
Abstract
Introduction
Immunocompromised patients infected with influenza exhibit prolonged viral shedding and higher risk of resistance. Optimized treatment strategies are needed to reduce the risk of antiviral resistance. This phase IIIb, randomized, double-blind study (NCT00545532) evaluated conventional-dose or double-dose oseltamivir for the treatment of influenza in immunocompromised patients.
Methods
Patients with primary or secondary immunodeficiency and influenza infection were randomized 1:1 to receive conventional-dose oseltamivir (75 mg adolescents/adults [≥ 13 years]; 30–75 mg by body weight in children [1–12 years]) or double-dose oseltamivir (150 or 60–150 mg, respectively), twice daily for an extended period of 10 days. Nasal/throat swabs were taken for virology assessments at all study visits. Co-primary endpoints were safety/tolerability and viral resistance. Secondary endpoints included time to symptom alleviation (TTSA) and time to cessation of viral shedding (TTCVS).
Results
Of 228 patients enrolled between February 2008 and May 2017, 215 (199 adults) were evaluable for safety, 167 (151 adults) for efficacy, and 152 (138 adults) for resistance. Fewer patients experienced an adverse event (AE) in the conventional-dose group (50.5%) versus the double-dose group (59.1%). The most frequently reported AEs were nausea, diarrhea, vomiting, and headache. Fifteen patients had post-baseline resistance, more commonly in the conventional-dose group (n = 12) than in the double-dose group (n = 3). In adults, median TTSA was similar between arms, while median TTCVS was longer with conventional dosing.
Conclusions
Oseltamivir was well tolerated, with a trend toward better safety/tolerability for conventional dosing versus double dosing. Resistance rates were higher with conventional dosing in this immunocompromised patient population.
Trial Registration
ClinicalTrials.gov identifier: NCT00545532.
Funding
F. Hoffmann-La Roche Ltd.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Oseltamivir is approved for the treatment of influenza in adults and children aged > 1 year in Europe and the United States, but there is concern about the development of oseltamivir resistance in immunosuppressed patients. |
As data from prospective, randomized studies of oseltamivir in immunocompromised patients are limited, we conducted a randomized phase IIIb study to evaluate the safety and tolerability of conventional-dose versus double-dose oseltamivir for the treatment of influenza in immunocompromised adults and children (> 1 year). |
What was learned from the study? |
Oseltamivir was well tolerated, with a trend toward a better safety/tolerability profile for conventional-dosing versus double-dosing in this immunocompromised patient population. |
This study, being the largest prospective study of a broad immunocompromised population and including the types of patients recommended for treatment in various national guidelines, serves to support long-standing recommendations for the use of oseltamivir in immunocompromised populations. |
Introduction
In immunocompromised patients, influenza infection is associated with substantial morbidity and mortality, including pulmonary and extrapulmonary complications and graft rejection [1, 2]. Reported rates of mortality in influenza-infected, untreated transplant recipients range from 25 to 40% [3], but appear to be lower in cohorts treated with antivirals [4,5,6]. Transplant recipients who are infected with influenza virus exhibit prolonged viral shedding, which is associated with a higher risk of resistance to antiviral drugs [5, 7]. Optimized treatment strategies are needed to reduce the risk of antiviral resistance and improve outcomes among immunocompromised patients.
Oseltamivir is a potent and selective inhibitor of influenza A and B neuraminidase enzymes [8] and is approved for the treatment of influenza in adults and children > 1 year in Europe and the United States [9, 10]. However, there is concern about the development of oseltamivir resistance in immunosuppressed patients, as prolonged viral replication and reduced immune-mediated virus clearance can result in a higher incidence of treatment-emergent drug-resistant viruses [11].
Data from prospective, randomized studies of oseltamivir in immunocompromised patients are limited. Therefore, we conducted a prospective, randomized phase IIIb study to evaluate the safety and tolerability of two dosing regimens and an extended 10-day dosing period for the treatment of influenza in immunocompromised adults and children (> 1 year), and to characterize oseltamivir resistance in this population.
Methods
Study Design
This randomized, double-blind, multicenter study (NCT00545532) investigated conventional-dose versus double-dose oseltamivir for the treatment of influenza in immunocompromised patients enrolled between February 2008 and May 2017.
