARS-CoV-2. The patients were considered eligible if admitted to the hospital with moderate-to-severe COVID-19, defined as oxygen saturation (SaO2) of ≤ 94% while breathing ambient air or significant clinical symptoms with chest x-ray changes. As an eligibility criterion, patients were enrolled within 10 days of disease onset. Patients were excluded if they had a history of myocardial infarction or an irregular rhythm/QTc (corrected QT interval) in the baseline electrocardiogram (ECG) of > 490 ms. The exclusion criteria also included major comorbidities such as hematologic malignancy, advanced (stage 4–5) chronic kidney disease or dialysis therapy, severe liver damage (Child-Pugh score ≥ C, AST > five times the upper limit), or HIV. Written informed consent was obtained from all patients or their legal representatives if they could not provide consent.
Randomization
Randomization was conducted using SAS v9.4 PROC PLAN to generate random allocation schedules. The randomization schedule was uploaded through an online web-based system in a 1:1 ratio to receive standard care (control group) or standard care plus the favipiravir and hydroxychloroquine combination (treatment group). It was stratified by enrolling center and by being admitted to ICU or on mechanical ventilation. The investigator disclosed the arm after all information regarding patient enrollment was recorded in the online system. The trial was open-label, and the delegated hospital pharmacy dispensed the study medications.
Trial Procedures
Daily assessment of the admitted patients and eligibility screening was done for possible study participants within the first 72 h of hospital admission. Once eligible, informed consent was obtained, and randomization was conducted. In addition to other standard medical treatments for COVID-19, per local protocols, patients randomized to the combination therapy were started on favipiravir for 10 days and hydroxychloroquine for 5 days. Favipiravir was given at a loading dose of 1800 mg (9 tablets) by mouth twice on day 1 followed by 800 mg (4 tablets) twice daily. Hydroxychloroquine dose was 400 mg twice daily on day 1 and then 200 mg twice daily. Other aspects of COVID-19 management were at the discretion of the treating physicians. The use of glucocorticoids, other immunomodulators, and antibiotic agents was allowed, and all were recorded throughout the hospitalization. As a safety consideration, the trial team assessed the QTc interval every 24 h while the patients were receiving the combination of the trial drugs.
Observation of possible adverse events (AEs) was done while using the investigational medications until day 14, including regular laboratory monitoring. In case of any concerns that might require holding/stopping medication, the principal investigator conducted a causality assessment, including a consultation with an independent clinician. One point contact at 28 and 90 days was planned to assess the vital status.
Outcomes
The primary endpoint was the time to clinical improvement, defined as the time from randomization to an improvement of two points on a seven-category ordinal scale or live discharge from the hospital, whichever came first. The seven-category ordinal scale consists of the following categories: a score of 1 indicated not hospitalized with the resumption of normal activities; 2, not hospitalized, but unable to resume normal activities; 3, hospitalization, not requiring supplemental oxygen; 4, hospitalization, requiring supplemental oxygen; 5, hospitalization, requiring nasal high-flow oxygen therapy and/or non-invasive mechanical ventilation; 6, hospitalization, requiring extracorporeal membrane oxygenation (ECMO) and/or invasive mechanical ventilation; 7, death.
The key secondary efficacy outcomes included the progress in clinical status and the requirement of ICU admission or mechanical ventilation within 14 days. The 28- and 90-day mortality was also monitored. In addition, the RT-PCR test conversion from positive to negative, length of hospital stay, and status at day 14 post-randomization were also collected. SARS-COV-2 RT-PCRs from a naso-/oropharyngeal swab or lower respiratory samples were obtained on days 1, 5, 10, 14, 21, and 28 or until the test was negative, whichever came first. All PCR testings are done in Central Laboratories, where all procedures and kits were approved by the Saudi FDA and CDC. These include: Perkin Elmer chemagic 360 Instrument (chemagic™ Viral DNA/RNA Extraction Kit), Roche MagnaPure96 (SV kit), Co-Diagnostics (Logix-Smart™-COVID-19), and Real Star SARS-CoV-2 RT-PCR Kit.
