
Abstract
Introduction
Deutetrabenazine is a vesicular monoamine transporter 2 inhibitor used to treat tardive dyskinesia (TD) and chorea associated with Huntington disease (HD). To enhance detection of safety signals across individual trials, integrated safety analyses of deutetrabenazine in TD and HD chorea were conducted.
Methods
For TD, safety data were integrated from two 12-week pivotal studies (ARM-TD and AIM-TD) and through week 15 of the open-label extension (OLE) study (RIM-TD). Data were analyzed by deutetrabenazine treatment group and placebo. For HD, safety data were integrated from the 12-week pivotal study (First-HD) and through week 15 of the OLE study (ARC-HD) for patients previously receiving placebo. Integrated deutetrabenazine data were compared with placebo from the pivotal study.
Results
For TD, deutetrabenazine (n = 384) was generally well tolerated compared with placebo (n = 130). Adverse event (AE) incidence was numerically higher in the response-driven deutetrabenazine vs the fixed-dose deutetrabenazine and placebo groups, respectively (any AE, 59.5% vs 44.4–50.0% and 53.8%; treatment-related AE, 38.1% vs 18.1–25.0% and 30.8%). Serious AEs were reported for 2.8–8.3% of patients in the deutetrabenazine groups and 6.9% in the placebo group. Common AEs (≥ 4%) included headache, somnolence, nausea, anxiety, fatigue, dry mouth, and diarrhea. AE incidence was higher during the titration vs maintenance periods. For HD, AE incidence was numerically higher with deutetrabenazine (n = 84) vs placebo (n = 45; any AE, 64.3% vs 60.0%; treatment-related AE, 38.1% vs 26.7%); serious AEs were reported for similar proportions for the deutetrabenazine and placebo groups, 2.4% and 2.2%, respectively. Common AEs (≥ 4%) included irritability, fall, depression, dry mouth, and fatigue.
Conclusions
Data from an integrated analysis of studies in TD and an integrated analysis of studies of chorea in HD showed that deutetrabenazine has a favorable safety profile and is well tolerated across indications.
Trial Registration
ClinicalTrials.gov identifiers, NCT02291861, NCT02195700, NCT01795859, NCT02198794, NCT01897896.
Plain Language Summary
Unintended movements are often the first sign of Huntington disease. This type of unintended movement is called chorea in Huntington disease. Tardive dyskinesia causes unintended body movements. Deutetrabenazine is a medicine used to treat both types of movements. This report summarizes deutetrabenazine safety across five clinical studies. Safety was assessed via adverse events (side effects). Adverse events were compared between deutetrabenazine and inactive treatment (placebo). Serious adverse events were also compared. Serious adverse events cause substantial impairment or disruption. In tardive dyskinesia and chorea in Huntington disease studies, most patients kept taking deutetrabenazine. Adverse events were not a common reason to stop treatment. For tardive dyskinesia, adverse event rates were similar between deutetrabenazine (≤ 60%) and placebo (54%). Serious adverse event rates were also similar for deutetrabenazine (≤ 8%) and placebo (7%). Adverse events tended to be reported earlier in treatment. Common adverse events were headache, sleepiness, nausea, anxiety, fatigue, dry mouth, and diarrhea. For chorea in Huntington disease, adverse event rates were similar for deutetrabenazine (64%) and placebo (60%). Serious adverse event rates were also similar for deutetrabenazine (2%) and placebo (2%). Irritability, fall, depression, dry mouth, and fatigue were common adverse events. Adverse events were similar between deutetrabenazine and placebo in both conditions. Deutetrabenazine was well tolerated for patients with either tardive dyskinesia or chorea in Huntington disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
Hyperkinetic movement from tardive dyskinesia (TD) and chorea associated with Huntington disease (HD) can have a severely negative impact on patient quality of life; deutetrabenazine is approved for the treatment of adults with these conditions. |
An integrated safety assessment from multiple pooled trials investigating deutetrabenazine in patients with TD or chorea in HD was conducted to gain additional insight into its adverse event (AE) profile. |
What has been learned from this study? |
Deutetrabenazine was well tolerated in adults with TD or chorea associated with HD. |
Although AEs may be initially reported for a higher proportion of patients during deutetrabenazine dose optimization (compared with placebo), proportions tended to decrease as patients continued treatment on a stable dose up to week 15. |
This integrated analysis of AEs provides additional information to help guide treatment decisions and discussions between physicians and their patients with TD or chorea associated with HD. |
Introduction
Tardive dyskinesia (TD) and Huntington disease (HD) are hyperkinetic movement disorders that negatively impact quality of life and can be socially stigmatizing [1,2,3]. TD is caused by dopamine receptor antagonists (DRAs). The estimated frequency of TD is approximately 20–30% for patients who use DRAs, but treatment and risk factors influence the development of TD [4,5,6,7]. TD is characterized clinically by varied types of involuntary movements, such as stereotypic or choreiform movements of the tongue, lower face, jaw, and extremities, although sometimes the pharyngeal, diaphragmatic, or trunk muscles are involved [1, 4, 8]. HD is a progressive disorder characterized by a triad of motor, cognitive, and psychiatric symptoms that impair a person’s activities and interactions [9, 10]. Chorea is a common motor manifestation of HD, affecting approximately 90% of patients [11].
