
Abstract
Laboratory experiments conducted for long time on stream (TOS: 14,350 h) provide information on Fischer–Tropsch synthesis (FTS) that is representative of time scales of industrial operations. Operation conditions that deliver desirable conversion and product distribution were investigated. Low gas hourly space velocity (GHSV) gave the highest conversion of 20.97% with the highest C5+ selectivity achieved was 59.77%, which was obtained at the highest GHSV level. A one-way ANOVA, followed by a post-hoc Bonferroni correction test, indicated a significant difference in response to GHSV with P(T <=t) two-tail values ranging from 1.5 × 10−4 to 2.7 × 10−35. The optimum condition for paraffin production is high pressure and low GHSV: in our experiments, this corresponded to 20.85 bar (abs): 648 h−1. Conversely, olefins production is favored low pressure and low GHSV [1.85 bar (abs): 648 h−1]. C5+ production was favored at high GHSV (2592 h−1) and was very sensitive to GHSV, as the sensitivity to C5+ products dropped sharply when the GHSV decreased to low values (from 1296 to 648 h−1); furthermore, the selectivity to C5+ was found to be independent of pressure. The pressure effect on selectivity is complex and selectivity toward overall gaseous (paraffin + olefin) hydrocarbons and C5+ does not seem to be significantly affected by variations in pressure. Long TOS FTS runs are possible ca. 14,500 h though product distribution trends tend to be changed. The catalyst survived long runs, though the selectivity to FTS became comparatively less favored than WGS with increasing TOS. Our findings may have useful implication for the design of a mobile small-scale biomass/waste to liquid process that would last for period similar to that of an industrial plant.
Similar content being viewed by others


Introduction
Fisher Tropsch synthesis (FTS) is an established technology that produces synthetic petroleum fuels (synfuels), chemical feedstock and pipeline gas. The most commonly used active metals catalyst in FTS are iron and cobalt, as these provide technical and economic advantages. Synthesis gas (syngas), which is a mixture of carbon monoxide and hydrogen (the stoichiometric ratio required is around 1:2), is used for FTS.
The composition of the feed syngas is important as it affects the activity and selectivity of catalyst and even the presence of inert gases such as nitrogen in the feed can affect the reaction. Muleja et al. [1] reported that when nitrogen is co-fed to fixed bed FT reactor loaded with a cobalt catalyst, the selectivity to C5+ (particular C5–C19) fraction is enhanced. Syngas can also be used for the reduction/activation of FT catalyst [2], but the composition of the syngas can influence the selectivity and activity of the catalyst. Chun et al. [3] conducted experiments on a highly selective iron-based FT catalysts activated by CO2-containing syngas. They found that the activation with this gas suppressed the production of undesired products, namely CH4 and C2–C4 hydrocarbons; and facilitated the production of valuable products the C5+ hydrocarbons. FTS is a chemical process that depends on operational conditions, including time on stream (TOS), space velocity (SV), pressure and temperature. Researchers [2,3,4,5] have reported on these factors, and how they influence the product distribution of hydrocarbons during FTS. One of the drawbacks to this research is that the experiments were conducted under conditions that do not reflect the real pilot or industrial plant operations, as the laboratory data are usually reported for relatively short TOS.
The literature is rich with information on the effect of process parameters on FTS at short TOS, for example, Bukur, ran up to TOS 140 h [4], Todic et al. [5] ran longer for TOS of up to 654 h; therefore, the findings may not hold for extended reaction times that are used industrially. The phenomenon of catalyst speciation (phase changes of the active catalyst to an inactive phase) or simply deactivation, which is a function of TOS, has a major influence on activity and product distribution. Hence, an understanding of the catalyst performance at long TOS is important.
The relationship between SV, pressure P, temperature T and contact time θ is as shown by the following equation:
In most reported cases, either reducing the SV or increasing pressure increases the production rate, which results from an increase in θ. One study [6] indicated that lowering the SV to prolong the interaction between the reactants and the catalyst led to further hydrogenation and oligomerization of short-chain hydrocarbons, which resulted in an increase in long chain (C5+) products that were more paraffinic.
Frequent shut-downs and start-ups of industrial FT reactors to replace spent catalyst adversely affect the profitability of an FT production plant. Implementing control system to alter the operating parameters, i.e., pressure and SV, to maintain the conversion (or product formation rate) is sometimes implemented. The choice of the parameters, either individual parameters, or in combinations, to maintain the yield and delay the replacement of the catalyst, is therefore of interest.