The safety outcomes included treatment-related adverse events (AEs), serious adverse events (SAEs), and premature discontinuations of the study medications. SAEs, defined as untoward medical events leading to death, a life-threatening experience, prolongation of hospitalization, or persistent or significant disability or incapacity in the judgment of the site investigator, were also reported.
Statistical Analysis
Under the classical two-arm parallel design, the total adequate sample size required was 472 subjects (236 per group). The estimated sample size achieved 85% power to detect a minimum of a 30% reduction in the median time to clinical improvement by at least two points or hospital discharge in the treatment group compared to the control group. The trial's overall sample size was estimated as 520 (260 per group), including 10% drop-out rate. A sample size adjustment was planned to be considered after the interim analysis of 60% of the subjects' recruitment and the DSMB recommendation. The interim analysis was designed to test for early stopping due to futility or efficacy and the sample size re-estimation. The interim analysis and final analysis plan were based on the method of the sum of the stage-wise p-value discussed in Chow and Chang, 2008. We performed several sensitivity analyses to determine the effect of different assumptions on the sample size. (Details are provided in the statistical analysis plan in Online Appendix 1).
The study's primary endpoint was the median time to clinical improvement by at least 2 points or live discharge from the hospital. It was analyzed using the log-rank test to compare median time to clinical improvement through day 14 between the two groups. The hazard ratios and 95% confidence interval (CI) were estimated using the proportional hazards regression model with treatment as the explanatory variable. The decision rule based on the study protocol were (1) stop the trial for early efficacy if the interim analysis p-value < 0.01, (2) stop the trial for futility if the interim analysis p-value ≥ 0.25, or (3) declare the trial significant if the sum of the interim analysis and final stage p-values < 0.1832. Similar analyses were performed for the secondary endpoints, including hospital discharge and mortality based on the log-rank test and the proportional hazards regression. For the hospital discharge, patients who were alive through day 14 were included. For the mortality, we compared the two treatment groups through day 28.
Subgroup analyses were performed for the primary endpoint and secondary endpoints to test the treatment effect across subgroups of age, gender, corticosteroid use, the baseline score of the seven-category ordinal scale at admission, and the time between onset to enrollment. The analyses were based on the proportional hazards regression with interaction terms of the treatment and subgroup variables. The results were reported in terms of hazard ratios, the 95% CI for each subgroup, and the interaction p-value. Further compressions and analyses of the secondary endpoints were applied using the chi-square or the Wilcoxon signed-rank tests whenever appropriate and indicated in result tables.
The analysis of the AEs data was descriptive and excluded patients who did not receive treatment. The proportions of the participants experiencing AEs were compared between the randomized groups using a Fisher's exact test.
No specific statistical handling of missing values was performed. Statistical significance was set at p < 0.05. Statistical analyses were done using SAS V9.4.
Stopping the Trial
The DSMB members voted for an early interim analysis after recruiting > 50% of the sample size based on the trial steering committee's concern about the low recruitment rate due to the decreased numbers of the new COVID-19 cases. On 26 January 2021, the DSMB conducted a closed session to review the interim data analysis and recommended aborting the trial because of futility. The patients' enrollment in the trial was stopped immediately, and the site's principal investigators, IRB, and Saudi FDA were notified of the termination.
Result
Patients
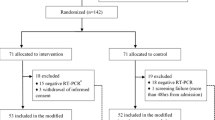
From 21 May 2020 to 26 January 2021, > 2800 patients were screened, of whom 268 were randomized. Participants were randomly assigned to the standard-of-care group (n = 136) or the standard-of-care plus the combination of favipiravir and hydroxychloroquine group (n = 132), of whom 254 participants (125 in the treatment group and 129 in the standard of care) were included in the primary outcome analysis. Seven patients withdrew consent, and another seven were not eligible and were excluded after randomization; all 14 patients did not receive treatment and were not included in the intention-to-treat population (Fig. 1).