Eligible adults (≥ 18 years) and children (1–17 years) had primary or secondary immunodeficiency and influenza infection confirmed by rapid test, reverse-transcription polymerase chain reaction (RT-PCR), or viral culture. Onset of influenza-like illness must have occurred ≤ 96 h before dosing. Patients with history, or symptoms suggestive of, renal failure were required to have creatinine clearance > 60 mL/min (> 18 years) or > 60 mL/min/1.73 m2 (< 18 years) within the last 3 months. Additional eligibility criteria are described in the Supplementary Methods.
Patients were randomized 1:1 to receive conventional-dose or double-dose oseltamivir and stratified according to transplant status (yes/no), time between symptom onset and treatment initiation (up to 96 h: ≤ 48, > 48 h), current influenza vaccination (yes/no), and age (≤ 12, > 12 years). Adults and adolescents (≥ 13 years) received 75 or 150 mg twice-daily (BID) oseltamivir. Children (1–12 years) received 30–75 mg BID (conventional) or 60–150 mg (double) oseltamivir according to body-weight (≤ 15 kg, 30 or 60 mg; > 15–23 kg, 45 or 90 mg; > 23–40 kg, 60 or 120 mg; > 40 kg, 75 or 150 mg). Patients receiving the conventional-dose received dose-matched placebo. All patients received oseltamivir BID as outpatients for 10 days at 12-h intervals followed by 30-day follow-up.
The study was performed in compliance with the principles of the 1964 Declaration of Helsinki and its amendments and in accordance with International Conference on Harmonization Good Clinical Practice guidelines. The study protocol was approved by all relevant institutional review boards and ethics committees. The primary independent ethics committee was Lithuanian Bioethics Committee Didzioji str. 22, Vilnius 01128, Lithuania. All patients or their legal guardians provided written informed consent before participating.
Assessments
Study visits took place on days 1, 2 or 3, 6, 8, 11, 15, and 40. Patients recorded influenza symptoms, temperature, and date/time of oseltamivir dose in diaries BID on days 1–10, and once daily thereafter. Adult and adolescent patients (≥ 13 years) completed a symptom scorecard comprising seven symptoms of influenza; parents/guardians of children < 13 years completed a diary comprising 18 symptoms of influenza, based on the Canadian Acute Respiratory Illness and Flu Scale (CARIFS) [12]. Safety assessments included adverse events (AEs), clinical laboratory evaluations, physical examination, vital signs and rejection and/or graft versus host disease. AEs were coded using Medical Dictionary for Regulatory Activities v.20.0 and tabulated according to severity and relationship to study treatment.
Two nasal and one throat swab were taken during all study visits for virology assessments and combined into one transport medium. All swabs were sent to a central laboratory (Viroclinics Biosciences, Rotterdam, The Netherlands) for RT-PCR, viral culture testing, assessment of influenza virus shedding, and viral resistance monitoring (see Supplementary Methods). The resistance monitoring plan included both phenotyping and population sequencing (genotyping) when possible, within the detection limit of both assays. Post-baseline (treatment-emergent) resistance assessment was performed using population sequencing and/or phenotyping assays. For some patients, resistance monitoring was incomplete (i.e., only phenotypic or genotypic resistance assessment were completed/available) or not performed (especially in the early influenza seasons of the study).
All viral isolates were cultured in Madin-Darby Canine Kidney cells and phenotyping was performed on all culture-positive samples. Phenotypic characterization of influenza isolates was performed by NA-Star assay (Applied Biosystems, Foster City, CA, USA) of cultured samples, and neuraminidase inhibition by oseltamivir was expressed in IC50 (concentration of drug required to inhibit neuraminidase activity by 50%). Definitions of phenotypic extreme outliers and phenotypic outliers were adapted from recent surveillance studies (Supplementary Methods) [13, 14].
Full-length Sanger population sequencing was performed for the hemagglutinin and neuraminidase genes after viral RNA extraction from the nasopharyngeal swab samples and RT-PCR. Sequencing was performed at least on the baseline sample and the last RT-PCR-positive sample, defined as the last sample with a cycle threshold < 32, indicating sufficient viral RNA. The presence of known oseltamivir resistance mutations (see Supplementary Methods) was assessed in both baseline and post-baseline samples.
Analysis Populations
Four analysis populations were defined. The intent-to-treat (ITT) population included all randomized patients; the ITT infected (ITTi) population included all randomized patients with confirmed influenza infection (excluding patients infected with oseltamivir-resistant influenza at baseline); the modified ITT infected (mITTi) population comprised those patients in the ITTi who received ≥ 1 dose of study drug. The safety population comprised all patients who received study drug and had a post-randomization safety assessment.