In an earlier investigation done by the researchers of this study [2], the results showed that FTS using an iron catalyst at low pressure (1.85 bar absolute) has fairly significant catalytic activity for the synthesis of hydrocarbons for TOS up to 1000 h at a temperature of 250 °C. In this study, the FT reaction was studied over a long period of time (more than 19 months), during which time the gas hourly space velocity (GHSV) and the pressure were varied and the catalyst activity and selectivity monitored. Three different experiments were run in parallel: an iron catalyst was reduced with three different reducing gases, namely CO, H2 and syngas. The activity and selectivity of the three catalysts were monitored for around 14,000 h TOS to ascertain how the three differently reduced iron catalysts responded to changes in operating condition over an extended period of time.
There is no, or very little, data available publicly on catalyst performance for long TOS, and thus these data should be of general interest. We are in particular interest to use these data for the conceptual design of a small-scale mobile waste-to-liquid plant, as the performance of the catalyst for long TOS is one of the most important parameters in determining the economics of such processes.
Experimental procedure
The experimental procedure presented in this study is similar to that used in the previous study done by the researchers [2]. A propriety iron-based catalyst (FeCuKSiO2) of the Materials and Process Synthesis research group (MaPS) which is now known as Institute for the Development of Energy for African Sustainability (IDEAS) at the University of South Africa was used. The BET characteristics of the catalyst are indicated in Table 1.
The gases required for FT synthesis, were supplied by African Oxygen (AFROX Ltd), in standard gas cylinders (40 kg) for use in the laboratory. This included the carrier gases and the auxiliaries (argon, helium, hydrogen and air) used for gas chromatography (GC) operations, which required ultra-high purity (UHP) grades (> 99.9997%). Three types of catalyst-reducing gases were used for the catalyst activation: (1) UHP H2; (2) UHP CO; (3) syngas, with a molar composition of H2/CO/N2 = 60%/30%/10%. The syngas was also used as feed to the FT reactors during synthesis. The online GC was calibrated by means of a gas mixture comprising H2, CO, CO2, N2, CH4, C2H4, and C2H6.
Reactor system
The experimental set-up consisted of three laboratory scale fixed bed reactors in a parallel configuration (see Fig. 1). An important aspect of the parallel concept was that it enabled all three reactors to share the same feed cylinder, nitrogen and analysis equipment, thereby reducing the possibility of errors. The same feed (syngas) was distributed to the three reactors using Brooks mass flow controllers (Brooks Instrument 5850). A non-return valve was mounted after each mass flow control (MFC) channel to prevent the products from flowing back to the MFC. Besides the lines used to feed the system with syngas, other lines were available to supply gases (such as nitrogen and reducing gases) to the reactors. Back pressure regulators were manually controlled to keep the reactor pressure at a desired set point.

Flow scheme of the laboratory scale Fischer–Tropsch rig with three fixed reactors in parallel
Characterization
The samples were characterized by X-ray powder diffraction (XRD) and High Resolution Transmission Electron Microscopy (HRTEM). XRD patterns were obtained using a Rigaku XRD instrument equipped with a scintillation counter detector (40 kV, 30 mA). The powder samples were scanned in the 0°–75° 2θ range at the rate of 0.2°/min. High resolution transmission electron micrographs were collected using an FEI Tecnai G2 20 field emission gun (FEG) TEM, which was operated in bright field mode at an accelerating voltage of 200 kV. The analysis was performed on the fresh, unreduced catalyst and on all three spent catalysts.
Catalyst reduction procedure
1 gram of the iron-based catalyst was loaded into each of the three reactors. Before catalyst reduction, the catalyst in each of the reactors was dried under the flow of nitrogen at gas hourly space velocity (GHSV) of 2592 h−1, at a temperature of 120 °C, and at atmospheric pressure (0.85 bar). This was done for 2 h, to remove any moisture in the catalyst. After the drying was completed, catalyst reduction began. Three different reducing agents were used for the catalyst reduction, i.e., syngas for the catalyst reduction in reactor 1 (Reac-Syn); H2 for catalyst in reactor 2 (Reac-H2); CO for reactor 3 (Reac-CO). The three types of reducing gases were introduced into the three reactors, respectively, at a GHSV of 2592 h−1, at atmospheric pressure (0.85 bar), and the temperature was increased from 120 °C (drying temperature) to 250 °C (reduction temperature) at a heating rate of 1 °C/min. The system was left at 250 °C in the atmosphere of reducing agents for 48 h, prior to running the FT reaction.