Trial profile
The mean age was 52 years (± 13), and 103 (40.5%) patients were women. The two groups were balanced for most demographic characteristics (Table 1). A history of diabetes was present in 107 (42.13%) patients and hypertension in 104 (40.94%). At randomization, six (2.3%) patients were on invasive mechanical ventilation, 229 (90.15%) required supplemental oxygen, and 19 (7.48%) were receiving neither. Fever, dry cough, shortness of breath, fatigue, and gastrointestinal symptoms were the most common at the time of hospital presentation (Table S1). The mean time from symptom onset to starting study drug was around 5.85 (± 2.06) days.
Of the treatment group, 117 (93.6%) patients received > 10 of 20 doses of favipiravir, and 72 (57.6%) completed the whole course of therapy. The treatment was discontinued before completing 50% of the favipiravir course in eight patients for reasons other than death or hospital discharge. Among those eight patients, four refused to continue because of the pill burden, and medications were stopped by the primary physician in the other patients as a personal preference and because of lack of observed clinical improvement. Hydroxychloroquine was given for the full course days in 99 (79%) patients, and only 7 received < 5 of the 10 doses of the medication. Early hydroxychloroquine stopping was mainly due to ECG changes, or the patients/primary physician elected not to continue the rest of the course. One patient did not receive any doses of either medication and was not included in the safety analysis.
Antivirals were used in the control group as part of the local standard-of-care primary team management. Concomitant treatment, including antibiotics and systemic glucocorticoid therapy, were similar in the two groups (Table1). Requiring more than one antibiotic occurred more in the control group (Supplementary Table S2). The details of the antibiotics prescribed are available in the supplementary material (Table S3).
Efficacy
In both groups, 173 (68.11%) patients showed clinical improvement of at least 2 points on the seven-category ordinal scale (from the status at randomization) by day 14. The time to clinical improvement was not significantly different between the two groups, with a median of 9 (95% CI: 8, 12) days in the treatment group and 7 (95% CI: 6, 10) days in the control group (HR: 0.845; 95% CI 0.617–1.157; p-value = 0.29) (Fig. 2A). The distribution of the patient's status on the seven-category ordinal scale at day 14 is shown in Table 2. After 14 days of enrollment, in the treatment group, 76 patients were discharged from the hospital and 84 patients in the control group, while 8 patients died in the treatment group compared to 5 in the control group. The median duration of hospitalization among patients discharged on or before day 14 was 9 (95% CI 8, 12) days for the treatment group and 8 (95% CI 7, 10) days for the control group with HR = 0.877; 95% CI 0.638–1.207; p-value = 0.42 (Fig. 2B). Twenty-eight-day mortality was not different between the two groups; 9 (7.63%) died in the treatment group vs. 13 (10.32%) in the control group; p-value = 0.45 (Fig. 2C). All-cause 90-day mortality was also assessed and showed similar results; 14 (12.61%) died in the treatment group vs. 15 (13.76%) in the control group; p-value = 0.91 (Table 2).

