
Abstract
Cardiac amyloidosis is a life-threatening disease that occurs when amyloid proteins, most commonly immunoglobulin light chain or transthyretin, mutate or become unstable, misfold, deposit as amyloid fibrils, and accumulate in the myocardium. Early diagnosis of cardiac amyloidosis is hindered by insufficient awareness, specifically regarding clinical red flags and diagnostic pathways. Cardiac amyloidosis diagnosis comprises two important phases, clinical suspicion (phase one) followed by definitive diagnosis (phase two). Each phase is associated with specific clinical techniques. For example, clinical features, electrocardiography, echocardiography, and cardiac magnetic resonance imaging serve to raise suspicion of cardiac amyloidosis and facilitate early diagnosis, whereas laboratory tests (i.e., blood or urine electrophoresis with immunofixation), biopsy, scintigraphy-based nuclear imaging, and genetic testing provide a definitive diagnosis of cardiac amyloidosis. In Egypt, both the lack of cardiac amyloidosis awareness amongst healthcare providers and the unavailability of clinical expertise for the use of diagnostic techniques must be overcome to improve the prognosis of cardiac amyloidosis in the region. Previously published diagnostic algorithms for cardiac amyloidosis have amalgamated techniques that can raise clinical suspicions of cardiac amyloidosis with those that definitively diagnose cardiac amyloidosis. Though such algorithms have been successful in developed countries, diagnostic tools like echocardiography, scintigraphy, and cardiac magnetic resonance imaging are not ubiquitously available across Egyptian facilities. This review presents the current state of knowledge regarding cardiac amyloidosis in Egypt and outlines a new diagnostic algorithm which leverages regional nuclear imaging expertise. Importantly, the proposed diagnostic algorithm guides accurate amyloid-typing to mitigate misdiagnosis and erroneous treatment selection and improve the cardiac amyloidosis diagnostic accuracy in Egypt.
Plain Language Summary
Diagnostic algorithms are useful tools for guiding clinical diagnosis by summarizing diagnostic approaches and defining the patient pathway. The diagnostic algorithms for cardiac amyloidosis amalgamate techniques that raise suspicion of the disease with those that can definitively diagnose the disease. These algorithms, for the early detection and diagnosis of cardiac amyloidosis, are designed in accordance with developed healthcare systems that have the resources and infrastructure for diagnostic equipment and clinical expertise. There are limited financial resources across healthcare facilities in Egypt for diagnostic equipment like echocardiograms (ECHO), scintigraphy, and cardiac magnetic resonance imaging (cMRI), and the required clinical training for the diagnosis of cardiac amyloidosis. This reduces the possibility of early diagnosis of the disease and subsequent early intervention. Evidently, there is a significant unmet clinical need to develop an algorithm for the diagnosis of cardiac amyloidosis in accordance with the Egyptian healthcare system. This review article details the current awareness regarding the diagnosis of cardiac amyloidosis and the associated challenges in Egypt. Accordingly, a diagnostic algorithm that leverages nuclear imaging expertise to guide accurate amyloid-typing in order to mitigate misdiagnosis and erroneous treatment, and also improve the diagnostic accuracy of cardiac amyloidosis, has been proposed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Diagnostic algorithms for cardiac amyloidosis amalgamate techniques that raise suspicion with those that definitively diagnose cardiac amyloidosis. |
Limited resources reduce the possibility of early accurate diagnosis and prompt management of cardiac amyloidosis in Egypt. |
There is a need for local initiatives to raise awareness regarding the prevalence and diagnostic pathways of cardiac amyloidosis among healthcare providers in the region. |
A diagnostic algorithm for cardiac amyloidosis that leverages regional nuclear imaging expertise in the context of the locally available infrastructure and resources of the Egyptian healthcare system has been proposed in this paper. |
Additional initiatives have also been proposed to track disease management and the prevalence of cardiac amyloidosis within the region. |
Introduction
Cardiac amyloidosis (CA) is a life-threatening disease that occurs due to the extracellular deposition of aggregated misfolded amyloid proteins in the myocardium leading to increased diastolic chamber stiffness and ventricular wall thickening [1,2,3,4,5,6,7]. While there are over 30 known types of proteins that can cause amyloidosis, the most significant are amyloidogenic immunoglobulin light chains (AL), transthyretin (TTR), and amyloid-protein A [5, 8]. AL-CA, caused by an underlying plasma cell dyscrasia, and TTR amyloid (ATTR)-CA, caused by amyloid originating from TTR, account for over 95% of all CA [7, 9]. Wild-type ATTR (ATTRwt)-CA is caused by the deposition of TTR that has become unstable due to aging while ATTRv-CA is caused by a genetic TTR mutation [7, 10].
Though no treatment options were available historically, novel treatments have been identified over the years for ATTR-CA and treatment strategies have improved over the past decade for the management of AL-CA [7]. However, optimal benefit can only be achieved by the diagnosis and treatment of CA during the early stages of the disease [11]. Early diagnosis of CA is hindered by insufficient awareness of the disease, its regional prevalence, clinical red flags and diagnostic pathways, and the lack of diagnostic capabilities and resources.
Several diagnostic algorithms that recommend the use of specialized interdisciplinary clinical teams and diagnostic equipment for the early detection and diagnosis of CA have previously been published [1, 12,13,14,15,16,17,18]. In Egypt, however, structured interdisciplinary care is underdeveloped, the gold standard diagnostic tools for CA diagnosis (i.e., endomyocardial biopsy; EMB) and specialized laboratory equipment are not ubiquitously available, and logistic failures concerning sample deliveries are common. There is a significant unmet clinical need for a diagnostic algorithm for CA in the context of the locally available infrastructure and resources of the Egyptian healthcare system.
The aim of this expert review is to detail the current state of knowledge regarding CA diagnosis, identify diagnostic challenges in Egypt, and propose a new diagnostic algorithm in accordance with the Egyptian clinical infrastructure.
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors. Therefore, ethical clearance approval from an Ethical Committee was not obtained.
Clinical Presentation and Prevalence
Though CA was previously considered a rare disease, there has been a marked increase in prevalence over the years [19]. For example, between 2000 and 2012, the prevalence and incidence of CA among Medicare beneficiaries in the United States (US) increased from 8–17 per 100,000 person-years to 18–55 per 100,000 person-years, respectively [20]. The incidence of CA significantly varies by geography and patient demographics [15]. Limited prevalence data on CA is available from Egypt and the rest of the Gulf region due to the lack of awareness of the disease, poor diagnostic efficiencies, and the lack of standardized reporting [21, 22].