Experimental conditions
The FT operating conditions were initially kept at a reactor pressure of 1.85 bar (absolute), a temperature of 250 °C and syngas gas feed at a GHSV of 2592 h−1 for about 5000 h TOS. Thereafter, various changes were made to the GHSV and pressure at different TOS. The aim was to test the responses of the catalysts to changes in the operating conditions at long TOS. The series of tests intended to identify the best reducing agent in terms of long-term catalyst activity, selectivity, stability and resistance to deactivation. The conditions at various TOS and selected results are summarized in Table 2. Data were obtained at three GHSV (2592–1296 h−1, then 648 h−1) and three pressures [P = 1.85, 10.85 and 20.85 bar (abs)].
The data obtained from 0 to 1000 h TOS were previously reported by the researchers [2]; the inclusion of these data in this article is intended to provide readers with a full picture of the effect of operating conditions on FTS over a TOS from 0 to ca. 14,500 h.
Product separation and analysis
The products exiting the reactor were cooled and collected as different fractions at different temperatures. The wax products were collected in the hot trap maintained at 150 °C. The liquid products were accumulated in the cold trap, which was kept at room temperature. The components that were not condensed, i.e., were gaseous at room temperature, were directed to the online gas chromatography (GC) for analysis and/or venting.
The product analysis was done through three different detectors: a flame ionization detector (FID) and two thermal conductivity detectors (TCDs). Of the two TCD detectors used in the online GC, TCD-A was used to analyze CH4, CO2, N2 and CO with UHP He as a carrier gas, while TCD-B was used to analyze H2 with UHP Ar as a carrier gas using Teknokroma molecular sieve columns.
Varian capillary columns were used in this FID system for the analysis of gaseous olefin and paraffin products (C1–C5). Samples from the tail gas were taken every 83 min via valves from the sampling loop. The excess gas from the sampling loop passed through a bubble meter to the vent. The tail gas from the three reactors was sampled cyclically (reactor 1–2–3:1–2–3 cycles).
CO conversions, %CO, were calculated as follows:
where Xco,in and Xco,out are the molar fractions of CO in the reactor inlet and outlet, respectively, Fin and Fout are the molar flow rates of gas in and out of the reactor, respectively.
The CO consumption rate, (mol/(min·gcat)), was calculated as follows:
where mcat is the mass of the catalyst used in this reaction, in grams.
The rate of formation of product θi, mol/(min·gcat) is given by
where \(X_{{\varphi_{i} ,{\text{out}}}}\) is the molar fraction of \(\varphi_{i}\) in the reactor outlet gas stream.The product selectivity for species \(\varphi_{i}\), \({\text{Sel}}(\varphi_{i} )\) is calculated on a moles of carbon basis, as follows:
where \([nC]_{{\varphi_{i} }}\) are the moles of carbon in species \(\varphi_{i}\) contained in a sample of the exit of reactor collected over time t.
Results and discussion
Characterization of fresh and spent catalysts
XRD
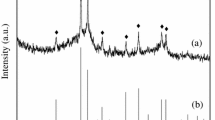
The XRD patterns of the three differently reduced spent catalysts at TOS ca. 14,500 h and that of the fresh catalyst are shown in Fig. 2. The spectra show the differences in the phases existing in each of the three differently reduced catalysts and also the phases that existed in the fresh catalyst.

XRD spectra of the fresh catalyst and spent catalyst from different reactors after ca 14,500 h of TOS
The fresh catalyst produced only one peak corresponding to magnetite, whereas the used catalyst indicated the presence of several phases, with graphitic carbon being dominant. Magnetite is a known water gas shift catalyst [7]. The iron carbide phases remaining in the deactivating catalyst might provide a reason for the rise in C1–C4 selectivities consequent on the decrease in GHSV, i.e., it may be attributable to a longer residence time. The presence of graphitic carbon might have hindered C5+ re-adsorption and diffusion, both inside and outside of the catalyst pores, hence a decrease in C5+ selectivity.