Safety and tolerability were analyzed in the safety-evaluable population, and resistance and efficacy (including rates of secondary illness) were analyzed in the mITTi population.
Analysis Method
Co-primary endpoints were safety/tolerability (AE profiles) and resistance development. Time to alleviation of all symptoms (TTAS), time to cessation of viral shedding (TTCVS), and the incidence of secondary illness were secondary endpoints.
Recruitment of 166 patients was planned, with ≥ 50 transplant recipients and ≥ 15 children. The sample size was chosen to provide an adequate number of patients with influenza A infections, in order to estimate rate of resistance and AEs with reasonable precision. Assuming that 90% of enrolled patients had laboratory-confirmed influenza, 75 patients would be needed per treatment arm to be evaluable for resistance and 83 patients per treatment arm to be evaluable for the assessment of safety. The number of influenza A virus-infected patients and rate of development of resistance was monitored in a blinded fashion.
Formal hypothesis testing was not performed; the study was designed to compare outcomes between treatment groups in terms of point estimates and confidence intervals (CIs). Median (95% CI) TTAS was determined for each treatment group. TTAS was defined as the time from treatment initiation to start of the 24-h period in which all seven influenza symptoms (adults/adolescents) or 18 CARIFS symptoms items (children) had scores ≤ 1 (mild or minor/no problem) and remained ≤ 1 for ≥ 21.5 h.
Results
Patients
Between February 2008 and May 2017, 843 patients were screened, with 228 enrolled into the study at 62 centers across 19 countries. Most participants were from the United States (n = 45), South Africa (n = 40), and Belgium (n = 33). Patients were randomized to receive conventional-dose (n = 113) or double-dose (n = 115) oseltamivir (Fig. 1). Eight patients were randomized but not dosed, and five patients had no post-randomization safety assessment, leaving 215 patients (199 adults) in the safety population (Supplementary Fig. 1). In addition, 45 patients were influenza negative, four had phenotypic baseline resistance, two had genotypic baseline resistance, and two were not immunocompromised, leaving 167 patients (151 adults) in the mITTi population.

Disposition of patients (mITTi, adults and children combined). Intent-to-treat infected population: all patients randomized and with central laboratory confirmation of influenza infection, excluding patients infected with oseltamivir-resistant influenza at baseline. mITTi population: all patients randomized to a particular treatment, regardless of whether they received that treatment or not, and received at least one dose of study drug and with central laboratory confirmation of influenza infection, excluding patients infected with oseltamivir-resistant influenza at baseline. mITTi modified intent-to-treat infected
Baseline demographic and disease characteristics were generally balanced between the treatment groups, with the exception of mean time from symptom onset to treatment, which was 47 h (conventional dose) and 54 h (double dose), respectively (Table 1). The conventional- and double-dose groups were generally balanced in terms of the proportion of patients infected with H1 (24.7% vs 19.8%), H3 (44.4% vs 46.5%), and influenza B subtypes (29.6% vs 31.4%, respectively). Five patients (4 adults aged > 18 years and 1 child < 13 years) had mixed influenza infections.
More adults in the double-dose group (62.4%) received concomitant immunosuppressive treatments compared with the conventional-dose group (54.1%). The most frequently received immunosuppressive treatments in adults were corticosteroids (38.8% conventional dose vs 41.6% double dose), and other immunosuppressants (37.8% vs 30.7%), including ciclosporin, mycophenolate mofetil, and tacrolimus. In children < 18 years, six patients (66.7%) in the double-dose group received concomitant immunosuppressive treatments compared with four patients (57.1%) in the conventional-dose group. The most frequently received immunosuppressive treatments were corticosteroids (2 patients [28.6%] conventional dose vs 3 patients [33.3%] double dose) and antimetabolites (1 patient [14.3%] vs 2 patients [22.2%]).
In adults, the most frequently received concomitant medications (> 30% of patients in either dose group) were corticosteroids (43.9% in the conventional-dose group vs 47.5% in the double-dose group). In children < 18 years, most patients received corticosteroids: four patients (57.1%) in the conventional-dose group and four patients (44.4%) in the double-dose group.