According to Kumar et al. in 2018, the 20-year global prevalence rate of AL-CA (also known as ‘primary’ amyloidosis) was estimated to be 51.27 cases per million population with a crude annual incidence rate of 10.44 cases per million population [23]. AL-CA resembles acute myocarditis characterized by early symptoms and rapid progression to end-stage heart failure caused by the toxic effects of AL fibrils [24]. Typical clinical manifestations of AL-CA include heart failure with multi-organ involvement including nephrotic syndrome, orthostatic hypotension, diarrhea, bladder disorder, peripheral neuropathy, macroglossia, and periorbital purpura [11]. Untreated AL-CA patients have a poor prognosis with a median survival of 6–12 months [25, 26]. Promisingly, early diagnosis and prompt treatment has significantly increased survival with one in five patients having a longevity of 10 years, and approximately 30% surviving 15–20 years following stem cell transplantation [27]. Considering the poor prognosis associated with AL-CA, the first step when amyloidosis is suspected is to rule out AL-CA via serum free light chain (FLC) assay and serum or urine protein electrophoresis with immunofixation (immunoelectrophoresis; see section “Laboratory tests”) [12].
Based on observations from a 20-year study, Ioannou et al. reported that 1967 patients were diagnosed with ATTR-CA [28]. The disease behaves as a progressive cardiomyopathy characterized by slow amyloid deposition within the atria, ventricles, and conduction system [24]. For this reason, ATTR-CA symptoms, onset, and clinical identification typically occur late in the course of the disease (more so for ATTRwt-CA as compared to ATTRv-CA). ATTRwt-CA (previously termed ‘senile’ amyloidosis) commonly occurs in patients of advanced age. Based on autopsy studies of patients over 80 years of age (n = 256), Tanskanen et al. reported that approximately 25% of patients had ATTRwt-CA infiltration in the myocardium that was either mild (78%), moderate (11%), or severe (11%), regardless of the presence of symptoms [29, 30]. Between 1987 and 2009, less than 3% of patients were diagnosed with ATTRwt-CA, 14% between 2010 and 2015, and 25% between 2016 and 2019 [19]. ATTRwt-CA manifestations include heart failure, conduction disorders, atrial fibrillation, aortic stenosis, cardiac impairment, carpal tunnel syndrome, lumbar spinal stenosis, and traumatic biceps tendon rupture [5]. ATTRv-CA (previously termed ‘familial’ amyloidosis) is an autosomal dominant disease with marked phenotypic heterogeneity [17]. Interestingly, more than 120 pathogenic TTR mutations with varied phenotypic presentations have been identified [5]. Clinical manifestations associated with ATTRv-CA include conduction disorders, heart failure, infrequent atrial fibrillation, ascending bilateral sensory-motor polyneuropathy, orthostatic hypotension, diarrhea, constipation, erectile dysfunction, glaucoma, intravitreal deposition, and scalloped pupils. Cardiac impairment in ATTRv-CA varies depending on the mutation. Further, the age of onset is broad, ranging from 20 to 70 years, and the offspring of an ATTRv-CA carrier have a 50% chance of inheriting single nucleotide polymorphism mutations [1].
In Saudi Arabia, data mining identified that p.Val142Ile was the most common variant associated with amyloidosis (allele frequency 0.001) [22]. The variant c.238A>G(p.Thr80Ala) was also found in the Saudi population (allele frequency of 0.00004), which was similarly common in 1.1% of the population in northwest Ireland [31]. Authors reported that globally common variants that were not present in Saudi Arabia included p.Val50Met (common in western Europe), p.Leu131Met (common in Denmark), p.Ile88Leu (common in Italy), p.Ser70Arg (common in Mexico), as well as p.Val50Ala, p.Ala117Ser, and p.Gly103Arg (all common in China) [32,33,34,35,36]. A recent review recommended the genetic screening of at-risk family members in order to facilitate early diagnosis of ATTRv [37]. However, genetic screening relies on a comprehensive understanding of the regional ATTRv prevalence based on ATTR databases, for which Arab populations are markedly underrepresented.
Though the prevalence of CA in Egypt is currently unknown, based on global estimates, we anticipate a higher CA prevalence than previously assumed. The diagnostic CA algorithm for Egypt has been developed in alignment with the regional context of diagnostic advances and availability of targeted treatment options.
Diagnostic Challenges
A patient-experience survey identified that 37% of patients had symptoms for more than a year prior to CA diagnosis, 32% have seen at least five physicians prior to diagnosis, 65% presented to a primary care physician first for AL-CA amyloidosis, and only 19% were correctly diagnosed [38]. There are three key challenges leading to the underdiagnosis or misdiagnosis of CA. Firstly, CA typically presents with nonspecific signs and symptoms, especially in the early stages of disease progression, with complex phenotypic and genotypic heterogeneity. Additionally, knowledge and awareness regarding CA is fragmented among healthcare professionals (HCPs). Furthermore, real-world clinical applications of biopsy (CA diagnostic gold standard) are limited since this technique is expensive, requires special expertise, and is associated with significant procedural risks [39].
Due to the high cost and the lack of specialized transportation logistics and local expertise to handle associated procedural risks and mitigate sampling errors, EMB is not available in Egypt. However, scintigraphy can diagnose CA without the need for a biopsy and can therefore be leveraged for diagnosis. Laboratory functions such as serum and urine testing capabilities are not ubiquitously available in Egypt. Moreover, there is a lack of structured, interdisciplinary collaboration of specialists in the Egyptian healthcare system (e.g., neurology, hematology, gastroenterology, pathology, genetics, and cardiology teams). There is also a lack of specialized facilities for cardiovascular disease management, clearly defined patient pathways, access to diagnostic tools, and clinical expertise. Local experts attribute these barriers to the lack of interest among HCPs in learning about CA red flags and existing misconceptions among HCPs that CA is incurable, uncommon in Egypt, and very expensive to treat.
The challenges in Egypt are not necessarily reflective of those in Western countries. Until now, published diagnostic algorithms have mainly been based on the healthcare systems of developed nations, which use a wide range of clinical, laboratory, and imaging indicators. Diagnosis comprises two important phases, clinical suspicion (phase one) followed by definitive diagnosis (phase two). Each phase is associated with specific clinical diagnostic techniques. For example, clinical features, electrocardiography (ECG), echocardiography (ECHO), and cardiac magnetic resonance imaging (cMRI) serve to raise suspicion of CA and facilitate early diagnosis, whereas laboratory tests (i.e., blood or urine electrophoresis with immunofixation), biopsy, scintigraphy-based nuclear imaging, and genetic testing serve to definitively diagnose CA.
Raising Clinical Suspicion Prior to Definitive Diagnosis
‘Red flags’ that raise suspicion of CA can be garnered from three key areas: clinical features, laboratory biomarkers, and imaging techniques such as ECG, ECHO, and cMRI.
Clinical Clues
Clinical clues may present as cardiac manifestations or non-cardiac manifestations, or can be garnered from the patient history as non-presenting symptoms (Fig. 1). Although identifying clinical features are an important first step in the diagnostic workup of CA, they are insufficient for accurate diagnosis and should be considered alongside laboratory biomarkers and imaging techniques [40].

Clinical clues which may raise suspicion of cardiac amyloidosis [14, 41]. CA cardiac amyloidosis, AL amyloidogenic immunoglobulin light chain, ATTRv hereditary transthyretin-mediated amyloidosis, ATTRwt amyloid transthyretin-wild-type, HF heart failure, HFpEF heart failure with preserved ejection fraction
Imaging
Multimodal imaging techniques, such as ECG, ECHO, and cMRI, are valuable screening tools for the early diagnosis of CA in high-risk individuals [16].