Deutetrabenazine is a novel vesicular monoamine transporter 2 (VMAT2) inhibitor used for the treatment of TD and chorea associated with HD in adults [12]. The pivotal studies included two 12-week, double-blind, randomized, placebo-controlled trials of patients with TD (ARM-TD and AIM-TD) [13, 14] and a 12-week, double-blind, randomized, placebo-controlled trial of patients with HD (First-HD) [15]. Open-label extension (OLE) studies in TD (RIM-TD) and HD (ARC-HD) were also conducted to evaluate the long-term safety and efficacy of deutetrabenazine treatment [16, 17].
As individual clinical trials enroll relatively small samples of patients, some safety signals may not be readily apparent. By pooling and analyzing data from multiple trials, an integrated safety analysis can supplement individual trials to help uncover rare adverse events (AEs) or unexpected safety trends. This study is an integrated safety analysis of deutetrabenazine combining data across separate studies in TD (3 studies, 514 patients included) and HD (2 studies, 129 patients included) to further assess the safety of deutetrabenazine and identify any potentially emerging safety signals within the first 12–15 weeks of exposure. Safety data beyond week 15 from the OLE studies have been published previously [16, 17] and were not included in these integrated safety analyses. Because of differences between patients with HD and patients with TD, data from each indication were analyzed separately but presented side by side, because physicians may find it informative when considering potential risks of treatment.
Methods
Ethics approval and informed consent were not required for this integrated analysis as they were obtained for the original clinical studies previously published [13,14,15,16,17], which were in accordance with the Declaration of Helsinki.
Tardive Dyskinesia Study Designs
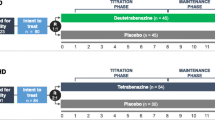
Three studies evaluated the efficacy, safety, and tolerability of deutetrabenazine in patients with TD: two 12-week pivotal studies (ARM-TD and AIM-TD) and one long-term OLE (RIM-TD) (Fig. 1). ARM-TD was a 12-week trial in which patients were randomized (1:1) to receive either response-driven dosing of deutetrabenazine (starting at 12 mg/day with weekly increments of 6 mg/day until adequate dyskinesia control, a clinically significant AE, or maximal allowable dose of 48 mg/day) or placebo [13]. AIM-TD was a 12-week trial in which patients were randomized (1:1:1:1) to receive either fixed-dose deutetrabenazine (12 mg/day, 24 mg/day, or 36 mg/day) or placebo [14]. Both studies included a 1-week washout period with a follow-up visit after the 12-week treatment period was completed. Patients who completed either ARM-TD or AIM-TD were eligible to enroll in the RIM-TD study to receive deutetrabenazine using a response-driven dosing regimen (maximum daily dosage of 48 mg with a body weight < 100 kg or 60 mg with a body weight ≥ 100 kg) over 3 years [14, 16].

Study design
Data were integrated from the 12-week study periods of the two pivotal studies and through week 15 of the RIM-TD study for patients who received placebo in the parent study. Data were analyzed by treatment group and included fixed-dose deutetrabenazine (12 mg/day, 24 mg/day, 36 mg/day) or response-driven deutetrabenazine (12 mg/day to 48 mg/day) compared with placebo. The response-driven deutetrabenazine group included pooled data from patients who received deutetrabenazine in the ARM-TD study and patients in the RIM-TD study who were previously in the placebo arm of either ARM-TD or AIM-TD and received their first dose of deutetrabenazine in RIM-TD. Patients who received placebo in the parent study contributed to the summaries both under placebo and under the OLE. Integrated data were also analyzed to compare safety outcomes during the titration vs maintenance periods. In the fixed-dose pivotal study (AIM-TD), the titration period was 4 weeks; in the ARM-TD study and the RIM-TD study, the titration period was 6 weeks, thus the maximum allowable daily dosage in the titration phase was 42 mg. The maintenance period evaluated for this analysis was 6–9 weeks.
Huntington Disease Study Designs
First-HD was a 12-week trial that randomized patients (1:1) to receive either response-driven dosing of deutetrabenazine (maximum daily dosage of 48 mg) or placebo to evaluate the efficacy and safety of deutetrabenazine in patients with HD [15]. ARC-HD was a single-arm study designed to evaluate the long-term safety of deutetrabenazine (maximum daily dosage 48 mg) in patients with HD in two cohorts of patients: the rollover cohort and the switch cohort [17]. The rollover cohort included patients who completed First-HD and underwent a 1-week washout prior to initiating deutetrabenazine in the long-term safety study. The switch cohort included patients who were receiving a stable dose of tetrabenazine (outside of a clinical trial) and were converted overnight to deutetrabenazine at a dose predicted to provide comparable systemic exposure.
Data were integrated upon first exposure to deutetrabenazine through week 12 of the First-HD study and through week 15 of the ARC-HD study for patients in the rollover cohort who previously received placebo in the First-HD study, in order to align with the 12-week exposure in the pivotal trial (Fig. 1). Integrated safety data for deutetrabenazine from both the First-HD and ARC-HD studies were compared with the placebo arm in First-HD. Patients from the placebo arm of First-HD who enrolled in ARC-HD contributed data to summaries under both placebo in the parent study and deutetrabenazine in the long-term safety study for these integrated analyses. Integrated data were also analyzed to compare safety outcomes during the titration vs maintenance periods. The titration period was 8 weeks in First-HD and ARC-HD, thus the maximum allowable daily dosage in the titration phase was 48 mg. The maintenance period was evaluated only up to 7 weeks from the long-term safety study.