Safety and Tolerability
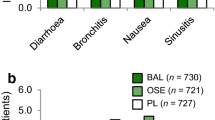
The overall safety profiles for conventional- and double-dose oseltamivir are shown in Table 2. The proportion of patients with AEs was lower in the conventional-dose group (50.5%) versus the double-dose group (59.1%). The most frequently reported AEs were nausea, diarrhea, vomiting, and headache (Table 2). Serious AEs (SAEs) were similar between groups (Supplementary Table 1).
The majority of on-treatment AEs in adults were mild to moderate in intensity. More adult patients in the conventional-dose group (13.2% [5/38]) had severe AEs compared with the double-dose group (6.4% [3/47]). Two adults (4.3% [2/47]) in the double-dose group had events that were considered life threatening and were reported as SAEs (bronchopulmonary aspergillosis and sepsis). No life-threatening events were reported with conventional dose oseltamivir. Pneumonia occurred in 4.1% (4/98) of adults in the conventional-dose group and in 1.0% (1/101) of adults in the double-dose group.
No severe or life-threatening events were reported in adolescents or children during the on-treatment phase. One child in the conventional-dose group and two children in the double-dose group experienced severe AEs. There were no reported cases of pneumonia in children < 18 years.
One adult patient in the double-dose group died during the study due to an off-treatment AE (Grade 5; recurrent leukemia). The event was considered unrelated to oseltamivir by the investigator. Two SAEs (renal failure and sepsis) in a single patient in the double-dose group were considered remotely related to treatment; all other SAEs were considered unrelated to treatment. All reported SAEs in adults had resolved by the end of the study, except for one event in the conventional-dose group (metastases to meninges [unresolved]) and three events in the double-dose group (recurrent leukemia [fatal], asthenia [unresolved], and sepsis [unresolved]).
Among adults (> 18 years), the incidence of secondary illnesses was similar in the double-dose group (5.1% [4/78]) and in the conventional-dose group (8.2% [6/73]) (see Supplementary Results).
Among adults in the ITTi population (n = 152), the proportion who required hospitalization between treatment initiation and the end of the study was comparable between treatment groups (6.8% [5 patients] in the conventional-dose group and 7.7% [6 patients] in the double-dose group). The median duration of hospitalization was also comparable between the treatment groups, at 7.0 days and 6.5 days, respectively. Among children < 18 years in the ITTi population (n = 17), only one patient in the conventional-dose group was hospitalized (for a period of 5 days) during the study. This patient experienced an event of oral herpes, which was considered unrelated to study treatment by the investigator.
Viral RNA Load
In adults, median baseline viral RNA load was similar in the conventional-dose (6.47 log10 vp/mL) and double-dose (6.52 log10 vp/mL) groups. Median change in viral RNA load from baseline to end of treatment (EOT) was − 3.5 log10 vp/mL in the conventional-dose group and − 3.0 log10 vp/mL in the double-dose group. At end of study, most patients were RT-PCR-negative. In children, median baseline viral RNA load was similar between the conventional-dose (5.9 log10 vp/mL) and double-dose (6.0 log10 vp/mL) groups. At EOT, only two patients in the conventional-dose group and one in the double-dose group were still influenza-positive by RT-PCR, so no meaningful comparison could be made.
Antiviral Resistance
Due to the emergence of naturally occurring oseltamivir resistance isolates during the first two influenza seasons of the trial (2008–09 and 2010–11), a H274Y mutation-specific RT-PCR was used to test baseline isolates, to help clinical sites determine the most appropriate treatment options for individual patients infected by H1N1 viral strains. Taken together, five adult patients (> 18 years) and one adolescent (aged 17 years) were infected at baseline with an oseltamivir-resistant virus; further characterization of baseline resistance is reported in the Supplementary Results.
Although resistance monitoring aimed to be as complete as possible, both assays have detection limits that can prevent analysis of samples for each time point for every patient. It was also necessary to have one baseline and at least one post-baseline sample to allow a patient to be analyzed for treatment-emergent resistance. Therefore, the incidence of post-baseline resistance was calculated only on patients for whom baseline resistance data as well as more than one post-baseline resistance assessment (genotypic or phenotypic) were available (Table 3). Treatment-emergent oseltamivir resistance was detected in 15 patients: 12 in the conventional-dose group (10 adults, 2 children) and three in the double-dose group (2 adults, 1 child) (Table 3). All 15 patients with treatment-emergent resistance had completed the full 10 days of study treatment; of these, 11 had received a transplant (5 solid organ transplant, 6 hematopoietic stem cell transplant [HSCT]).