Electrocardiography
Aggregation of amyloids deposited within cardiac tissue is associated with electrical disturbances. In a report of 127 patients with biopsy-proven AL-CA, the most common ECG findings were low voltage (46%) and the presence of pseudo-infarct patterns (47%) [42]. Additional ECG findings associated with CA include left anterior hemiblock, T-wave abnormalities (ischemic or nonspecific), and atrial fibrillation [3, 43]. Interestingly, ECG findings appear to be somewhat specific to the type of CA. A retrospective study comparing ECG abnormalities between AL-CA and ATTR-CA in 244 patients (AL-CA, n = 106; ATTRwt, n = 108; ATTRv, n = 30) reported that atrial fibrillation presented in over 33% of ATTRwt-CA patients, compared to 20% and 6% for ATTRv-CA and AL-CA, respectively [24]. Furthermore, ATTR-CA patients commonly presented with AV block, and intraventricular delay, whereas AL-CA patients presented with low-voltage sinus rhythm. In addition, QRS voltages are a typical finding in the early stages of cardiac involvement [44]. Early identification of lowered QRS voltages or fragmented QRS are associated with poor prognosis and should prompt a consideration of CA, warranting further investigations. It is important to note that ECG is limited by low-sensitivity and specificity, due in part to interference from non-cardiac comorbidities such as obesity. ECG findings should therefore be considered together with information yielded from ECHO and cMRI [4].
Echocardiography
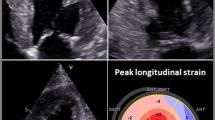
ECHO is an important part of the CA diagnostic work up and is widely available in most medical facilities both globally and locally within Egypt. Classic cardiac involvement is typically observed via ECHO in the advanced stages of CA. Characteristic ECHO findings include increased cardiac wall thickness (typically 12 mm or greater) in the absence of a known cause (e.g., hypertension) and is often less severe in AL-CA than in ATTR-CA [41]. Other features include increased left and right ventricular (LV, RV) wall thickening with granular sparkling, normal or mildly decreased LV chamber volume with atrial enlargement, atrial septal thickening, valve thickening, signs of elevated filling pressures as well as restrictive diastolic filling (grade II-IV diastolic dysfunction), preserved or moderately decreased LV ejection fraction (LVEF) with poor longitudinal function, CA characteristic LV strain pattern with apex preservation (termed ‘bull’s eye’ or ‘cherry-on-top’) and finally, low tissue Doppler velocities 5′5′5′ sign [3, 43, 45]. Abnormal RV or LV strain is considered an early diagnostic clue associated with CA [46]. Atrial thrombi are commonly detected in 35% of AL-CA [47]. Notably, the severity and magnitude of ECHO abnormalities together with the rapid rate of progression are associated with the worst prognosis [48]. Importantly, not all patients manifest the aforementioned characteristic ECHO features. Some patients may present with heart failure alongside normal LV wall thickness, which is characteristically increased in CA patients. This may be attributable to circulating light chain-mediated cardiotoxicity causing myocardial dysfunction [49, 50]. Current consensus identifies that ECHO is limited by a lack of sensitivity in the early stages of the disease and is therefore employed to raise clinical suspicion and not a definitive diagnosis [51].
Cardiac Magnetic Resonance Imaging
cMRI holds an important place in the non-invasive diagnostic workup of ATTR-CA [52]. cMRI can be employed to determine amyloid burden via accurate measurement of cardiac structure, function, and pathology [53]. Specifically, cMRI can assess morphological characteristics associated with CA such as increased myocardial thickness (greater in ATTR-CA than AL-CA), preserved ejection fraction, diastolic dysfunction, and ventricular strain patterns [52]. Tissue characterization on the other hand is typically evaluated using late gadolinium enhancement (LGE) and T1 mapping techniques [54].
Gadolinium (Gd)-enhanced cMRI can quantify changes in myocardial extracellular spaces that are observed in CA patients [55]. For example, in a healthy individual, the myocardial extracellular space is approximately 25%, compared with 60% in ATTR-CA patients [56]. Gd accumulates exclusively in the myocardial extracellular space since it is bound to a macromolecular chelator which restricts the molecule from passing through the entire cell membrane. Using characteristic LGE patterns on Gd-enhanced cMRI, Vogelsberg et al. demonstrated that the CA diagnostic sensitivity and specificity of cMRI are 80% and 94%, respectively [54]. In addition, the use of LGE patterns for early CA diagnosis has been proposed since cMRI patterns can present before a significant increase in mass size [52, 57]. Notably, the clinical applications of linear Gd-based contrast agents in CA patients are limited in that it is contraindicated in patients with estimated glomerular filtration rate (eGFR) below 30 ml/min/1.73 m2, which is a common finding in AL-CA [52]. Medical agencies emphasize the need to assess the risks and benefits of using Gd-based contrast agents in patients with chronic kidney disease against diagnostic challenges of performing a non-contrast scan and recommend using the least possible dose of Gd-based contrast agents if a cMRI is needed [58, 59]. Alternatively, low doses of high-relaxivity Gd-based contrast agents can be used [59]. However, limitations associated with Gd-enhanced cMRI LGE may, in some cases, be overcome by using T1 mapping.
T1 mapping can detect CA, track progression over time, and may help to determine the type of CA (i.e., AL-CA or ATTR-CA) [60]. T1 mapping quantifies the longitudinal relaxation time of myocardial signals pre-contrast (native T1) or post-contrast by color coding image pixels to generate a “map” of myocardial intrinsic signals during a single breath-hold. Clinically, the progression of diffuse fibrosis to scar tissue and later amyloid with edema can be visualized via native T1 imaging. This provides an early ‘red flag’ indicator since increased native T1 is typically detected before LGE, biomarker elevation, or LV hypertrophy can be observed. Cut-off values have been developed for T1 maps generated by the scanner at 1.5 T. Suspicion of CA may be excluded with 98% negative predictive value when T1 is below 1036 ms and confirmed with 98% positive predictive value when T1 is greater than 1164 ms [61].
Definitive Diagnosis
Upon clinical suspicion of CA, patients should undergo a definitive diagnostic work up. It is therefore fundamental to patient prognosis that HCPs have sound clinical understanding of CA and the associated diagnostic tools. In a recent online survey, CA awareness, knowledge of disease manifestations, and approach to diagnosis were evaluated in 272 physicians practicing across United Arab Emirates, Bahrain, Qatar, Oman, and Kuwait [62]. Data revealed that 17% of responders did not consider themselves to be familiar with the signs and symptoms of CA. Data also suggested that 41% of responders would not consider CA in patients with heart failure and 61% would not consider CA in patients with severe aortic stenosis, both of which are known to be associated with CA. Data further suggested that many responders did not find diagnostic utility in scintigraphy with bone-seeking radiotracers (40%) or clinical biomarkers (69%).