Safety Assessments
Safety was assessed via AE, vital signs, and laboratory parameters. Serious adverse events cause substantial impairment or disruption. AEs of interest in these populations were analyzed using Standardized Medical Dictionary for Regulatory Activities (MedDRA) Queries (SMQs) for depression, suicidality/self-injury, akathisia, Parkinson-like events, and Torsades de pointes/QT prolongation (Supplementary Table S1), or selected preferred terms, including somnolence and sedation. For patients with TD, parkinsonism was further assessed using the Unified Parkinson’s Disease Rating Scale (UPDRS) motor score, which consists of 27 tasks, each rated on a 5-point scale (0–4), with higher values indicating increasing severity. For patients with HD, parkinsonism was assessed using the Unified Huntington’s Diseases Rating Scale (UHDRS) parkinsonism subscale, which is the sum of scores for finger tapping, pronation/supination of the hands, rigidity of the arms, bradykinesia, and retropulsion pull test; the UHDRS has a range of 0–40, and a higher score indicates greater parkinsonism. Observed electrocardiogram (ECG) parameters and shifts from screening for clinically significant abnormal findings were also assessed, including the number of patients having an on-treatment Fridericia’s corrected QT interval (QTcF) > 450 ms, > 480 ms, or > 500 ms, or a change from baseline in QTcF of > 30 ms or > 60 ms. Suicidal ideation, suicidal behavior, and self-injurious behavior without suicidal intent were assessed using the Columbia Suicide Severity Rating Scale (C-SSRS). The C-SSRS is a US Food and Drug Administration (FDA)-endorsed questionnaire where affirmative responses to questions indicate either suicidal ideation or suicidal behavior (Supplementary Table S2). Depression was assessed using the Hospital Anxiety and Depression Scale (HADS) depression subscale score with seven items; higher scores reflect greater frequency or severity of depression symptoms over the preceding week.
Statistical Analysis
Demographic and baseline characteristics were summarized using descriptive statistics, counts, or proportions. AEs were coded using MedDRA version 17.0. Incidences of AEs were summarized using descriptive statistics by system organ class, preferred term, and SMQ. Patients were counted only once in each applicable system organ class category, SMQ category, and/or preferred-term category. Treatment-related AEs were defined as those with possible, probable, definite, or missing relationships to study drug.
Results
Safety Assessments in Patients with Tardive Dyskinesia
Patients
In the TD analyses, patients received either fixed-dose (12 mg/day, 24 mg/day, or 36 mg/day) or response-driven deutetrabenazine (mean dose, 38.4 mg/day), or placebo. Across groups, the majority of patients were female and White (Table 1). Comorbid psychotic disorders were reported for a numerically higher proportion of patients compared with mood disorders (Table 1). Approximately half of patients were receiving concomitant antidepressant therapy, and more than three-quarters were receiving DRAs (Table 1). Mean body mass index (BMI) ranged from 27.8 to 30.5 kg/m2 across groups (Table 1). The proportions of patients who discontinued treatment prior to week 12 or week 15 were 11%, 12%, and 14% in the fixed-dose deutetrabenazine groups (12 mg/day, 24 mg/day, and 36 mg/day, respectively), 10% in the response-driven deutetrabenazine group, and 11% in the placebo group.
Incidence of AEs
In this integrated analysis of patients with TD, deutetrabenazine was generally well tolerated compared with placebo. Among patients receiving fixed-dose deutetrabenazine or response-driven deutetrabenazine vs placebo, the incidence of any AE ranged from 44.4% to 59.5% across deutetrabenazine groups vs 53.8% in the placebo group, and the incidence of serious AEs was 2.8–8.3% vs 6.9% (Fig. 2a). Treatment-related AEs were reported in 15.3–25.0% of patients receiving fixed-dose deutetrabenazine, 38.1% of patients receiving response-driven deutetrabenazine, and 30.8% of patients receiving placebo. The incidence of AEs leading to discontinuation from the study ranged from 2.8% to 5.6% across deutetrabenazine groups and was 3.1% in the placebo group. Common AEs across groups were somnolence, headache, anxiety, diarrhea, fatigue, nausea, and insomnia (Table 2).

a Overall AEs and b AEs of interesta in the TD group during the overall treatment period. AE adverse event, MedDRA Medical Dictionary for Regulatory Activities, SMQ standardized MedDRA queries, TD tardive dyskinesia. aAEs are based on SMQs defined in Supplementary Table S1
The incidence of AEs of interest (analyzed by SMQs), including depression, suicidality/self-injury, akathisia, Parkinson-like events (preferred terms reported in this study: bradykinesia, cogwheel rigidity, drooling, gait disturbance, hypokinesia, musculoskeletal stiffness, parkinsonian gait, parkinsonism, tremor), and Torsade de pointes/QT prolongation, was generally low across groups (Fig. 2b). The incidence of AEs by preferred term was also generally low, except for the combined somnolence and sedation terms, which were reported by 12.5% of patients in the response-driven deutetrabenazine group vs 6.9% of patients in the placebo group. Parkinson-like events were reported for 1.4% of patients in the 12 and 24 mg/day groups, 6.0–6.9% of patients in the 36 mg/day and response-driven groups, and 1.5% of patients in the placebo group. Of note, AEs associated with the SMQ of Torsade de pointes/QT prolongation were reported for four patients (1 [1.4%] each in the fixed-dose deutetrabenazine 12 mg/day, 24 mg/day, and 36 mg/day groups and 1 [0.8%] in the placebo group); however, none of these events categorized using this SMQ were AEs with preferred term of Torsade de pointes. Data for the QTcF interval for overall treatment period are summarized in Supplementary Table S3. Overall, 6–13% of patients in the deutetrabenazine groups compared with 9% of patients in the placebo group experienced a QTcF > 450 ms; 0–3% of patients in the deutetrabenazine groups and < 1% of patients in the placebo group experienced a QTcF of either > 480 ms or > 500 ms. Approximately 6–13% of patients across groups experienced a QTcF change > 30 ms, and one patient each in the response-driven deutetrabenazine and placebo groups experienced a QTcF change > 60 ms. The onset, resolution status, and mean duration of AEs of interest are provided in Supplementary Table S4.