Thirteen of 15 patients (87%) with treatment-emergent oseltamivir resistance were infected with influenza type A; eight were infected with the A/H1N1-subtype (1 A/H1N1, 7 A/H1N1-pdm09; 6 in the conventional-dose group; 2 in the double-dose group) and five with A/H3N2 (4 in the conventional-dose group; 1 in the double-dose group). Two adult patients in the conventional-dose group were infected with the influenza type B strain and had an outlier phenotype only (with a 2.3- and 2.7-fold increase in IC50 from the baseline mean); no population sequencing was performed. For most patients, oseltamivir resistance was detected for the first time at day 6 (8 patients) and day 8 (3 patients), with the last two patients harboring oseltamivir-resistant virus for the first time at day 11.
Efficacy and Impact of Resistance
The proportion of adults shedding virus at baseline was similar between the conventional- (100%) and double-dose (97.4%) groups. The proportion of patients continuing to shed virus at EOT was also similar in the conventional- (25.4%) and double-dose (21.9%) groups, and fell steadily to 1.5% of patients at the end of study. All children < 18 years were shedding virus at baseline, and similar to adults, by EOT the proportion of children shedding virus was 28.6% in the conventional-dose group and 14.3% in the double-dose group; all had stopped shedding virus by end of study on day 40. In adults, 10.6% of patients in the conventional-dose group and 9% of patients in the double-dose group were still influenza-positive (by RT-PCR) on day 15. Corresponding values in patients aged < 18 years, were 42.5% and 0%, respectively. Persistent shedding (viral load reduction < 1 log10 vp/mL at EOT compared with baseline) was observed in one child in the conventional-dose group and four adults in the double-dose group.
In adults aged ≥ 18 years, TTCVS was 23.9 h shorter in the double-dose group (154.1 h) than in the conventional-dose group (178.0 h), although 95% CIs overlapped. Furthermore, when patients with post-baseline resistance were excluded, TTCVS in adults was similar for the two dosing groups (Fig. 2a). Ten of 12 adult patients with a treatment-emergent resistant virus had prolonged viral shedding of > 10 days; of these, eight were in the conventional-dose group and were responsible for the longer TTCVS seen in that group. All had stopped shedding virus by end of study.

Evaluating the impact of treatment-emergent resistance on a time to cessation of viral shedding and b time to alleviation of symptoms (median [95% CI]; mITTi, adults only). All patients with baseline resistance were excluded from this analysis. *Patients who were still shedding virus (TTCVS)/symptomatic (TTAS) at end of study were censored from the analysis. CI confidence interval, mITTi modified intent-to-treat infected, TTAS time to symptom alleviation, TTCVS time to cessation of viral shedding
TTCVS in pediatric patients was similar between conventional- (n = 8) and double-dose groups (n = 8) (181.0 vs 180.5 h, respectively).
TTAS in adults aged ≥ 18 years was comparable between treatment groups (103.3 vs 103.6 h; Fig. 2b), with overlapping 95% CIs. Similar results were observed for pooled adult and adolescent (≥ 13 years) data (data not shown). In both groups, faster symptom resolution was observed in patients who received oseltamivir < 48 h after disease onset (median [95% CI] TTAS: conventional dose, 86.2 h [40.0, 107.2]; double dose, 74.0 h [45.4, 120.3]) versus patients who received oseltamivir > 48 h after disease onset (median [95% CI] TTAS: conventional dose, 122.7 h [44.2, 182.3]; double dose, 103.6 h [52.0, 169.7]). TTAS in the conventional-dose adult group was longer when patients with post-baseline resistance were included versus excluded (103 vs 95 h; Fig. 2b); however, 95% CIs overlapped.
In pediatric patients < 13 years, TTAS was longer in the double-dose versus conventional-dose group (115.9 vs 32.1 h), although interpretation is limited by small patient numbers.
Discussion
This prospective randomized study evaluated conventional-dose versus double-dose oseltamivir when given for an extended 10-day period for the treatment of influenza in immunocompromised adults and children. Notably, this is the largest prospective study in a population of immunocompromised patients with influenza. Conventional-dose oseltamivir showed a trend for slightly better tolerance versus double-dose oseltamivir. No remarkable AE findings were noted and the most frequently reported AEs were nausea, diarrhea, vomiting, and headache consistent with the established safety profile for oseltamivir. Other AEs observed reflected influenza complications, comorbidities, and co-treatments typical of this patient population. The incidence of secondary illnesses was similar in the double-dose group compared with the conventional-dose group.