Laboratory Tests
Upon suspicion of CA, the presence of AL-CA should be assessed via the measurement of serum free light chain (FLC) assay and serum or urine protein electrophoresis with immunofixation (immunoelectrophoresis) [12]. Immunoelectrophoresis, in combination with serum FLC assay, has 99% sensitivity for the diagnosis of AL amyloidosis [63]. Notably, electrophoresis must be performed in combination with immunofixation, since immunofixation identifies the presence of monoclonal bands and electrophoresis determines their type. In the presence of monoclonal proteins, an immediate patient referral to hematology should be registered. In addition to serum or urine protein immunoelectrophoresis, bone marrow biopsy is recommended to confirm plasma cell dyscrasia and determine the percentage and type of kappa- or lambda-producing plasma cells [12].
Scintigraphy-Based Nuclear Imaging
Nuclear imaging with bone seeking tracer is a non-invasive technique to diagnose ATTR-CA in patients who do not have a monoclonal gammopathy (i.e., monoclonal bands on serum or urine immunofixation, or an abnormal FLC ratio) [18]. Bone scintigraphy has a sensitivity of more than 99% and a specificity of 86% for ATTR-CA. In Western counties, the most common scintigraphy bone imaging agents are 99mTc-pyrophosphate (PYP), 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid (DPD) or 99mTc-hydroxymethylene diphosphonate (HMDP) [4], whereas in Egypt, 99mTc-PYP is more commonly used due to regional availability. Scans are graded according to the uptake of the bone tracer that binds to TTR amyloid. The following grading criteria has been established: grade 0, no uptake; grade 1, uptake but less than ribs; grade 2, uptake equal to that of ribs; grade 3, uptake greater than that of ribs. Grade 2 or 3, along with the absence of monoclonal gammopathy, has a 100% positive predictive value for ATTR-CA. False-positive scans for AL can occur, as it is difficult to differentiate between a grade 1 and a grade 2 scan in clinical practice. However, the risk of false-positive scans due to blood pooling can be reduced by delaying imaging by 3 h. It is important to note that it is possible for a patient to have early amyloidosis even with a negative scintigraphy scan. In this case, a biopsy will be required to confirm diagnosis. Where biopsy is not possible, experts note that it is acceptable to closely monitor the patient and repeat scintigraphy scans within 6–12 months to confirm diagnosis and initiate treatment.
Biopsy
EMB with histopathological evaluation is the gold standard diagnostic tool for CA [18]. However, EMB is associated with several limitations such as high cost, the requirement of expertise to perform EMB surgery, significant procedural risks, and a high risk of sampling errors [64]. As such, in real-world clinical practice, CA is typically diagnosed using cMRI, scintigraphy, and positron emission tomography, while EMB is reserved for clinically ambiguous cases where patients present with mild or moderate uptake of 99mTc-PYP. Alternatively, biopsy samples taken from less invasive regions can spare patients from EMB, where possible [65, 66].
According to the Mayo Clinic, abdominal fat aspirate can be used to determine the type of amyloid deposit with reduced surgical risk compared to EMB [67]. Surrogate sites (e.g., abdominal fat) and extracardiac biopsies are often clinically preferred in order to make a timely diagnosis when EMB requires patient referral to specialized and experienced centers [65]. Following sample collection, diagnosis is determined via Congo red staining analysis, which produces a pathognomonic green birefringence under cross-polarized light in the presence of amyloid fibrils [3]. Notably, abdominal fat aspirate with Congo red staining is preferred, as it is sensitive for AL amyloid (~ 70%), ATTRv amyloid (~ 67%), and ATTRwt amyloid (14%) [43]. Subsequently, amyloid typing using techniques like immunohistochemical staining can be used to identify the type of deposited amyloid fibrils [29].
Genetic Testing
Genetic testing is important in the context of strong suspicions of ATTRv to determine disease prognosis and verify the eligibility for novel ATTR-targeted therapies [65]. ATTRv gene mutations are influenced by age, mutation type, and geographical location. For example, the penetrance of the c.148G>A(p.(Val50Met)) variant in patients aged 50 years is markedly higher in Portugal (80%) than in France (18%) and Sweden (11%). Moreover, penetrance considerably increases with advanced age. In Portugal, France, and Sweden, penetrance in patients aged 70 years is increased by 11%, 32%, and 25%, respectively, compared to patients aged 50 years [68]. Genetic testing should be performed parallelly alongside genetic counselling and are recommended in the following scenarios:
-
To verify ATTR-CA when clinical features, ECHO, ECG, and cMRI have raised a high index of suspicion in combination with unexplained LV hypertrophy in the absence of monoclonal gammopathy.
-
To differentiate between ATTR variants (ATTRv and ATTRwt) when scintigraphy-based nuclear imaging is grade 2 or higher in the absence of monoclonal gammopathy [65].
-
Genetic screening for first-degree adult biological relatives of indexed patients with proven ATTRv-CA [4]. Screening is crucial for early diagnosis of asymptomatic ATTRv-CA mutation carriers. Although the genetic testing of minors is not typically recommended since ATTRv is associated with adult onset, it may be indicated in minors with a positive family history of CA.
Diagnostic Algorithm
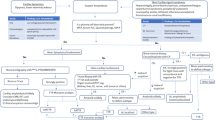
Previously published diagnostic algorithms for CA have amalgamated diagnostic screening techniques that raise suspicion of CA with those that definitively diagnose CA [1, 12,13,14,15,16,17,18, 69, 70]. This approach is successful in countries where diagnostic facilities are equipped with specialist expertise and advanced diagnostic equipment, and where cost is not a concern. However, techniques such as ECHO and cMRI that raise CA suspicion are not ubiquitously available across Egyptian facilities. Figure 2 details an adapted algorithm for application in Egypt that isolates the definitive diagnostic techniques necessary to accurately diagnose CA and does not include diagnostic screening tests that raise the suspicion of CA, which are the initial steps in other published diagnostic algorithms. This is not to say that techniques associated with raising clinical suspicion are not valuable. Rather, such techniques should be employed, where possible, to yield the most comprehensive patient disease profile and facilitate early diagnosis. On the other hand, diagnostic techniques that raise the suspicion of CA must not be prioritized above, or delay access to, using definitive diagnostic techniques or add to already existing health care costs. Importantly, this clinical algorithm guides appropriate amyloid-typing to mitigate misdiagnosis, erroneous treatment selection, and improve overall diagnostic accuracy in Egypt.

Diagnostic algorithm for Egypt. AL immunoglobulin light chain, ATTR amyloid transthyretin, ATTRv hereditary transthyretin-mediated amyloidosis, ATTRwt amyloid transthyretin-wild-type, CA cardiac amyloidosis, CM cardiomyopathy, FLC serum free light chain immunoglobulin assay, Tc-PYP technetium pyrophosphate, TTR transthyretin. *If AL-CA is confirmed by organ biopsy, treat it as a medical emergency. If ATTR-CA is confirmed by organ biopsy, confirm sub-type and initiate sub-type specific treatment. Note: Organ biopsy in the algorithm includes endomyocardial biopsy
Both invasive and non-invasive diagnostic techniques have applications in the diagnosis of CA. Notably, invasive diagnostic criteria apply to all forms of CA (i.e., AL-CA and ATTR-CA), yet non-invasive criteria apply only to ATTR-CA [4]. Further, serum or urine immunofixation should be conducted, where possible, in parallel with scintigraphy and bone marrow or fat aspiration. Regional experts have called for national improvements regarding serum or urine testing capabilities in Egypt since these techniques are not ubiquitously available across all Egyptian facilities.