Safety During the Titration vs Maintenance Periods
Among patients with TD, the incidence of any AE was higher during titration vs maintenance periods across all groups (fixed-dose deutetrabenazine: 12 mg/day, 33.3% vs 27.8%; 24 mg/day, 36.1% vs 22.2%; 36 mg/day, 38.9% vs 29.2%; response-driven deutetrabenazine: 49.4% vs 32.7%; placebo: 43.1% vs 25.4%). Similarly, the incidence of serious AEs, treatment-related AEs, and AEs leading to discontinuation of study drug was generally higher during the titration period vs maintenance period (Fig. 3a). The most common AEs (reported for ≥ 4% of patients in any group) during the titration period were somnolence, headache, diarrhea, fatigue, dry mouth, hypertension, nasopharyngitis, depression, and nausea; only 1 AE (headache) met this threshold during the maintenance period (Fig. 3b). Rates of somnolence and sedation and Torsade de pointes were higher in the titration period than in the maintenance period, but other specific AEs were low and comparable during the titration and maintenance periods (Table 3).

a Overall AEs and b common AEs (≥ 4%) in the TD group by preferred term during the titration period and the maintenance period. In ARM-TD, titration was from the first dose of treatment until the time point before the first dose of study drug received at the week 6 visit (up to 42 mg/day) and maintenance was from the first dose time point of treatment received at the week 6 visit until the week 12 visit. In AIM-TD, dose escalation was from the first dose of treatment until the day before the week 5 telephone contact or until randomized dose was achieved and maintenance was from the day of the week 5 telephone contact until the day of the week 12 visit. In OLE, titration was from the first dose of treatment until 7 days after the week 6 visit (up to 42 mg/day) and maintenance (for this analysis) was from 8 days after the week 6 visit until the day of the week 15 visit. AE adverse event, DTBZ deutetrabenazine, OLE open-label extension, TD tardive dyskinesia
Metabolic Parameters
There were no notable changes in BMI among patients who received deutetrabenazine vs placebo (Supplementary Fig. S1A). No meaningful changes in serum glucose, serum cholesterol, or serum triglycerides were observed across groups (Supplementary Fig. S1B). Of note, the proportion of patients taking blood glucose-lowering drugs (excluding insulins) at baseline ranged from 13% to 18% across groups. Patients taking insulins and insulin analogues at baseline ranged from 1% to 10%, and the percentage of patients taking lipid-modifying agents at baseline ranged from 19% to 28%.
Other Safety Assessments
Patient-reported assessments did not suggest an increase in depression or suicidality with deutetrabenazine treatment. In addition to the passive collection of AEs for suicidality/self-injury, patients were actively assessed via C-SSRS for suicidal ideation or behavior. At any time post-baseline, suicidal ideation was reported for 1 (1%), 4 (6%), 2 (3%), 3 (2%), and 5 (4%) patients in the fixed-dose deutetrabenazine 12 mg/day, 24 mg/day, and 36 mg/day, response-driven deutetrabenazine, and placebo groups, respectively. Suicidal intent was only reported for 2 (3%) patients in the deutetrabenazine 24 mg/day group. Suicidal behavior was reported for 1 (1%) patient in the fixed-dose deutetrabenazine 24 mg/day group and 1 (< 1%) patient in the placebo group, and self-injurious behavior without suicidal intent was reported in 1 (1%) patient in the fixed-dose deutetrabenazine 24 mg/day group. In addition, changes from baseline in the depression subscale of the HADS were − 0.5, − 0.5, 0.6, 0.2, and − 0.4, and likely not clinically meaningful, with fixed-dose deutetrabenazine 12 mg/day, 24 mg/day, and 36 mg/day, response-driven deutetrabenazine, and placebo, respectively. Mean (SD) baseline UPDRS motor scores were 7.2 (8.54), 9.9 (12.14), 10.6 (12.60), 8.0 (9.83), and 9.7 (10.47) for fixed-dose deutetrabenazine 12 mg/day, 24 mg/day, and 36 mg/day, response-driven deutetrabenazine, and placebo, respectively. There was no worsening of UPDRS motor score observed across groups. UPDRS motor scores decreased in each group (indicating improvement); however, changes were not clinically meaningful (− 2.9, − 2.6, − 1.9, − 0.7, and − 3.2 with fixed-dose deutetrabenazine 12 mg/day, 24 mg/day, and 36 mg/day, response-driven deutetrabenazine, and placebo, respectively) [18].
Safety Assessments in Patients with Huntington Disease
Patients
The majority of patients with HD in both treatment groups were male and White (Table 4). Mean baseline BMI was 25.5 kg/m2 in the deutetrabenazine group and 26.0 kg/m2 in the placebo group (Table 4). The majority of patients were receiving concomitant medications at baseline, with more than half of patients in both groups receiving concomitant antidepressant therapy (Table 4). Of note, patients using antipsychotics at baseline were not eligible to participate in either study; however, a small proportion of patients (approx. 5%) received antipsychotics during the trial, and these were considered protocol deviations.