Resistance was a co-primary endpoint and a lower resistance rate was observed in adult patients in the double-dose group (2.8%) than in the conventional-dose group (14.9%); a similar trend was seen in children, although the number of patients with resistance was small. Resistance rates observed for adults were higher than in historical studies in otherwise healthy (OwH) adult patients, but were within ranges previously reported for immunocompromised patients [15,16,17]. In immunocompromised children, resistance rates were of a similar level to those previously reported for immunocompetent young children (aged < 5 years), a population known to exhibit higher resistance rates than adults [18, 19]. Notably, most of the adults and children who developed resistance were transplant recipients. The rates of treatment-emergent resistance reported here, and in previous studies in patients with hematologic malignancy and recipients of an allogeneic HSCT, emphasize the significant unmet need for optimized treatments to improve outcomes for this vulnerable patient population [15, 20, 21]. Of note, previous studies of oseltamivir for the treatment of uncomplicated influenza reported no apparent clinical or virologic advantage of double- versus conventional-dosing [22,23,24].
Prolonged viral shedding is often reported in immunocompromised patients [17, 25] so evidence that a higher dose might reduce shedding would have been of interest. Although the study was not powered to assess any differences in efficacy between the conventional- and double-dose oseltamivir groups, TTCVS and TTAS were investigated as secondary objectives. In adults, median TTCVS was shorter in the double-dose versus conventional-dose group. Interestingly, when the patients who developed post-baseline resistance were excluded from the analyses, median TTCVS in adults was similar between treatment groups, suggesting that the difference in the overall adult population may be due to the larger number of patients in the conventional-dose group who developed treatment-emergent resistance and had corresponding prolonged viral shedding. In contrast, the clinical efficacy of the two dosage regimens, as measured by TTAS in adults, was similar, suggesting that doubling the oseltamivir dose is unlikely to provide an efficacy benefit over the conventional dose. Symptom resolution was faster in patients who received oseltamivir within 48 h compared with > 48 h (although 95% CIs overlapped), suggesting that early treatment with either conventional- or double-dose oseltamivir is favorable.
It was not possible to include a placebo control group in the present study for ethical reasons, as most treatment guidelines recommend oseltamivir for immunocompromised patients. While it is not recommended to make comparisons across clinical trials due to differences in trial design, to provide some context, we compared these data with clinical trials of OwH patients who were not immunocompromised [26,27,28,29]. Median TTAS for the pooled immunocompromised patients (both dose groups), appeared to be shorter versus OwH placebo-treated patients but longer versus OwH oseltamivir-treated patients. In addition, the 95% CI for treated immunocompromised patients overlapped substantially with those for OwH-treated patients, indicating that, while the median is higher, the range of duration of symptoms was similar, a finding that could be consistent with treatment-related reduction in TTAS in these immunocompromised patients. There are limitations in these comparisons—e.g., influenza studies conducted in different seasons, even for the same population, can yield differing measures of symptom or shedding duration. Furthermore, the 9-year study duration likely resulted in infection with viruses of varying pathogenicity and standards of care for immunocompromised patients may have changed over this period affecting the precision of the median estimates for duration of symptoms or virus shedding.
Current clinical practice guidelines recommend the use of oseltamivir in immunocompromised populations [30,31,32]. This study, being the largest prospective study of a broad immunocompromised population and including the types of patients recommended for treatment in various national guidelines, serves to support these long-standing recommendations. Although the study did not compare the longer 10-day dosing with the usual 5-day dosing, longer dosing did not appear to be associated with any increased tolerability burden and is already often recommended by treatment guidelines.
Conclusion
We report that oseltamivir was well tolerated, with a trend toward a better safety/tolerability profile for conventional- versus double-dosing in immunocompromised patients. The clinical impact of the higher resistance incidence is unknown; most patients cleared the infection by the end of treatment, and those who did not cleared the infection during the follow-up period.
References
Ison MG, Hayden FG. Viral infections in immunocompromised patients: what’s new with respiratory viruses? Curr Opin Infect Dis. 2002;15:355–67.
Kumar D, Michaels MG, Morris MI, et al. Outcomes from pandemic influenza A H1N1 infection in recipients of solid-organ transplants: a multicentre cohort study. Lancet Infect Dis. 2010;10:521–6.
Ison MG. Influenza, including the novel H1N1, in organ transplant patients. Curr Opin Infect Dis. 2010;23:365–73.