Findings of scintigraphy-based nuclear imaging, a non-invasive diagnostic technique, may be evaluated via quantitative or semi-quantitative assessment of myocardial tracer uptake. Quantitative evaluation of cardiac retention is achieved by calculating the heart-to-contralateral lung uptake ratio (H/CL) at 1 h. A H/CL ratio greater than 1.5 with intensely diffuse myocardial tracer retention has a 97% sensitivity and 100% specificity for identifying ATTR-CA [71]. Semi-quantitative evaluation is achieved by visual comparison of bone uptake at 3 h [69]. Visual scores of 2 or higher on planar or single-photon emission computerized tomography (SPECT) images at 3 h are classified as ATTR-CA positive, and scores of 1 or lower, as ATTR-CA negative. A grade of 2 or higher, in the absence of monoclonal gammopathy, has a 100% positive predictive value for ATTR-CA. Grade 0 indicates no uptake or normal bone uptake; grade 1 indicates uptake less than that of rib; grade 2 indicates uptake equal to that of rib; grade 3 indicates uptake greater than that of rib with mild or no rib uptake.
EMB, an invasive diagnostic technique, is included within the algorithm since it is the established gold standard diagnostic tool for CA, and to highlight the urgent unmet need to facilitate biopsy testing in Egypt. Until then, regional experts outlined that when biopsy is not possible, it is acceptable to closely monitor the patient and repeat scintigraphy assessments after 1 year to confirm diagnosis before initiating treatment.
TTR gene sequencing is indicated in suspected ATTR-CA since it is impossible to distinguish between ATTRv and ATTRwt on clinical results alone [72]. Further, TTR amyloid can infiltrate the tendons in distal biceps and cause the tendons in proximal biceps to rupture. This is common in patients with ATTR-CA. Geller et al. found that 33% (37 of 111) of patients with ATTRwt had distal bicep tendon rupture [73]. Similarly, TTR amyloid may deposit in the lumbar spinal area, rotator cuff, large joints of the hip and knee, as well as the carpal tunnel. During carpal tunnel release, distal bicep tendon rupture, and spinal stenosis surgery, tissue is typically removed and discarded without pathological evaluation. This presents a window of opportunity for pathological examination and early diagnosis of ATTR-CA in Egypt. Sperry et al. demonstrated that 10.2% (10 of 98) of men undergoing carpal tunnel release surgery tested positive for amyloidosis based on Congo red staining [74]. Furthermore, carpal tunnel syndrome is thought to precede CA manifestations by approximately 5–10 years [75].
Staging and Prognosis
Staging criteria that estimate the mean survival for AL-CA and ATTR-CA based on the elevation of various biomarkers such as cardiac troponin T (cTnT), N-terminal pro-B-type-natriuretic peptide (NT-proBNP), difference in serum free light chains (dFLC), and eGFR have been proposed (Table 1) [76,77,78].
The most commonly used staging criteria for AL-CA patients was developed by the Mayo Clinic, which associated patients having cTnT of 0.025 ng/ml or higher, NT-proBNP more than 1800 pg/m, and dFLC more than 18 mg/dl with the worst prognosis (6 months; Table 1) [76]. Interestingly, the extent of cardiac involvement in AL-CA may be determined by calculating the kappa–lambda ratio, which, if lesser than 0.26 and greater than 1.65, indicates monoclonal lambda gammopathy and monoclonal kappa gammopathy, respectively [76].
The prognostic profile of ATTR-CA is markedly more favorable compared to AL-CA and is associated with longer patient survival [41]. Regarding ATTRv, prognosis is determined primarily by the genetic mutation profile and those that are classified as having myocardial involvement (e.g., V122I) tend to have shorter survival rates compared to those that are classified as having neuropathic involvement (e.g., V30M) or ATTRwt [79, 80]. Grogan et al. proposed a staging criterion for ATTRwt patients, which associated patients with NT-proBNP over 3000 ng/l and cTnT greater than 0.05 µg/l with the worst prognosis of 20 months [81]. However, more recently, Gillmore et al. proposed staging criterion for both ATTRv and ATTRwt, which associated patients with NT-proBNP greater than 3000 ng/l and eGFR below 45 ml/min/1.73 m2 with the worst prognosis of 24.1 months [77].
In addition to biomarker-based staging methods, which are fast and low-cost indicators of survival, ECHO, 99mTc-PYP scintigraphy, and cMRI also hold prognostic value [82,83,84]. Specifically, apical sparing with reduced LVEF, myocardial contraction fraction, global longitudinal strain, and stroke volume index on ECHO are associated with poor prognosis [80]. Further, heart-to-contralateral ratio of 1.6 or higher assessed via 99mTc-PYP scintigraphy, as well as decreased indexed ejection volume and increased LGE, extracellular volume, and native T1 assessed via cMRI are also associated with poor disease prognosis.
Discussion
Globally, the prognosis for CA patients has improved significantly with increasing disease awareness and educational initiatives, specialized referral centers, and Centers of Excellence, as well as the advent of diagnostic techniques (particularly non-invasive techniques) and diagnostic algorithms. In Egypt, the lack of CA awareness amongst HCPs regarding prevalence, red flags, diagnostic pathways, as well as the unavailability of clinical expertise associated with CA diagnosis must be overcome to improve CA prognosis in this region.
CA awareness and educational initiatives for HCPs are vital for better CA prognosis. A recent survey conducted in the Unites States (US) identified that 85% of primary care providers were not confident in differentiating between ATTRv and ATTRwt, 74% were not familiar with various mutations of ATTRv, 74% were not confident with genetic testing techniques, 67% were not confident identifying soft tissue red flags and 63% were not confident differentiating between ATTR-CA and AL-CA [85]. We hypothesize that these knowledge gaps also apply to the HCPs in Egypt, since the availability of prevalence data, medical infrastructure, and access to diagnostic equipment are notably limited compared to the US. Experts in Egypt underline the need for local initiatives to focus on CA prevalence, red flags, diagnostic techniques, implementation of the Egyptian diagnostic algorithm in clinical practice, and the establishment of a multidisciplinary healthcare structure. Furthermore, awareness initiatives must emphasize the importance of accurately reporting clinical cases in order to reliably track disease management and disease prevalence within the region. The latter may be supported by the integration of an online consultation platform to bridge the gap between the patient and specialist HCPs.
To further support the success and impact of the proposed initiatives, there is an unmet need to establish specialized referral centers and Centers of Excellence across Egypt. Centers of Excellence offer an established multidisciplinary HCP structure, as well as the availability of specialized laboratories and diagnostic equipment within one clinical facility. Experts within Egypt further emphasize the need to develop protocols and checklists as educational tools to guide HCPs in CA case reporting. Experts also detail the need to establish a system to ensure quality control of the serum or urine immunofixation assays, which are an integral diagnostic test in the workup of CA.