Incidence of AEs
In the HD analyses, at least one AE was reported for 54 (64.3%) patients receiving deutetrabenazine and 27 (60.0%) patients receiving placebo (Fig. 4a). The incidences of serious AEs were 2.4% and 2.2% and the incidences of AEs leading to discontinuation from the study were 1.2% and 2.2% with deutetrabenazine and placebo, respectively (Fig. 4a). The incidence of treatment-related AEs was higher in patients receiving deutetrabenazine vs placebo (38.1% vs 26.7%) (Fig. 4a). The most common treatment-emergent AEs included fall, somnolence, insomnia, irritability, and dry mouth (Table 5). The incidence of AEs of interest (analyzed by SMQs or selected preferred terms) was generally low, regardless of treatment group (Fig. 4b). Parkinson-like events were reported for 5 (6.0%) patients in the deutetrabenazine group and none in the placebo group (Fig. 4b). No patients in either group reported AEs associated with the SMQ of Torsade de pointes/QT prolongation (Fig. 4b). The onset, resolution status, and mean duration of AEs of interest are provided in Supplementary Table S5.

a Overall AEs and b AEs of interesta in the HD group during the overall treatment period. AE adverse event, HD Huntington disease, MedDRA Medical Dictionary for Regulated Activities. aAEs are based on standardized MedDRA queries defined in Supplementary Table S1
Safety During the Titration vs Maintenance Periods
The incidence of any AE was higher during the titration vs maintenance periods in patients with HD (deutetrabenazine, 57.1% vs 28.9%; placebo, 55.6% vs 20.0%) (Fig. 5a). Similarly, the incidences of treatment-related AEs and AEs leading to discontinuation of study medication were generally higher in both groups during the titration period vs the maintenance period, but incidence of serious AEs was similar for deutetrabenazine and higher in the placebo group during the maintenance period (Fig. 5b–d). Rates of specific treatment-emergent AEs were < 5% and comparable during the titration and maintenance periods, except for somnolence and sedation, which was 9.5% in the titration period for the deutetrabenazine group, and Parkinson-like events, which only occurred in the deutetrabenazine group (Table 6).

a–d AEs in the HD group during titration period and maintenance periods
Metabolic Parameters
Patients in the deutetrabenazine group experienced an increase from baseline in BMI compared with patients in the placebo group, who had a modest decrease in BMI (Supplementary Fig. S2). At baseline, there was one patient (1.2%) in the deutetrabenazine group and no patients in the placebo group who received blood glucose-lowering drugs (i.e., metformin). The proportion of patients taking lipid-modifying agents at baseline was 19.0% and 26.6% among patients receiving deutetrabenazine and placebo, respectively.
Other Safety Assessments
Separate from the passively collected AEs, active C-SSRS assessment showed no indication of suicidal ideation, suicidal behavior, or self-injurious behavior without suicidal intent in any patient at any time point post-baseline in the deutetrabenazine group; in the placebo group, one patient had suicidal ideation and no patients had suicidal behavior or self-injurious behavior without suicidal intent. Baseline mean (SD) in the depression subscale of the HADS was 2.2 (2.50) with deutetrabenazine and 3.2 (2.57) with placebo, and mean (SD) changes from baseline to week 12/15 were considered not clinically meaningful (− 0.1 [2.30] and − 0.4 [2.30], respectively). Baseline and change from baseline mean (SD), which were not clinically meaningful, in the parkinsonism subscale of the UHDRS for the sum of the finger taps, pronate/supinate, rigidity, bradykinesia, tandem walking, and retropulsion pull test scores were 10.4 (5.65) and − 0.5 (3.50) with deutetrabenazine and 11.7 (5.64) and − 0.5 (3.05) with placebo, respectively.
Discussion
Treatment options previously available to manage hyperkinetic movement disorders, including TD and HD, were limited, and some have undesirable side effects that may make them less appealing. It is important for clinicians to be aware of the safety and tolerability profiles of deutetrabenazine in both TD and HD; however, non-specialists may be less aware of the safety profile of deutetrabenazine and how it compares with other agents. These integrated safety analyses used multiple studies to provide a collective perspective on safety upon first exposure to deutetrabenazine in TD and HD within the first 12–15 weeks of exposure to deutetrabenazine, when AEs are most likely to occur. Analysis of complete long-term safety data has been previously published [16, 17].
In these analyses, treatment with deutetrabenazine was shown to be generally well tolerated across studies for both indications. In the separate analyses of patients with TD or HD, the incidence rates of any AE, serious AEs, and AEs leading to discontinuation were generally comparable between deutetrabenazine treatment arms and placebo. Patients with TD in the response-driven deutetrabenazine group had a numerically higher incidence of any AE and treatment-related AEs compared with the fixed-dose deutetrabenazine and placebo groups, but there was no meaningful increase in study drug discontinuation in the response-driven deutetrabenazine group. In patients with HD, the incidence of treatment-related AEs was numerically higher among patients receiving deutetrabenazine than placebo, but there was no increase in study drug discontinuation attributable to AEs. The lack of increase in study discontinuation despite the increase in AEs with higher doses can be interpreted as the benefit seen with improved motor control in a response-driven manner outweighing the adverse experience in patients who experienced AEs.