Ison MG, Sharma A, Shepard JA, Wain JC, Ginns LC. Outcome of influenza infection managed with oseltamivir in lung transplant recipients. J Heart Lung Transplant. 2008;27:282–8.
Nichols WG, Guthrie KA, Corey L, Boeckh M. Influenza infections after hematopoietic stem cell transplantation: risk factors, mortality, and the effect of antiviral therapy. Clin Infect Dis. 2004;39:1300–6.
Machado CM, Boas LS, Mendes AV, et al. Use of oseltamivir to control influenza complications after bone marrow transplantation. Bone Marrow Transplant. 2004;34:111–4.
Boivin G, Goyette N, Bernatchez H. Prolonged excretion of amantadine-resistant influenza a virus quasi species after cessation of antiviral therapy in an immunocompromised patient. Clin Infect Dis. 2002;34:E23–5.
Hayden FG, Atmar RL, Schilling M, et al. Use of the selective oral neuraminidase inhibitor oseltamivir to prevent influenza. N Engl J Med. 1999;341:1336–43.
Tamiflu® (oseltamivir phosphate) Summary of product characteristics, Roche. http://www.mhra.gov.uk/home/groups/comms-ic/documents/websiteresources/con046637.pdf. Accessed 26 June 2019.
Bohannon RW, Smith MB. Interrater reliability of a modified Ashworth scale of muscle spasticity. Phys Ther. 1987;67:206–7.
Ison MG, Gubareva LV, Atmar RL, Treanor J, Hayden FG. Recovery of drug-resistant influenza virus from immunocompromised patients: a case series. J Infect Dis. 2006;193:760–4.
Jacobs B, Young NL, Dick PT, et al. Canadian Acute Respiratory Illness and Flu Scale (CARIFS): development of a valid measure for childhood respiratory infections. J Clin Epidemiol. 2000;53:793–9.
Sheu TG, Deyde VM, Okomo-Adhiambo M, et al. Surveillance for neuraminidase inhibitor resistance among human influenza A and B viruses circulating worldwide from 2004 to 2008. Antimicrob Agents Chemother. 2008;52:3284–92.
Okomo-Adhiambo M, Sleeman K, Ballenger K, et al. Neuraminidase inhibitor susceptibility testing in human influenza viruses: a laboratory surveillance perspective. Viruses. 2010;2:2269–89.
Tramontana AR, George B, Hurt AC, et al. Oseltamivir resistance in adult oncology and hematology patients infected with pandemic (H1N1) 2009 virus, Australia. Emerg Infect Dis. 2010;16:1068–75.
Alonso M, Rodriguez-Sanchez B, Giannella M, et al. Resistance and virulence mutations in patients with persistent infection by pandemic 2009 A/H1N1 influenza. J Clin Virol. 2011;50:114–8.
van der Vries E, Stittelaar KJ, van Amerongen G, et al. Prolonged influenza virus shedding and emergence of antiviral resistance in immunocompromised patients and ferrets. PLoS Pathog. 2013;9:e1003343.
Stephenson I, Democratis J, Lackenby A, et al. Neuraminidase inhibitor resistance after oseltamivir treatment of acute influenza A and B in children. Clin Infect Dis. 2009;48:389–96.
Lina B, Boucher C, Osterhaus A, et al. Five years of monitoring for the emergence of oseltamivir resistance in patients with influenza A infections in the influenza resistance information study. Influenza Other Respir Viruses. 2018;12:267–78.
Hurt AC, Chotpitayasunondh T, Cox NJ, et al. Antiviral resistance during the 2009 influenza A H1N1 pandemic: public health, laboratory, and clinical perspectives. Lancet Infect Dis. 2012;12:240–8.
Iioka F, Sada R, Maesako Y, Nakamura F, Ohno H. Outbreak of pandemic 2009 influenza A/H1N1 infection in the hematology ward: fatal clinical outcome of hematopoietic stem cell transplant recipients and emergence of the H275Y neuraminidase mutation. Int J Hematol. 2012;96:364–9.
Dixit R, Khandaker G, Hay P, et al. A randomized study of standard versus double dose oseltamivir for treating influenza in the community. Antivir Ther. 2015;20:689–98.
South East Asia Infectious Disease Clinical Research Network. Effect of double dose oseltamivir on clinical and virological outcomes in children and adults admitted to hospital with severe influenza: double blind randomised controlled trial. BMJ. 2013;346:f3039.