This review presents the current state of knowledge regarding CA in Egypt and outlines a new diagnostic algorithm that leverages regional expertise in nuclear cardiology and isolates definitive diagnostic techniques for early detection and diagnosis of CA in the region.
References
Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286–300.
Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–16.
Martinez-Naharro A, Hawkins PN, Fontana M. Cardiac amyloidosis. Clin Med (Lond). 2018;18(Suppl 2):s30–5.
Garcia-Pavia P, Rapezzi C, Adler Y, Arad M, Basso C, Brucato A, et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2021;42(16):1554–68.
Gonzalez-Lopez E, Lopez-Sainz A, Garcia-Pavia P. Diagnosis and treatment of transthyretin cardiac amyloidosis. Progress and hope. Rev Esp Cardiol (Engl Ed). 2017;70(11):991–1004.
Kittleson MM, Maurer MS, Ambardekar AV, Bullock-Palmer RP, Chang PP, Eisen HJ, et al. Cardiac amyloidosis: Evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation. 2020;142(1):e7–22.
Versteylen MO, Brons M, Teske AJ, Oerlemans M. Restrictive atrial dysfunction in cardiac amyloidosis: differences between immunoglobulin light chain and transthyretin cardiac amyloidosis patients. Biomedicines. 2022;10(8):1768–78.
Kastritis E, Palladini G, Minnema MC, Wechalekar AD, Jaccard A, Lee HC, et al. Daratumumab-based treatment for immunoglobulin light-chain amyloidosis. N Engl J Med. 2021;385(1):46–58.
Donnelly JP, Hanna M. Cardiac amyloidosis: an update on diagnosis and treatment. Cleve Clin J Med. 2017;84(12 Suppl 3):12–26.
See ASY, Ho JS-Y, Chan MY, Lim YC, Yeo T-C, Chai P, et al. Prevalence and risk factors of cardiac amyloidosis in heart failure: a systematic review and meta-analysis. Heart Lung Circ. 2022;31(11):1450–62.
Oerlemans M, Rutten KHG, Minnema MC, Raymakers RAP, Asselbergs FW, de Jonge N. Cardiac amyloidosis: the need for early diagnosis. Neth Heart J. 2019;27(11):525–36.
Grogan M, Dispenzieri A, Gertz MA. Light-chain cardiac amyloidosis: strategies to promote early diagnosis and cardiac response. Heart. 2017;103(14):1065–72.
Gertz MA. Immunoglobulin light chain amyloidosis diagnosis and treatment algorithm 2018. Blood Cancer J. 2018;8(5):1–8.
Donnelly JP, Hanna M, Sperry BW, Seitz WH Jr. Carpal tunnel syndrome: a potential early, red-flag sign of amyloidosis. J Hand Surg Am. 2019;44(10):868–76.
Yilmaz A, Bauersachs J, Bengel F, Buchel R, Kindermann I, Klingel K, et al. Diagnosis and treatment of cardiac amyloidosis: Position statement of the German Cardiac Society (DGK). Clin Res Cardiol. 2021;110(4):479–506.
Papathanasiou M, Carpinteiro A, Rischpler C, Hagenacker T, Rassaf T, Luedike P. Diagnosing cardiac amyloidosis in every-day practice: a practical guide for the cardiologist. Int J Cardiol Heart Vasc. 2020;28: 100519.
Kitaoka H, Izumi C, Izumiya Y, Inomata T, Ueda M, Kubo T, et al. JCS 2020 guideline on diagnosis and treatment of cardiac amyloidosis. Circ J. 2020;84(9):1610–71.
Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133(24):2404–12.
Ravichandran S, Lachmann HJ, Wechalekar AD. Epidemiologic and survival trends in amyloidosis, 1987–2019. N Engl J Med. 2020;382(16):1567–8.
Gilstrap LG, Dominici F, Wang Y, El-Sady MS, Singh A, Di Carli MF, et al. Epidemiology of cardiac amyloidosis-associated heart failure hospitalizations among Fee-for-Service Medicare beneficiaries in the United States. Circ Heart Fail. 2019;12(6): e005407.
Al Badarin F, Al-Humood K, Bader F, Alsaid S, Sulaiman K, Alzadjali M, et al. Physician knowledge and awareness about cardiac amyloidosis in the Middle East and Gulf region. JACC CardioOncol. 2022;4(3):421–4.
Abouelhoda M, Mohty D, Alayary I, Meyer BF, Arold ST, Fadel BM, et al. Established and candidate transthyretin amyloidosis variants identified in the Saudi population by data mining. Hum Genom. 2021;15(1):52.
Kumar N, Zhang NJ, Cherepanov D, Romanus D, Hughes M, Faller DV. Global epidemiology of amyloid light-chain amyloidosis. Orphanet J Rare Dis. 2022;17(1):278.
Cappelli F, Vignini E, Martone R, Perlini S, Mussinelli R, Sabena A, et al. Baseline ECG features and arrhythmic profile in transthyretin versus light chain cardiac amyloidosis. Circ Heart Fail. 2020;13(3): e006619.
Siddiqi OK, Sanchorawala V, Ruberg FL. Echocardiography and survival in light chain cardiac amyloidosis. Circ Cardiovasc. 2018;11(5): e007826.
Rossi M, Varrà GG, Porcari A, Saro R, Pagura L, Lalario A, et al. Re-definition of the epidemiology of cardiac amyloidosis. Biomedicines. 2022;10(7):1566.
Staron A, Zheng L, Doros G, Connors LH, Mendelson LM, Joshi T, et al. Marked progress in AL amyloidosis survival: a 40-year longitudinal natural history study. Blood Cancer J. 2021;11(8):139.
Ioannou A, Patel RK, Razvi Y, Porcari A, Sinagra G, Venneri L, et al. Impact of earlier diagnosis in cardiac ATTR amyloidosis over the course of 20 years. Circulation. 2022;146(22):1657–70.
Sabbour H, Hasan KY, Al Badarin F, Alibazoglu H, Rivard AL, Romany I, et al. From clinical clues to final diagnosis: the return of detective work to clinical medicine in cardiac amyloidosis. Front Cardiovasc Med. 2021;8: 644508.
Tanskanen M, Peuralinna T, Polvikoski T, Notkola IL, Sulkava R, Hardy J, et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann Med. 2008;40(3):232–9.
Reilly MM, Staunton H, Harding AE. Familial amyloid polyneuropathy (TTR ala 60) in north west Ireland: a clinical, genetic, and epidemiological study. J Neurol Neurosurg Psychiatry. 1995;59(1):45–9.
Damy T, Kristen AV, Suhr OB, Maurer MS, Planté-Bordeneuve V, Yu C-R, et al. Transthyretin cardiac amyloidosis in continental Western Europe: an insight through the Transthyretin Amyloidosis Outcomes Survey (THAOS). Eur Heart J. 2019;43:391–400.