A potential concern for patients and clinicians is whether certain AEs emerge after a stable dose of deutetrabenazine is achieved. Analysis of AEs during titration and maintenance periods in both TD and HD demonstrated that deutetrabenazine was generally well tolerated, with AE rates similar to placebo during both phases. Furthermore, AE rates were generally higher during titration and decreased during maintenance. These data demonstrate that AEs occurring early after treatment initiation tend to resolve after achieving a stable dose, which may be related to dose adjustments. These data provide important context for the dosing schedule to providers and patients and reassurance that most initial AEs are transient in nature.
The incidences of AEs of interest (analyzed by SMQs or selected preferred terms) were generally low across groups for both TD and HD. In particular, Parkinson-like events (per the SMQ that included preferred terms bradykinesia, cogwheel rigidity, drooling, gait disturbance, hypokinesia, musculoskeletal stiffness, parkinsonian gait, parkinsonism, and tremor) were reported for higher proportions of patients in the higher-dose deutetrabenazine groups vs placebo or low-dose deutetrabenazine groups for patients with TD, and was higher in deutetrabenazine vs placebo for patients with HD. Additionally, UPDRS motor score improvements tapered off with higher doses of deutetrabenazine among patients with TD; UHDRS parkinsonism subscale scores were similar among patients with HD treated with deutetrabenazine and placebo. However, Parkinson-like events are a class-wide AE for VMAT2 inhibitors, as VMAT2 inhibition results in dopamine depletion in the nerve terminal [19, 20]. Treatment decisions should use a patient-first, holistic approach that considers the risks and the benefits of available treatments.
Data from this analysis show no difference in the change in QTcF between deutetrabenazine and placebo in patients with TD, which supports that the degree of QT prolongation is not clinically meaningful for the recommended dosage range. Importantly, there were no reports of preferred-term AEs of Torsade de pointes in patients with TD despite most patients receiving concomitant DRAs.
In patients with TD, the incidence of depression defined by the SMQ was numerically higher in patients receiving deutetrabenazine than placebo, but there were no notable differences in HADS depression subscale scores across groups. Separately, AEs in the SMQ of suicidality/self-injury were generally low across groups. In addition, C-SSRS data indicated that suicidal ideation was generally low but comparable across groups and suicidal and self-injurious behavior was infrequent. The incidence of depression commonly associated with HD was low with treatment during the study time frame; there were no notable differences between groups in patients with HD. AEs were passively collected (without a prompt), while assessments such as C-SSRS and HADS actively collect data on these symptoms, and therefore may be more sensitive. This analysis of the safety of deutetrabenazine treatment and a cross-sectional study of the Enroll-HD database [21] suggest AEs of depression and suicidality are not common with either agent, despite the boxed warnings for both tetrabenazine [20] and deutetrabenazine [12] products.
Deutetrabenazine treatment did not appear to substantially affect BMI or other metabolic parameters. Notably, BMI changes in both patient populations were modest compared to those seen with other medications commonly used in these populations (e.g., antipsychotics), and these changes did not appear to affect other indicators of metabolic health, such as cholesterol, glucose, or triglycerides in patients with TD. Importantly, patients with HD often experience weight loss [22], and, in this regard, weight gain may be desirable in patients with HD. Therefore, a modest amount of weight gain, as observed in this analysis, should not be perceived as an adverse safety outcome for these patients.
Limitations
Integrated safety analyses increase the sample size by combining studies that may have different designs (inclusion criteria, prior medication exposure, presence of placebo control, masking, duration between office visits). Even with the sample sizes in these analyses, AEs were expected in a small number of patients. Therefore, these analyses could not be powered for statistical analyses.
Conclusion
Overall, the safety of deutetrabenazine treatment was demonstrated in patients with TD and HD taking a broad range of concomitant medications and with underlying health conditions. Although some antipsychotics have been developed to reduce the risk, TD continues to develop in some patients. Modification of the antipsychotic agent or dose may reduce TD symptoms; however, VMAT2 inhibitor treatment may allow for uninterrupted use of concomitant DRAs for underlying psychiatric conditions. Furthermore, the benefits of treatment appear to be meaningful to patients who may experience initial AEs while optimizing deutetrabenazine dose, as patients who continued treatment on a stable dose up to week 15 tend to report fewer side effects. By integrating safety data across separate studies in patients with TD and HD, this study provides important evidence for clinicians and patients that may inform treatment decisions and help anticipate potential safety concerns upon first exposure to deutetrabenazine and throughout the course of treatment. In particular, integrating safety data across separate studies provides data from more patients and presents a more robust summary of safety regarding treating two unique hyperkinetic movement disorders with deutetrabenazine over the first 12–15 weeks of therapy. Taken together, these data may aid conversations around treatment selection between clinicians and patients with these movement disorders and may improve patient outcomes.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Lerner PP, Miodownik C, Lerner V. Tardive dyskinesia (syndrome): current concept and modern approaches to its management. Psychiatry Clin Neurosci. 2015;69(6):321–34.
Ho AK, Hocaoglu MB. Impact of Huntington’s across the entire disease spectrum: the phases and stages of disease from the patient perspective. Clin Genet. 2011;80(3):235–9.
Thorley EM, Iyer RG, Wicks P, et al. Understanding how chorea affects health-related quality of life in Huntington disease: an online survey of patients and caregivers in the United States. Patient. 2018;11(5):547–59.