A–C Kaplan-Meier estimates of cumulative recoveries. Time to clinical improvement in the intention-to-treat population (A) and the number of patients without clinical improvement were still included in the number at risk. The hospital discharge curves are cumulative incidence curves of alive hospital discharge (B). All the patients were followed up daily to discharge or 14 days after randomization. A patient was considered discharged from the hospital once discharged from the index hospitalization. Transferring to another hospital is not considered alive hospital discharge; rehospitalizations were not considered in this analysis, and deaths before day 14 were excluded. The survival curves are survival function (Kaplan-Meier) curves with a p-value calculated by the log-rank test. (C) Time to death in the intention-to-treat population. Patients were followed up for death until day 28 following randomization using in-hospital records and phone follow-up. Data on patients who died on or before day 28 were censored at day 28. Nine patients had unknown vital status at 28 days. There was no difference between the treatment and control groups in clinical improvement, time to discharge, or survival
For the 211 patients who were not in the ICU at recruitment, the incidence of ICU admission was not different in the treatment group compared with the control group [33 (26.40%) vs. 26 (20.16%), p-value = 0.23]. The incidence of new mechanical ventilation or ECMO use in the 248 patients who did not receive these interventions at enrollment was 21 (16.80%) in the treatment group and 20 (15.50%) in the control group, with a p-value of 0.77 (Table 2). No significant differences were observed between the two groups in the days from randomization to discharge and the incidence of hospital readmission. The RT-PCR for SARS-Cov-2 was positive for all patients prior to enrollment. Within 28 days, the rate of undetectable viral RNA in the treatment group was 25 (32.05%) and 23 (29.49%) in the control group. The follow-up PCR testing was not done in 98 patients, and only 12 patients were persistently positive by day 21.
We conducted a post hoc analysis to determine whether any subpopulation might have benefitted from receiving corticosteroids along with the combination therapy. However, it had no effect on the primary outcome or mortality. In the subgroup using the combination therapy within 5 days of the onset of symptoms, clinical improvement and the 28-day mortality were not significantly different between the groups (Fig. 3). The between-group differences in clinical improvement and mortality varied considerably according to baseline severity. There were no significant between-group differences concerning the disease complications outcomes of thromboembolism/coagulopathy, cardiac arrest, or acute kidney injury within 28 days (Table S4). Bacterial pneumonia and pneumothorax were documented more in the control group.

Time to clinical improvement according to subgroups
Safety
The AEs in the safety population are reported in Table 3. An elevation in the ALT levels was more frequent in patients receiving hydroxychloroquine plus favipiravir than in the standard-of-care group. ALT elevation in the treatment group was only mild or moderate, and levels improved with time. The bilirubin and AST levels were almost similar to those of the control group. One patient had grade 4 elevation in bilirubin, which returned to within the normal limits in the following days. Uric acid elevation was not a significant concern in the patients receiving favipiravir in our trial.
The prolongation of the QTc interval was more frequent in the patients receiving hydroxychloroquine plus favipiravir than in patients in the control group (Table 3); however, fewer patients in the control group had serial ECG studies performed during follow-up than did patients in the treatment groups. All 14 deaths during the observation period were judged by the site investigators to be unrelated to the intervention, and none was attributed to the treatment.
Discussion
This is the only randomized trial to assess the efficacy of a hydroxychloroquine and favipiravir combination therapy compared with standard of care in hospitalized patients with moderate-to-severe COVID-19. Our study showed that the combination of a 5-day course of hydroxychloroquine in addition to a 10-day course of favipiravir did not result in a better clinical outcome. Managing the COVID-19 pandemic is still challenging because of the lack of an antiviral agent with a mortality benefit. This finding was provided by the Solidarity Trial, investigating remdesivir, hydroxychloroquine, lopinavir, and interferon, in which all regimens had little or no effect on hospitalized patients with COVID-19 [12]. The finding in our trial is not an exception, and it confirms the absence of effective antiviral medication for SARS-CoV-2 even when combining two antivirals with favorable in vitro activity against SARS-CoV-2 [8, 13]. Remdesivir, has been licensed by the US FDA for emergency use in adults and pediatric patients for the treatment of COVID-19 requiring hospitalization, still an agent with conflicting effectivity reported in the literature [14,15,16].