Suhr OB, Svendsen IH, Andersson R, Danielsson A, Holmgren G, Ranlov PJ. Hereditary transthyretin amyloidosis from a Scandinavian perspective. J Intern Med. 2003;254(3):225–35.
Gagliardi C, Perfetto F, Lorenzini M, Ferlini A, Salvi F, Milandri A, et al. Phenotypic profile of Ile68Leu transthyretin amyloidosis: an underdiagnosed cause of heart failure. Eur J Heart Fail. 2018;20(10):1417–25.
González-Duarte A, Cárdenas-Soto K, Bañuelos CE, Fueyo O, Dominguez C, Torres B, et al. Amyloidosis due to TTR mutations in Mexico with 4 distinct genotypes in the index cases. Orphanet J Rare Dis. 2018;13(1):1–7.
Yin J, Xia X, Shi Y, Lu Y, Zhao C, Huang Z, et al. Chinese familial transthyretin amyloidosis with vitreous involvement is associated with the transthyretin mutation Gly83Arg: a case report and literature review. Amyloid. 2014;21(2):140–2.
Conceição I, Damy T, Romero M, Galán L, Attarian S, Luigetti M, et al. Early diagnosis of ATTR amyloidosis through targeted follow-up of identified carriers of TTR gene mutations. Amyloid. 2019;26(1):3–9.
Lousada I, Comenzo RL, Landau H, Guthrie S, Merlini G. Light chain amyloidosis: patient experience survey from the amyloidosis research consortium. Adv Ther. 2015;32(10):920–8.
Maurer MS. Noninvasive identification of ATTRwt cardiac amyloid: the re-emergence of nuclear cardiology. Am J Med. 2015;128(12):1275–80.
Hasib Sidiqi M, Gertz MA. Immunoglobulin light chain amyloidosis diagnosis and treatment algorithm 2021. Blood Cancer J. 2021;11(5):90.
Rapezzi C, Merlini G, Quarta CC, Riva L, Longhi S, Leone O, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120(13):1203–12.
Murtagh B, Hammill SC, Gertz MA, Kyle RA, Tajik AJ, Grogan M. Electrocardiographic findings in primary systemic amyloidosis and biopsy-proven cardiac involvement. Am J Cardiol. 2005;95(4):535–7.
Mohty D, Damy T, Cosnay P, Echahidi N, Casset-Senon D, Virot P, et al. Cardiac amyloidosis: updates in diagnosis and management. Arch Cardiovasc Dis. 2013;106(10):528–40.
Cipriani A, Michieli LD, Porcari A, Licchelli L, Sinigiani G, Tini G, et al. Low QRS voltages in cardiac amyloidosis: clinical correlates and prognostic value. JACC CardioOncol. 2022;4:458–70.
Dorbala S, Cuddy S, Falk RH. How to image cardiac amyloidosis: a practical approach. JACC Cardiovasc Imaging. 2020;13(6):1368–83.
Miller F Jr, Bellavia D. Comparison of right ventricular longitudinal strain imaging, tricuspid annular plane systolic excursion, and cardiac biomarkers for early diagnosis of cardiac involvement and risk stratification in primary systematic (AL) amyloidosis: a 5-year cohort study: reply. Eur Heart J Cardiovasc Imaging. 2013;14(1):91–2.
Feng D, Syed IS, Martinez M, Oh JK, Jaffe AS, Grogan M, et al. Intracardiac thrombosis and anticoagulation therapy in cardiac amyloidosis. Circulation. 2009;119(18):2490–7.
Aljaroudi WA, Desai MY, Tang WH, Phelan D, Cerqueira MD, Jaber WA. Role of imaging in the diagnosis and management of patients with cardiac amyloidosis: state of the art review and focus on emerging nuclear techniques. J Nucl Cardiol. 2014;21(2):271–83.
Suresh R, Grogan M, Maleszewski JJ, Pellikka PA, Hanna M, Dispenzieri A, et al. Advanced cardiac amyloidosis associated with normal interventricular septal thickness: an uncommon presentation of infiltrative cardiomyopathy. J Am Soc Echocardiogr. 2014;27(4):440–7.
Gertz MA, Grogan M, Kyle RA, Tajik AJ. Endomyocardial biopsy-proven light chain amyloidosis (AL) without echocardiographic features of infiltrative cardiomyopathy. Am J Cardiol. 1997;80(1):93–5.
Nakahashi T, Arita T, Yamaji K, Inoue K, Yokota T, Hoshii Y, et al. Impact of clinical and echocardiographic characteristics on occurrence of cardiac events in cardiac amyloidosis as proven by endomyocardial biopsy. Int J Cardiol. 2014;176(3):753–9.
Vergaro G, Aimo A, Barison A, Genovesi D, Buda G, Passino C, et al. Keys to early diagnosis of cardiac amyloidosis: red flags from clinical, laboratory and imaging findings. Eur J Prev Cardiol. 2020;27(17):1806–15.
Zhao L, Tian Z, Fang Q. Diagnostic accuracy of cardiovascular magnetic resonance for patients with suspected cardiac amyloidosis: a systematic review and meta-analysis. BMC Cardiovasc Disord. 2016;16(1):129.
Vogelsberg H, Mahrholdt H, Deluigi CC, Yilmaz A, Kispert EM, Greulich S, et al. Cardiovascular magnetic resonance in clinically suspected cardiac amyloidosis: noninvasive imaging compared to endomyocardial biopsy. J Am Coll Cardiol. 2008;51(10):1022–30.
Jurcut R, Onciul S, Adam R, Stan C, Coriu D, Rapezzi C, et al. Multimodality imaging in cardiac amyloidosis: a primer for cardiologists. Eur Heart J Cardiovasc Imaging. 2020;21(8):833–44.
Kotecha T, Martinez-Naharro A, Treibel TA, Francis R, Nordin S, Abdel-Gadir A, et al. Myocardial edema and prognosis in amyloidosis. J Am Coll Cardiol. 2018;71(25):2919–31.
Syed IS, Glockner JF, Feng D, Araoz PA, Martinez MW, Edwards WD, et al. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovasc Imaging. 2010;3(2):155–64.
Rezk T, Fontana M, Gillmore JD. A review of the criteria for non-invasive diagnosis of cardiac transthyretin amyloidosis. Expert Opin Orphan Drugs. 2021;9(3):87–94.
Hao J, Bourrinet P, Desché P. Assessment of pharmacokinetic, pharmacodynamic profile, and tolerance of gadopiclenol, a new high relaxivity GBCA, in healthy subjects and patients with brain lesions (phase I/IIa study). Investig Radiol. 2019;54(7):396–402.
Fontana M, Chung R, Hawkins PN, Moon JC. Cardiovascular magnetic resonance for amyloidosis. Heart Fail Rev. 2015;20(2):133–44.
Mongeon F-P, Jerosch-Herold M, Coelho-Filho OR, Blankstein R, Falk RH, Kwong RY. Quantification of extracellular matrix expansion by CMR in infiltrative heart disease. JACC Cardiovasc Imaging. 2012;5(9):897–907.