Waln O, Jankovic J. An update on tardive dyskinesia: from phenomenology to treatment. Tremor Other Hyperkinet Mov (NY). 2013;3. tre-03-161-4138-4131.
Bashir HH, Jankovic J. Treatment of tardive dyskinesia. Neurol Clin. 2020;38(2):379–96.
Savitt D, Jankovic J. Tardive syndromes. J Neurol Sci. 2018;389:35–42.
Carbon M, Hsieh CH, Kane JM, Correll CU. Tardive dyskinesia prevalence in the period of second-generation antipsychotic use: a meta-analysis. J Clin Psychiatry. 2017;78(3):e264-278.
Ward KM, Citrome L. Antipsychotic-related movement disorders: drug-induced parkinsonism vs. tardive dyskinesia—key differences in pathophysiology and clinical management. Neurol Ther. 2018;7(2):233–48.
Bates GP, Dorsey R, Gusella JF, et al. Huntington disease. Nat Rev Dis Primers. 2015;1:15005.
Ross CA, Aylward EH, Wild EJ, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. 2014;10(4):204–16.
Haddad MS, Cummings JL. Huntington’s disease. Psychiatr Clin North Am. 1997;20(4):791–807.
Austedo. Package insert. Teva Neuroscience, Inc; 2023.
Fernandez HH, Factor SA, Hauser RA, et al. Randomized controlled trial of deutetrabenazine for tardive dyskinesia: the ARM-TD study. Neurology. 2017;88(21):2003–10.
Anderson KE, Stamler D, Davis MD, et al. Deutetrabenazine for treatment of involuntary movements in patients with tardive dyskinesia (AIM-TD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Psychiatry. 2017;4(8):595–604.
Frank S, Testa CM, Stamler D, et al. Effect of deutetrabenazine on chorea among patients with Huntington disease: a randomized clinical trial. JAMA. 2016;316(1):40–50.
Hauser RA, Barkay H, Fernandez HH, et al. Long-term deutetrabenazine treatment for tardive dyskinesia is associated with sustained benefits and safety: a 3-year, open-label extension study. Front Neurol. 2022;13: 773999.
Frank S, Testa C, Edmondson MC, et al. The safety of deutetrabenazine for chorea in Huntington disease: an open-label extension study. CNS Drugs. 2022;36(11):1207–16.
Horváth K, Aschermann Z, Ács P, et al. Minimal clinically important difference on the Motor Examination part of MDS-UPDRS. Parkinsonism Relat Disord. 2015;21(12):1421–6.
Ingrezza. Package insert. Neurocrine Biosciences, Inc; 2021.
Xenazine. Package insert. Lundbeck; 2020.
Schultz JL, Killoran A, Nopoulos PC, Chabal CC, Moser DJ, Kamholz JA. Evaluating depression and suicidality in tetrabenazine users with Huntington disease. Neurology. 2018;91(3):e202–207.
Frank S. Tetrabenazine as anti-chorea therapy in Huntington disease: an open-label continuation study. Huntington Study Group/TETRA-HD Investigators. BMC Neurol. 2009;9:62.
Medical Writing and Editorial Assistance
Medical writing and editorial support for the development of this manuscript, under the direction of the authors, were provided by Melanie Chen, PharmD (Cello Health Communications/MedErgy), Jeffrey A. Blair, PhD, CMPP, Jennifer C. Jaworski, MS, BCMAS, CMPP, and Kelsey Gribbon, MS (Ashfield MedComms, an Inizio Company), and funded by Teva Branded Pharmaceutical Products R&D, Inc.
Funding
This study and the rapid service fee for publication was funded by Teva Branded Pharmaceutical Products R&D, Inc.
Author information
Authors and Affiliations
Contributions
Samuel Frank, Karen E. Anderson, Hubert H. Fernandez, Robert A. Hauser, Daniel O. Claassen, David Stamler, Stewart A. Factor, Joohi Jimenez-Shahed, Hadas Barkay, Amanda Wilhelm, Jessica K. Alexander, Nayla Chaijale, Steve Barash. Juha-Matti Savola, Mark Forrest Gordon, and Maria Chen meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, made substantial contributions to the drafting and revising of the manuscript, and have reviewed and approved the final manuscript for submission. Samuel Frank, Karen E. Anderson, Hubert H. Fernandez, Robert A. Hauser, Daniel O. Claassen, David Stamler, Stewart A. Factor, Joohi Jimenez-Shahed, Hadas Barkay, Amanda Wilhelm, Jessica K. Alexander, Nayla Chaijale, Steve Barash. Juha-Matti Savola, Mark Forrest Gordon, and Maria Chen contributed to the conception or design of this analysis and/or the acquisition, analysis, or interpretation of data.