HRTEM
To give further insight, both fresh and spent catalysts were subjected to characterization via HRTEM analysis. Figure 3 depicts images of (a) fresh catalyst, (b) spent catalyst from Reac-Syn, (c) spent catalyst from Reac-H2, (d) spent catalyst from Reac-CO, (e) HRTEM of the fresh catalyst and (f) the plot profile (ImageJ software) of the distance between two lattice fringes of the fresh catalyst. The HRTEM micrographs shown in Fig. 3a–d as inserted pictures allow for identification of the growth of graphitic carbon from all the spent catalysts in each of the three reactors (see dimming of the light in Fig. 4). The presence of carbon deposits is confirmed by the XRD patterns in Fig. 2. Carbon deposition is one of the supported metal catalyst deactivation routes; it is responsible for blocking of surface sites, metal crystallite encapsulation, plugging of pores, and destruction of catalyst pellets by carbon filaments [8, 9]. This deposition of carbon affects the diffusion of heavy olefins and paraffins both inside and outside the catalyst pores or surface and GHSV becomes an important factor. However, the effect may not be as pronounced in light hydrocarbons, which have almost the same volatilities and hence are not influenced by the diffusion limitations [10].

TEM images of a fresh catalyst, b spend catalyst from Reac-Syn, c spent catalyst from Reac-H2, d spent catalyst from Reac-CO, e HRTEM of the fresh catalyst and f plot profile of two lattice fringes from image e using ImageJ software

Selected area electron diffraction (SAED) pattern of a fresh catalyst, b Reac-Syn spent catalyst, c Reac-H2 spent catalyst and d Reac-CO spent catalyst
Furthermore, the crystallinity of the fresh iron catalyst is confirmed by the HRTEM image (Fig. 3e) which visibly displays the presence of lattice fringes distribution. The analysis of the lattice fringes was then rendered possible with ImageJ software. The measurement of the value between the two lattice fringes was obtained (0.251 nm) with the plot profile analysis using ImageJ software. The obtained value, 0.251 nm, approximates value reported in the literature of 0.253 nm [11]. Researchers [11] have reported that the value (approximately 0.25 nm) of the distance between two lattice fringes corresponds to the (311) plane of the magnetite crystal. XRD patterns have also confirmed the presence of magnetite in all four samples (fresh and spent catalysts). It is interesting to see that the selected area electron diffraction (SAED) patterns highlight the difference between the three used catalysts and the fresh catalyst when the analysis is focused on the appearance of the rings in Fig. 4 which indicate the polycrystalline phases of the particles. It is remarkable that the polycrystalline phases of the particles decrease in the spent catalysts, most likely due the deposition of carbon during FT reaction after a long TOS.
Effect of process parameters on the product distribution
The effect of process parameters on the activity and hydrocarbon selectivity was studied under the experimental conditions described in Table 2. We will consider the various conditions that are described in Table 2 separately and then later compare the results.
Effect of TOS on the CO conversation; TOS < 5000 h
In this study, the experiment included several stages. Initially, the synthesis was done at 250 °C for about 5000–6000 h TOS at fixed pressure (1.85 bar abs) and GHSV (2592 h–1); at this stage, the conversion for all three catalysts dropped from around 22–15% to Reac-syn—6.03%; Reac-H2—6.99%; Reac-CO 4.36%. The results of these experiments are presented in Fig. 5. The data show that the CO conversion for all three reduced catalysts decreases with an increase in TOS, indicating that the catalyst deactivated with time as expected.

Effect of TOS on CO conversion of differently reduced catalysts at a pressure of 1.85 bar (abs), GHSV of 2592 h−1 and temperature of 250 °C with standard deviation error bars
Effect of GHSV on conversion
The GHSV was then decreased from 2592 to 1296 h−1, while maintaining the pressure at 1.85 bar (abs) as described in Table 2. The three differently reduced catalysts responded positively to the reduction in feed gas flow rate as shown in Fig. 6, with conversion increasing in all three cases. However, the responses were of different magnitudes: Reac-Syn showed the highest response a 2.31-fold increase from GHSV 2592–1296 h −1; Reac-H2 showed a 1.77-fold increase; Reac-CO showed a 1.69-fold increase.