Al Badarin F, Al Ali J, Bader F, Shehab AM, Al Said S, Sulaiman K, et al. Knowledge of clinical presentation and diagnostic pathways for cardiac amyloidosis among physicians in the Middle East-Gulf region: on behalf of the Gulf Cardiac Amyloidosis Working Group. Circulation. 2020;142(Suppl 3): A16240-A.
Palladini G, Russo P, Bosoni T, Verga L, Sarais G, Lavatelli F, et al. Identification of amyloidogenic light chains requires the combination of serum-free light chain assay with immunofixation of serum and urine. Clin Chem. 2009;55(3):499–504.
Van Geluwe F, Dymarkowski S, Crevits I, De Wever W, Bogaert J. Amyloidosis of the heart and respiratory system. Eur Radiol. 2006;16(10):2358–65.
Varga C, Dorbala S, Lousada I, Polydefkis MJ, Wechalekar A, Maurer MS, et al. The diagnostic challenges of cardiac amyloidosis: a practical approach to the two main types. Blood Rev. 2021;45: 100720.
Porcari A, Baggio C, Fabris E, Merlo M, Bussani R, Perkan A, et al. Endomyocardial biopsy in the clinical context: current indications and challenging scenarios. Heart Fail Rev. 2022. https://doi.org/10.1007/s10741-022-10247-5.
Mayo Foundation for Medical Education and Research M. Amyloidosis 2021. https://www.mayoclinic.org/diseases-conditions/amyloidosis/diagnosis-treatment/drc-20353183.
Pueyo CL, Arregui MÁA, Gutierrez AG, Juana EB, Guillén SM. Estimating the prevalence of allelic variants in the transthyretin gene by analysing large-scale sequencing data. Eur J Hum Genet. 2019;27(5):783–91.
Garibaldi B, Zaas D. An unusual case of cardiac amyloidosis. J Gen Intern Med. 2007;22(7):1047–52.
Merlini G, Seldin DC, Gertz MA. Amyloidosis: pathogenesis and new therapeutic options. J Clin Oncol. 2011;29(14):1924.
Bokhari S, Castaño A, Pozniakoff T, Deslisle S, Latif F, Maurer MS. 99mTc-pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circ Cardiovasc. 2013;6(2):195–201.
Maurer MS, Elliott P, Comenzo R, Semigran M, Rapezzi C. Addressing common questions encountered in the diagnosis and management of cardiac amyloidosis. Circulation. 2017;135(14):1357–77.
Geller HI, Singh A, Alexander KM, Mirto TM, Falk RH. Association between ruptured distal biceps tendon and wild-type transthyretin cardiac amyloidosis. JAMA. 2017;318(10):962–3.
Sperry BW, Reyes BA, Ikram A, Donnelly JP, Phelan D, Jaber WA, et al. Tenosynovial and cardiac amyloidosis in patients undergoing carpal tunnel release. J Am Coll Cardiol. 2018;72(17):2040–50.
Boyle RP, Sharan J, Schwartz G. Carpal tunnel syndrome in transthyretin cardiac amyloidosis: Implications and protocol for diagnosis and treatment. Cureus. 2021;13(4): e14546.
Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012;30(9):989–95.
Gillmore JD, Damy T, Fontana M, Hutchinson M, Lachmann HJ, Martinez-Naharro A, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J. 2018;39(30):2799–806.
Grogan M, Scott CG, Kyle RA, Zeldenrust SR, Gertz MA, Lin G, et al. Natural history of wild-type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J Am Coll Cardiol. 2016;68(10):1014–20.
Siddiqi OK, Ruberg FL. Cardiac amyloidosis: an update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med. 2018;28(1):10–21.
Simões MV, Fernandes F, Marcondes-Braga FG, Scheinberg P, Correia EDB, Rohde LEP, et al. Position statement on diagnosis and treatment of cardiac amyloidosis—2021. Arq Bras Cardiol. 2021;117:561–98.
Grogan M, Dispenzieri A. Natural history and therapy of AL cardiac amyloidosis. Heart Fail Rev. 2015;20(2):155–62.
Rubin J, Steidley DE, Carlsson M, Ong M-L, Maurer MS. Myocardial contraction fraction by M-mode echocardiography is superior to ejection fraction in predicting mortality in transthyretin amyloidosis. J Cardiac Fail. 2018;24(8):504–11.
Castano A, Haq M, Narotsky DL, Goldsmith J, Weinberg RL, Morgenstern R, et al. Multicenter study of planar technetium 99m pyrophosphate cardiac imaging: predicting survival for patients with ATTR cardiac amyloidosis. JAMA Cardiol. 2016;1(8):880–9.
Fontana M, Pica S, Reant P, Abdel-Gadir A, Treibel TA, Banypersad SM, et al. Prognostic value of late gadolinium enhancement cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2015;132(16):1570–9.
Raichlin E, Sagalovich M. Survey conducted at an academic medical center revealed knowledge gaps of transthyretin amyloidosis cardiomyopathy predominantly in primary care providers. J Cardiac Fail. 2020;26(10):S53.
Acknowledgements
Funding
The journal’s Rapid Service Fee was funded by Pfizer, Egypt.
Medical Writing Assistance
Medical writing support was provided by Hannah Wilson-Nortey and Eric Mario of Connect Communications, Dubai, United Arab Emirates. It was funded by Pfizer, Egypt.
Author Contributions
The authors confirm contribution to the paper as follows: conception and design of work: Dr. Mohamed Abdelghany, Dr. Magdy Abdelhamid, Dr. Adel Allam, Dr. Adel El Etriby, Sherif Hafez, Dr. Hany Ragy, and Dr. Mohamed Sobhy; data sources: Dr. Mohamed Abdelghany, Dr. Magdy Abdelhamid, Dr. Adel Allam, Dr. Adel El Etriby, Dr. Hany Ragy, and Dr. Mohamed Sobhy; data interpretation: Dr. Mohamed Abdelghany, Dr. Magdy Abdelhamid, Dr. Adel Allam, Dr. Adel El Etriby, Sherif Hafez, Dr. Hany Ragy, and Dr. Mohamed Sobhy; critical review of previous versions of the article: Dr. Mohamed Abdelghany, Dr. Magdy Abdelhamid, Dr. Adel Allam, Dr. Adel El Etriby, Sherif Hafez, Dr. Hany Ragy, and Dr. Mohamed Sobhy. All authors reviewed and approved the final version of the manuscript.
Disclosures
Dr Magdy Abdelhamid reported speaker honoraria from Bayer, AstraZeneca, Novartis, and Boehringer-Ingelheim. Dr Hany Ragy reported speaker honorarium from Pfizer. Sherif Hafez is an employee at Pfizer, Egypt. Dr Mohamed Abdelghany, Dr Adel Allam, Dr Adel El Etriby, and Dr Mohamed Sobhy declared no competing interests.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors. Therefore, ethical clearance approval from an Ethical Committee was not obtained.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Abdelghany, M., Abdelhamid, M., Allam, A. et al. Detection and Diagnosis of Cardiac Amyloidosis in Egypt. Cardiol Ther 12, 197–213 (2023). https://doi.org/10.1007/s40119-022-00299-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40119-022-00299-x