Corresponding author
Ethics declarations
Conflict of Interest
Samuel Frank is principal investigator of First-HD and ARC-HD (Teva), consultant for Novartis, Sage Therapeutics, and uniQure, and has received research support from Cure Huntington’s Disease Initiative Foundation, Huntington’s Disease Society of America, the Huntington Study Group, Neurocrine, Roche/Genentech, and Triplet Therapeutics. Karen E. Anderson is global principal/co-principal investigator of AIM-TD and ARM-TD (Teva Pharmaceuticals; funds paid directly to her institution, Georgetown University), North American study co-principal investigator of LEGATO-HD (Teva), site principal investigator of Pride-HD, First-HD, and ARC-HD (Teva), ENROLL-HD (Cure Huntington’s Disease Initiative Foundation), Vaccinex, and site investigator for Generation HD studies (Roche Biogen). She is a consultant for NeuroNEXT 105 study (Azevan) and scientific advisor for Azevan, Biogen, Cure Huntington’s Disease Initiative Foundation, Novartis, Prana, and Roche. She has received salary support from The Griffin Foundation and Honoraria from Vindico Medical Education. Hubert H. Fernandez has received honoraria from AbbVie, Biogen, Carling Communications, GE Health Care, inVentiv, International Parkinson and Movement Disorders Society, Kyowa Hakko Kirin, Lundbeck, Medscape (speaker in CME events), Merz Pharmaceuticals, Pfizer (as a consultant), Prime Education, Sunovion, and Voyager Therapeutics. He has been provided grant and research support by AbbVie, Acadia, Biotie/Acorda, Civitas, Kyowa/ProStrakan, Michael J. Fox Foundation, Movement Disorders Society, National Institute of Health/National Institute of Neurological Disorders and Stroke, Parkinson Study Group, Rhythm Pharmaceuticals, Synosia, Teva Pharmaceuticals. He has received royalties from Demos Publishing (serving as a book author/editor) and contractual services with The Cleveland Clinic via a contract with Teva for Dr Fernandez’s role as co-principal investigator in SD-809 tardive dyskinesia global studies. Dr Fernandez also serves as a member of the publication committee for Acorda, but does not receive any personal compensation for this. He has received a stipend from the International Parkinson and Movement Disorders Society for serving as medical editor for the MDS website. Robert A. Hauser has served as a consultant with AbbVie, Academy for Continued Healthcare Learning, Acorda, Adamas Pharmaceuticals, AstraZeneca, Back Bay Life Science, Biotie, ClearView Healthcare Partners, ClinicalMind Medical and Therapeutic Communications, Cowen and Company, Cynapsus, eResearch Technology, Expert Connect, Gerson Lehrman Group, Guidepoint Global, Health Advances, HealthLogiX, Impax Laboratories, Kyowa Kirin Pharmaceutical Development, LCN Consulting, LifeMax, Lilly, Lundbeck, Michael J. Fox Foundation, National Institutes of Health, National Parkinson Foundation, Neurocrine, Neuropore, Outcomes Insights, PeerView Press, Pfizer, Pharma Two B Ltd., Prexton, Projects in Knowledge, Putnam Associates, Quintiles, RMEI Medical Education for Better Outcomes, Sarepta, Sunovion, Teva Pharmaceuticals, US WorldMeds, and Vista Research. Daniel O. Claassen has received research support from Griffin Foundation, Huntington’s Disease Society of America, Michael J. Fox Foundation, National Institute of Neurological Disorders and Stroke, and National Institute on Aging, and grant support from AbbVie, Acadia, Biogen, Bristol Myers Squibb, Cerecor, Jazz Pharmaceuticals, Lilly, Lundbeck, Teva Pharmaceuticals, Vaccinex, and Wave Life Sciences. Dr Claassen is a consultant/scientific advisor for Acadia, Adamas Pharmaceuticals, Alterity, Lundbeck, Neurocrine, Teva Pharmaceuticals, and Wave Life Sciences. David Stamler is a former employee of Teva Pharmaceuticals. He receives salary and benefits from Auspex Pharmaceuticals. Patents pending: US20160346270, US20160287574. Stewart A. Factor has received honoraria from Acorda, Biogen, CereSpir, Neurocrine, Lundbeck, Teva Pharmaceuticals, Avanir, UCB, US WorldMeds, Sunovion, and Adamas Pharmaceuticals. He has received research support from Biohaven, Boston Scientific, Impax, Ipsen, Jazz Pharmaceuticals, Lilly, Medtronic, National Institutes of Health (U10 NS077366), Neurocrine, Parkinson Foundation, Sun Pharmaceuticals Advanced Research Company, Voyager Therapeutics, Auspex, US WorldMeds, Pharm-Olam, Cynapsus/Sunovion, Vaccinex, Solstice, Cure Huntington’s Disease Initiative Foundation, Michael J. Fox Foundation. He has received royalties from Demos, Blackwell Futura, Springer (textbooks), Uptodate. He has other conflicts of interest with Signant Health (Bracket Global LLC) and CNS Ratings. Joohi Jimenez-Shahed has received consulting fees from AbbVie, Blue Rock Therapeutics, Bracket Global, Medtronic, Nuvelution, Photopharmics, St. Jude Medical, Teva Pharmaceuticals. Hadas Barkay is an employee of Teva Pharmaceutical Industries Ltd. Amanda Wilhelm and Jessica K. Alexander are former employees of Teva Branded Pharmaceutical Products R&D, Inc. Nayla Chaijale, Steve Barash, Mark Forrest Gordon, and Maria Chen are employees of Teva Branded Pharmaceutical Products R&D, Inc. Juha-Matti Savola is a former employee of Teva Pharmaceuticals International GmbH, Basel, Switzerland.
Ethical Approval
This article is based on previously conducted studies and did not require any approvals or informed consent; approval and consent were obtained with conduct of the initial studies, which were in accordance with the Declaration of Helsinki.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Frank, S., Anderson, K.E., Fernandez, H.H. et al. Safety of Deutetrabenazine for the Treatment of Tardive Dyskinesia and Chorea Associated with Huntington Disease. Neurol Ther 13, 655–675 (2024). https://doi.org/10.1007/s40120-024-00600-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40120-024-00600-1