Conversions of differently reduced reactors with changing GHSV: a Reac-Syn; b Reac-H2, c Reac-CO. Reactor temperature was maintained at 250 °C and reactor pressure at 1.85 bar (abs)
A further halving in flow rate to a GHSV of 648 h−1 did not yield quite as much of a difference in the conversion. For example, from 1296 to 648 h −1, the conversion of Reac-Syn increased from 9.86 to 17.20% (a 1.74-fold increase), Reac-H2 increased from 12.35 to 20.97% (a 1.70-fold increase); Reac-CO increased from 7.34 to 10.18% (1.39-fold increase). Thus, in terms of conversion, the performance of the Reac-CO reactor performed significantly worse than the other two reactors.
Table 3 provides a summary of one-way ANOVA results that compare CO conversion obtained from three reactors. To enable a comparison of the response to changes in GHSV, a post-hoc Bonferroni correction test was performed, and the results tabulated. As shown in all ANOVA tables, the P values are small, which provides strong evidence that the three reactors responded differently to changes in GHSV.
Effect of GHSV on hydrocarbon selectivity
The products’ distribution of hydrocarbons formed at the various GHSV are summarized in Table 4a–c for the light olefins, light paraffins, total (i.e., olefin and paraffin) light hydrocarbons and C5+ fraction, for all three reactors. The results are shown as averages in the tables as these are simple to see and interpret.
The selectivity to paraffins was observed to increase with the decrease in GHSV, while the selectivity towards olefins tended to follow the same pattern, except for the discrepancies of C2 olefin for Reac-Syn and Reac-H2. In general, the lighter hydrocarbons increased in selectivity with decrease in the GHSV, whereas the selectivity towards C5+ (heavy hydrocarbons) decreased with decreasing GHSV. The observed C5+ trend is different to the normal trend observed in the literature, for example [12]. As the GHSV is decreased, the reactant–catalyst contact time increased hence increasing time for secondary reactions to occur (Eq. 1) which would suggest that hydrogenation and chain growth is favored resulting in a heavier product distribution. This is not what is observed at long TOS. The water gas shift (WGS) reaction could be favored relative to FTS, as this reaction is known to be catalyzed by the magnetite [8]. This would result in a decrease in hydrocarbon selectivity with TOS.
This potentially has a big impact on the design of the proposed mobile small-scale biomass/waste to liquid process. When such a process is designed to run at low pressure [such as 1.85 bar(abs), as in this study], one will need to balance a higher conversion which is achieved at lower GHSV’s against the increase to lighter hydrocarbons that occurs at lower GHSV’s. The information in Table 4 shows that both olefin and paraffin selectivities increase with a decrease in GHSV.
Effect of GHSV on WGS and CH4 selectivity
The CH4 selectivity was not very sensitive to GHSV, whereas the CO2 selectivity increased significantly with decreasing GHSV (see Fig. 7). The averaged results of Fig. 7 are summarized in Table 5 for ease of interpretation.

CO2 and CH4 selectivities with reductions in GHSV at different TOS
Effect of pressure on the CO conversion
The system pressure was varied and the CO conversion was measured; the results are shown in Fig. 8 under the experimental procedure in Table 2 for TOS starting from 12,000 to 14,100 h. CO conversion increased with increasing pressure for all the differently reduced catalysts as expected. As summarized in Table 6, the CO conversion increased 2.54-fold, 3.81-fold and 5.34-fold for Reac-Syn, Reac-H2 and Reac-CO reduced reactors, respectively, when pressure was increased from 1.85 to 10.85 bar (abs). When pressure was further increased from 10.85 to 20.85 bar (abs), Reac-Syn showed the largest increase of 2.17-fold followed by the Reac-CO (with a 1.75-fold), and finally, Reac-H2 (with a 1.56-fold increase).

CO conversion versus TOS at pressures of 1.85, 10.85 and 20.85 bar (abs) at temperature 250 °C, GHSV of 2592 h−1, for TOS between 12,000 and 14,400 h
Effect of pressure on the selectivity to hydrocarbons
The results are summarized in Table 7. The effect of pressure on selectivity is complex and selectivity toward the total light hydrocarbons (paraffin + olefin) and C5+ does not seem to be significantly affected by variations in pressure.