In March 2020, when the trial was initiated, it was based on the promising in vitro results of favipiravir activity [9], and the initial broad exploration of the hydroxychloroquine effect in the pandemic [17, 18]. Although there was no difference in the primary or secondary endpoints between the treatment and standard-of-care groups, there was numerically less mortality on day 28, 9 (7.63%) in the treatment group vs. 13 (10.32%) in the control group, which was not statistically significant. There would be no expectation of additional expansion if the study was not stopped early for futility, especially with the 90-day mortality result recorded. Interestingly, examining the mortality rate reported in our trial, it has been nearly consistent between 10 and 15% in admitted patients with little variability compared to previously reported outcomes in severe COVID-19 cases [14,15,16]. The possible confounders, including steroid or tocilizumab [19], were studied in the subgroup analysis and did not reveal any new findings.
Examining the effect of the combination of hydroxychloroquine and favipiravir rather than alone for a possible additive effect was based on the initial studies which demonstrated activity against SARS-CoV-2 and the absence of a known drug interaction between the two medications, based on the PK-PD characteristics [13, 20, 21]. Currently, hydroxychloroquine is likely confirmed as not active and is probably associated with higher AEs in treating COVID-19 patients, especially with the higher dose regimen [22].
Although favipiravir had positive results in previous trials [23, 24], our trial is the largest to examine favipiravir's efficacy in the moderate-to-severe confirmed COVID-19 population, with no clinical benefit observed. In another large trial done comparing favipiravir to lopinavir/ritonavir regimen, there was no effect in reducing the number of ICU admissions, intubations, or in-hospital mortality [25]. In addition, it is undeniable that use of combination therapy was associated with a higher rate of AEs than in the control group. Stopping medications for possible cardiac toxicity has been shown in our trial, particularly with the concerns of using hydroxychloroquine [22, 26], limiting the possibility of exploring the same combination in the future again. Additionally, the favipiravir manufacturer suggests a dose of 1800 mg twice daily on day 1 and 800 mg twice daily for the following days based on pharmacokinetic studies to achieve plasma concentration equal to 60 µg/ml, which is higher than recommended for influenza virus [27]. This dosing regimen has resulted in 18 tablets of favipiravir alone on the first day and eight tablets daily for the following days. Despite the acceptable safety and tolerability profile of this dosing [10], the high pill burden was a major barrier to adherence.
This trial had several limitations. We used an open-label design without a placebo group, potentially leading to biases in patient care and data reporting. Second, the trial only included hospitalized patients, and findings may not be generalizable to other populations at the early stage of the disease. Another limitation is the high number of follow-up SARS-CoV-2 (RT-PCR) tests that were not obtained because of the limited resources and variable practices. Due to this limitation, the virologic outcomes, such as treatment effect on SARS-CoV-2 viral clearance, were not adequately measured. Lastly, our trial's premature termination could also have led to an increased data censoring related to the clinical outcome, but it would not change the overall finding.
Conclusion
In conclusion, the use of the hydroxychloroquine and favipiravir combination therapy plus standard of care did not achieve a higher efficacy than standard of care alone in patients hospitalized with moderate-to-severe COVID-19. Routine clinical use or further studies to evaluate this combination are discouraged for this patient population. Future studies to assess the efficacy of favipiravir in populations other than that of moderate-to-severe COVID-19 may be warranted.
References
World Health Organization. Coronavirus Disease (COVID-19) Situation Report. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports. Accessed 15 2021. (Published 15 April 2021).
Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. JAMA. 2020;323 (13):1239.
Rosenthal N, Cao Z, Gundrum J, Sianis J, Safo S. Risk factors associated with in-hospital mortality in a US national sample of patients with COVID-19. JAMA Netw Open. 2020;3 (12):e2029058.
Gupta S, Hayek SS, Wang W, et al. Factors associated with death in critically ill patients with coronavirus disease 2019 in the US. JAMA Intern Med. 2020;180 (11):1436.
Horby P, Lim WS, RECOVERY Collaborative Group, et al. Dexamethasone in hospitalized patients with Covid-19. N Engl J Med. 2021;384 (8):693–704.