Noel ZR, Bastin MLT, Montgomery AA, Flannery AH. Comparison of high-dose versus standard dose oseltamivir in critically ill patients with influenza. J Intensive Care Med. 2017;32:574–7.
Khanna N, Steffen I, Studt JD, et al. Outcome of influenza infections in outpatients after allogeneic hematopoietic stem cell transplantation. Transpl Infect Dis. 2009;11:100–5.
Treanor JJ, Hayden FG, Vrooman PS, et al. Efficacy and safety of the oral neuraminidase inhibitor oseltamivir in treating acute influenza: a randomized controlled trial. US Oral Neuraminidase Study Group. JAMA. 2000;283:1016–24.
Nicholson KG, Aoki FY, Osterhaus AD, et al. Efficacy and safety of oseltamivir in treatment of acute influenza: a randomised controlled trial. Neuraminidase Inhibitor Flu Treatment Investigator Group. Lancet. 2000;355:1845–50.
Martin C, Mahoney P, Ward P. Oral oseltamivir reduces febrile illness in patients considered at high risk of influenza complications. In: World Congress on Options for the Control of Influenza IV. Crete, Greece: Excerpta Medica. 2000:807–11.
Gibiansky L, Giraudon M, Rayner CR, et al. Population pharmacokinetic analysis of oseltamivir and oseltamivir carboxylate following intravenous and oral administration to patients with and without renal impairment. J Pharmacokinet Pharmacodyn. 2015;42:225–36.
Harper SA, Bradley JS, Englund JA, et al. Seasonal influenza in adults and children–diagnosis, treatment, chemoprophylaxis, and institutional outbreak management: clinical practice guidelines of the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:1003–32.
Engelhard D, Mohty B, de la Camara R, Cordonnier C, Ljungman P. European guidelines for prevention and management of influenza in hematopoietic stem cell transplantation and leukemia patients: summary of ECIL-4 (2011), on behalf of ECIL, a joint venture of EBMT, EORTC, ICHS, and ELN. Transpl Infect Dis. 2013;15:219–32.
Kumar D, Morris MI, Kotton CN, et al. Guidance on novel influenza A/H1N1 in solid organ transplant recipients. Am J Transplant. 2010;10:18–25.
Acknowledgements
We thank the investigators and patients who participated in this study.
Funding
This study was sponsored by F. Hoffmann-La Roche Ltd. F. Hoffmann-La Roche Ltd also funded the Rapid Service Fees.
Medical Writing and Editorial Assistance
Third-party medical writing assistance, under the direction of the authors, was provided by Nicole Antonio, of Gardiner-Caldwell Communications, and was funded by F. Hoffmann-La Roche Ltd.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Essack Mitha reports investigator fees paid by Roche during the conduct of the study. Frederique Jacobs was a consultant for MSD during the conduct of the study. Sally Alrabaa reports investigator fees paid by Roche during the conduct of the study. Sophie Le Pogam is an employee of Genentech and owns stock in F. Hoffmann-La Roche Ltd. Andrew Kenwright is an employee of and own stocks in Roche Products Ltd. Barry Clinch is an employee of and own stocks in Roche Products Ltd. Gergely Krivan, Arnon Nagler, Analia Mykietiuk and Loreta Vareikiene report no conflict of interest.
Compliance with Ethics Guidelines
The study was performed in compliance with the principles of the 1964 Declaration of Helsinki and its amendments and in accordance with International Conference on Harmonization Good Clinical Practice guidelines. The study protocol was approved by all relevant institutional review boards and ethics committees. The primary independent ethics committee was Lithuanian Bioethics Committee Didzioji str. 22, Vilnius 01128, Lithuania. All patients or their legal guardians provided written informed consent before participating.
Data Availability
Qualified researchers may request access to individual patient level data through the clinical study data request platform (www.clinicalstudydatarequest.com). Further details on Roche’s criteria for eligible studies are available here (https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Roche.aspx). For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here (https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm).
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Digital Features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.9977273.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any non-commercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Mitha, E., Krivan, G., Jacobs, F. et al. Safety, Resistance, and Efficacy Results from a Phase IIIb Study of Conventional- and Double-Dose Oseltamivir Regimens for Treatment of Influenza in Immunocompromised Patients. Infect Dis Ther 8, 613–626 (2019). https://doi.org/10.1007/s40121-019-00271-8
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-019-00271-8