Table 7 shows that the selectivity to olefins generally decreased with an increase in the pressure from 1.85 bar (abs) to 20.85 bar (abs), for all three reactors; the selectivity to paraffins in contrast increases with pressure. Observed anomalies occurred in Reac-H2, where C3 and C5 olefins do not follow this trend. The behavior that was noticed could be attributed to the hydrogenation of olefins to paraffins, as this was also observed in other studies [5, 13]. In general, the results confirm that when the FT reactor pressure is increased, the product selectivity generally increases towards paraffinic products. Paraffinic product means more wax product, and this finding agrees with findings proposed by Farias et al. [14]; they reported that high pressures (25–30 atm) favored the production of waxes, while moderate pressure (e.g., 20 atm) showed selectivity towards the diesel fraction.
When compared to the Reac-Syn and Reac-H2 reactors, Reac-CO always showed a larger scale change in terms of selectivity to olefins. The trend is expressed as
where the subscript o refers to olefin.
For paraffins, the following trend is observed:
where the subscript p refers to olefin.
For C5+, at all pressures, the trend is:
where the subscript C5+ refers to hydrocarbons of chain length 5 or longer.
Effect of pressure on the selectivity to CH4 and CO2 production
The effect of pressure on the selectivity of CH4 and CO2 was also measured for all three reactors. The results are displayed in Fig. 8a–c and the data are summarized in Table 8. The data show that CO2 selectivity increases slightly with increasing pressure; a slight, but obvious increase in selectivity was seen when the pressure was increased from 1.85 to 10.85 bar (abs), however, the magnitude of increase was much smaller when the pressure was increased from 10.85 to 20.85 bar (abs). The CH4 selectivity remains nearly constant almost the same, except for Reac-H2 which had a low CH4 selectivity at low pressure. Our finding patterns agree with statistical models based on experimental data from the literature [15] (Fig. 9).

The selectivity of CO2 and CH4 at different pressures, with GHSV = 2592 h−1 and temperature 250 °C for the three differently reduced catalyst
The sensitivity with respect to changes in GHSV and pressure for methane is a more difficult case to explain. Research suggests that the iron carbides favor the formation of methane [16, 17]. This prediction is supported based on the calculated reaction energies and effective barriers by [16] using spin-polarized density functional theory calculations that CH4 formation is more favorable on Fe5C2 and Fe2C. The XRD analysis of spent catalyst seen in Fig. 2 shows the presence of iron carbide remnants. With this observation, one would expect a significant variation in selectivity with decreasing GHSV and increasing pressure.
Conclusion
An iron catalyst was reduced with syngas, hydrogen or carbon monoxide. The three differently reduced catalysts were run for long TOS. The product distribution changed due to catalyst speciation as deactivation occurs. At long TOS (> 1000 h), a decrease in GHSV resulted in an increase in light paraffin and olefin selectivities (C1–C5) and a corresponding decrease in C5+ selectivity. This occurred in all three catalysts. This trend is the inverse to what was observed at shorter TOS and also what is reported in the literature. At reduced GHSV, the WGS reaction was enhanced for all three catalysts. The catalyst speciation that could be observed as the catalyst deactivated with TOS, resulted in the formation of magnetite, which is a water gas shift (WGS) active catalyst. Catalyst characterization results suggest that deactivation was caused by oxidation of the catalyst and carbon deposition. It was found that olefin content decreased and paraffin increased with increased pressure; methane selectivity was essentially independent of reaction conditions.
These changes could have an impact on the design of a small-scale gas-to-liquid process designed to run at low pressure. The three differently reduced catalysts had similar optimum operating conditions. The optimum condition for paraffin production is high pressure and low GHSV [20.85 bar (abs): 648 h−1]. For olefins, it is low pressure and low GHSV [1.85 bar (abs): 648 h−1], while C5+ production is favored at high GHSV and is not affected by pressure. Extended operation was achieved quite easily and catalyst performance was stable during extended operation.