Gordon AC, Mouncey PR, REMAP-CAP Investigators, et al. Interleukin-6 receptor antagonists in critically ill patients with Covid-19. N Engl J Med. 2021;384 (16):1491–502.
Delang L, Abdelnabi R, Neyts J. Favipiravir as a potential countermeasure against neglected and emerging RNA viruses. Antivir Res. 2018;153:85–94.
Furuta Y, Komeno T, Nakamura T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc Jpn Acad Ser B Phys Biol Sci. 2017;93 (7):449–63.
Chen C, Zhang Y, Huang J, et al. Favipiravir versus Arbidol for COVID-19: a randomized clinical trial [Internet]. Infect Dis (except HIV/AIDS). 2020. https://doi.org/10.1101/2020.03.17.20037432.
Udwadia ZF, Singh P, Barkate H, et al. Efficacy and safety of favipiravir, an oral RNA-dependent RNA polymerase inhibitor, in mild-to-moderate COVID-19: A randomized, comparative, open-label, multicenter, phase 3 clinical trial. Int J Infect Dis IJID Off Publ Int Soc Infect Dis. 2021;103:62–71.
Bosaeed M, Mahmoud E, Hussein M, et al. A Trial of Favipiravir and Hydroxychloroquine combination in Adults Hospitalized with moderate and severe Covid-19: a structured summary of a study protocol for a randomised controlled trial. Trials. 2020;21 (1):904.
Pan H, Peto R, WHO Solidarity Trial Consortium, et al. Repurposed antiviral drugs for Covid-19—interim WHO solidarity trial results. N Engl J Med. 2021;384 (6):497–511.
Yao X, Ye F, Zhang M, et al. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Clin Infect Dis. 2020;71 (15):732–9.
Beigel JH, Tomashek KM, Dodd LE, et al. Remdesivir for the treatment of Covid-19—final report. N Engl J Med. 2020;383 (19):1813–26.
Spinner CD, Gottlieb RL, Criner GJ, et al. Effect of remdesivir vs standard care on clinical status at 11 days in patients with moderate COVID-19: a randomized clinical trial. JAMA. 2020;324 (11):1048.
Wang Y, Zhang D, Du G, et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet Lond Engl. 2020;395 (10236):1569–78.
Boulware DR, Pullen MF, Bangdiwala AS, et al. A Randomized Trial of Hydroxychloroquine as Postexposure Prophylaxis for Covid-19. N Engl J Med. 2020;383 (6):517–25.
Rosenberg ES, Dufort EM, Udo T, et al. Association of treatment with hydroxychloroquine or azithromycin with in-hospital mortality in patients with COVID-19 in New York State. JAMA. 2020;323 (24):2493–502.
Salama C, Han J, Yau L, et al. Tocilizumab in patients hospitalized with Covid-19 pneumonia. N Engl J Med. 2021;384 (1):20–30.
Lemaitre F, Solas C, Grégoire M, et al. Potential drug–drug interactions associated with drugs currently proposed for COVID-19 treatment in patients receiving other treatments. Fundam Clin Pharmacol. 2020;34 (5):530–47.
Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30 (3):269–71.
Singh B, Ryan H, Kredo T, Chaplin M, Fletcher T. Chloroquine or hydroxychloroquine for prevention and treatment of COVID-19. Cochrane Database Syst Rev. 2021;2:CD013587.
Dabbous HM, Abd-Elsalam S, El-Sayed MH, et al. Efficacy of favipiravir in COVID-19 treatment: a multi-center randomized study. Arch Virol. 2021;166 (3):949–54.
Ivashchenko AA, Dmitriev KA, Vostokova NV, et al. AVIFAVIR for treatment of patients with moderate COVID-19: interim results of a Phase II/III multicenter randomized clinical trial. Clin Infect Dis Off Publ Infect Dis Soc Am. 2020;2:1176.
Solaymani-Dodaran M, Ghanei M, Bagheri M, et al. Safety and efficacy of Favipiravir in moderate to severe SARS-CoV-2 pneumonia. Int Immunopharmacol. 2021;95:107522.
Cavalcanti AB, Zampieri FG, Rosa RG, et al. Hydroxychloroquine with or without azithromycin in mild-to-moderate Covid-19. N Engl J Med. 2020;383 (21):2041–52.
Doni Y, Hibino M, Hase R, et al. A prospective, randomized, open-label trial of early versus late favipiravir therapy in hospitalized patients with COVID-19. Antimicrob Agents Chemother. 2020;64 (12):e01897-20.
Acknowledgements
We are grateful for funding for this trial through King Abdullah International Medical Research Center (KAIMRC), Ministry of National Guard—Health Affairs, Saudi Arabia. We also express our sincere gratitude to the site's team members for the great efforts and help to enroll participants (names are provided in Supplementary Appendix) and all the healthcare workers who took care of the patients during this trial. We also thank the DSMB members for their input and critical review of the data.
Funding
This work was funded by a grant from Abdullah International Medical Research Center, Saudi Arabia (RC20/17/R). The Journal’s Rapid Service Fee was funded by the authors.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
Study design: Mahmoud, A. Alharbi, H. Alqahtani, Alsaedy, AlShamrani, Aljeraisy, Bouchama, Arabi, Bosaeed. Implementation of the study, trial management or data collection: Altayib, Albayat, F. Alharbi, Ghalilah, Abdulm. Al Arfaj, AlJishi, Abdull. Alarfaj, Almaghaslah, Alyahya, Bawazir, AlEisa, M Alharbi, Al Johani, Alzahrani, Althaqafi, Almarhabi. Statistical analysis: Aldibasi, Bosaeed. Drafting the manuscript: Bosaeed, Mahmoud, A. Alharbi. Administrative, technical, or material support: Almutairi, Alotaibi, N. Alqahtani, Aljeraisy, Alaskar. All authors contributed to conducting the trial, critically reviewed and approved the final version of the manuscript.
Disclosures
Mohammad Bosaeed, Ebrahim Mahmoud, Ahmad Alharbi, Hadeel Altayib, Hawra Albayat, Faisal Alharbi, Khalid Ghalilah, Abdulmajid Al Arfaj, Jumana AlJishi, Abdullatif Alarfaj, Hajar Alqahtani, Badriah M. Almutairi, Manar Almaghaslah, Nawaf M. Alyahya, Abdullah Bawazir, Saud AlEisa, Abdulrahman Alsaedy, Abderrezak Bouchama, Malak Alharbi, Majid AlShamrani, Sameera Al Johani, Majed Aljeraisy, Mohammed Alzahrani, Abdulhakeem O. Althaqafi, Hassan Almarhabi, Athari Alotaibi, Nasser Alqahtani, Yaseen M. Arabi, Omar S. Aldibasi, and Ahmad Alaskar declare that they have nothing to disclose.
Compliance with Ethics Guidelines
Ethical approval was obtained from the Institutional Review Board (IRB) at the Ministry of National Guard-Health Affairs (MNGHA) and Ministry of Health (MOH). The trial was done according to the Declaration of Helsinki principles and the International Conference on Harmonization-Good Clinical Practice guidelines. All patients or legally acceptable representatives provided informed consent.
Data Availability
The study Statistical Analysis Plan is available with this publication as part of the supplementary material. The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. A data-sharing agreement will be needed.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Bosaeed, M., Mahmoud, E., Alharbi, A. et al. Favipiravir and Hydroxychloroquine Combination Therapy in Patients with Moderate to Severe COVID-19 (FACCT Trial): An Open-Label, Multicenter, Randomized, Controlled Trial. Infect Dis Ther 10, 2291–2307 (2021). https://doi.org/10.1007/s40121-021-00496-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-021-00496-6