References
Muleja AA, Yao Y, Glasser D, Hildebrandt D (2016) Effect of feeding nitrogen to a fixed bed Fischer-Tropsch reactor while keeping the partial pressures of reactants the same. Chem Eng J 293:151–160
Gorimbo J, Lu X, Liu X, Hildebrandt D, Glasser D (2017) A long term study of the gas phase of low pressure Fischer-Tropsch products when reducing an iron catalyst with three different reducing gases. Appl Catal A Gen 534:1–11
Chun DH, Park JC, Hong SY, Lim JT, Kim CS, Lee HT, Il Yang J, Hong S, Jung H (2014) Highly selective iron-based Fischer-Tropsch catalysts activated by CO2-containing syngas. J Catal 317:135–143
Bukur DB, Lang X, Akgerman A, Feng Z (1997) Effect of Process Conditions on Olefin Selectivity during Conventional and Super- critical Fischer−Tropsch Synthesis. Ind Eng Chem Res 36:2580
Todic B, Nowicki L, Nikacevic N, Bukur DB (2016) Fischer-Tropsch synthesis product selectivity over an industrial iron-based catalyst: effect of process conditions. Catal Today 261:28–39
Dictor RA, Bell AT (1986) Fischer-Tropsch synthesis over reduced and unreduced iron oxide catalysts. J Catal 97:121–136
Bukur DB, Nowicki L, Patel SA (1996) Activation studies with an iron Fischer-Tropsch catalyst in fixed bed and stirred tank slurry reactors. Can J Chem Eng 74:399–404
de Smit E, Weckhuysen BM (2008) The renaissance of iron-based Fischer-Tropsch synthesis: on the multifaceted catalyst deactivation behaviour. Chem Soc Rev 37:2758–2781
Tsakoumis NE, Voronov A, Rønning M, Van Beek W, Borg Ø, Rytter E, Holmen A (2012) Fischer–Tropsch synthesis : an XAS / XRPD combined in situ study from catalyst activation to deactivation. J Catal 291:138–148
Khodakov AY, Chu W, Fongarland P (2007) Advances in the development of novel cobalt Fischer−Tropsch Catalysts for synthesis of long-chain hydrocarbons and clean fuels advances in the development of novel cobalt Fischer−Tropsch catalysts for synthesis of long-chain hydrocarbons and clean fuel. Am Chem Soc 107:1692–1744
Mandal P, Chattopadhyay AP (2015) Excellent catalytic activity of magnetically recoverable Fe3O4–graphene oxide nanocomposites prepared by a simple method. Dalt Trans 44:11444–11456
Savost’yanov AP, Narochnyi GB, Yakovenko RE, Bakun VG, Zemlyakov ND (2014) Synthesis of high-molecular-weight hydrocarbons from CO and H2 over a cobalt catalyst. Catal Ind 6:292–297
Yan F, Qian W, Sun Q, Zhang H, Ying W, Fang D (2014) Product distributions and olefin-to-paraffin ratio over an iron-based catalyst for Fischer–Tropsch synthesis. React Kinet Mech Catal 113:471–485
Farias FEM, Sales FG, Fernandes FAN (2008) Effect of operating conditions and potassium content on Fischer-Tropsch liquid products produced by potassium-promoted iron catalysts. J Nat Gas Chem 17:175–178
Atashi H, Razmjooei S, Khorashadizadeh M, Shiva M, Tabrizi FF, Mousavi SAHS (2015) Effects of operating conditions on selectivity of Fe-Co-Mn/MgO at high temperature CO hydrogenation. J Taiwan Inst Chem Eng 54:83–90
Huo C-F, Li Y-W, Jianguo W, Haijun J (2009) Insight into CH4 formation in iron-catalyzed Fischer-Tropsch synthesis. J Am Chem Soc 131:14713–14721
Pérez De Berti IO, Bengoa JF, Stewart SJ, Cagnoli MV, Pecchi G, Marchetti SG (2016) Effect of activation atmosphere in the Fischer–Tropsch Synthesis using a “quasi-model” catalyst of γ-Fe2O3 nanoparticles supported on SBA-15. J Catal 335:36–46
Acknowledgements
The authors are grateful for financial support provided by the University of South Africa, National research foundation of South Africa and the Institute for the Development of Energy for African Sustainability (IDEAS) research unit at UNISA.
Authors’ contributions
JG and AM did the laboratory work. DH and XL supervised the work. All the authors discused the results and the story line. JG wrote the first draft.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gorimbo, J., Muleja, A., Liu, X. et al. Fischer–Tropsch synthesis: product distribution, operating conditions, iron catalyst deactivation and catalyst speciation. Int J Ind Chem 9, 317–333 (2018). https://doi.org/10.1007/s40090-018-0161-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40090-018-0161-4