
Abstract
Nemaline myopathy (NM) is a congenital myopathy of great heterogeneity, characterized by the presence of rods in the cytoplasm of muscle fibers. The samples of 16 nemaline myopathy patients diagnosed by characteristically pathological features went through whole exon sequencing. Clinico-pathological and genetic features of the cases were systematically analyzed. According to the classification of nemaline myopathy by ENMC, 8 cases are typical congenital subtype, 6 cases are childhood/juvenile onset subtype and 2 case are adult onset subtype. In histological findings, characteristic purple-colored rods are discovered under modified gömöri trichrome staining (MGT). Electron microscopy revealed the presence of high electron-dense nemaline bodies around the submucosa and the nucleus nine patients (9/16 56.3%) were detected pathogenic causative mutations, among whom mutations in the NEB gene were the most frequent (6 patients, 66.7%). KBTBD13 gene mutation was discovered in two patients and ACTA1 gene mutation was discovered in 1 patient. Nemaline myopathy is a congenital myopathy with highly clinico-pathological and genetic heterogeneity. NEB gene mutation is the most common mutation, in which splicing change c.21522 +3A > G is hotspot mutation in Chinese NM patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Nemaline myopathy (NM) is a common type of congenital myopathy, which is named by the presence of rod-like structures in skeletal muscle fibers [1, 2]. In 2000, European Neuromuscular Center (ENMC) International Consortium defined six types of NM according to different clinical manifestations [3]: severe, intermediate, typical congenital, childhood/juvenile onset, adult onset and other unusual forms. In nemaline myopathy, various genetic mutations cause structural component changes of skeletal muscle, leading to hypotonia or general weakness predominantly affecting facial, axial, and proximal limb muscles. Until now, 13 mutant genes have been discovered [4,5,6,7,8,9,10,11,12,13]: TPM3, NEB, ACTA1, TPM2, TNNT1, CFL2, KBTBD13, KLHL40, KLHL41, LMOD3, MYPN, MYO18B, TNNT3. In our previous study, we have systematically reviewed the clinical and pathological features of nemaline myopathy patients in China [14]. But there is no systemic research on the genetic features of nemaline myopathy patients in China until now. Here, in order to investigate the clinical features and mutational spectrum of nemaline myopathy in China, we present 16 nemaline myopathy cases from our neuromuscular disease database with clinico-pathological and genetic data.
Patients and methods
Patients
A total of 16 patients were included in the study. From April 1986 to October 2018, 5429 muscle biopsies were performed on patients with suspected myopathy in our department, and 16 patients (0.29%) were diagnosed nemaline myopathy according to characteristic pathological features. Written informed consent was obtained from each patient. The study was approved by the institutional Review Board of Chinese PLA General Hospital.
Clinical information and Histological analysis
Detailed clinical data were collected from each patient, which included detailed personal history (age at onset, initial symptom or sign, motor milestone delay, clinical course and family history); clinical features (distribution of muscle weakness and atrophy, progression pattern, dysphagia and mental retardation); dysmorphic features (pseudo hypertrophy, high arched palate and foot, elongated face and spinal deformity); and muscle power (manual muscle testing by physicians).
Blood samples of the nemaline myopathy patients were also collected and the level of serum creatine kinase (CK) was test by ELISA according to manufacturer’s protocols (Sinopharm Chemical Reagent, Beijing, China). For electromyography and nerve conduction velocity tests, different muscle parts of patients including bilateral deltoid muscle, biceps brachii muscle, quadriceps femoris muscle and anterior tibial muscle were tested by the keypoint 2000 equipment (Dantec Dynamics, Skovlunde, Denmark). After inserting electrode into muscles, the neurologist recorded signals form the muscle at rest and contraction. The nerve velocity was measured by the distance and the time that the signal transmitted along nerves which is recorded by keypoint 2000.
Open muscle biopsy was performed on the clinically affected limb muscles (biceps brachii or quadriceps femoris mostly) under local anesthesia. Following removal, muscle samples were immediately frozen in isopentane cooled with liquid nitrogen and were stored at − 80 °̊C. Transversal serial 5-μm frozen muscle sections were stained with hematoxylin and eosin staining (HE), modified gömöri trichrome staining (MGT), reduced nicotinamide adenine dinucleotide dehydrogenase-tetrazolium reductase staining (NADH-TR). Morphometric evaluation of the muscle specimens was performed under a light microscope (B × 51, Olympus, Tokyo, Japan). 0.5 × 0.5 × 1.0 cm-size muscle sample fixed in 2% glutaraldehyde, post fixed with 1% osmium tetroxide, was embedded in araldite. Ultrathin sections were stained with uranyl acetate and lead citrate and were viewed and photographed with a JEM1230 electron microscope (JEOL, Tokyo, Japan). All reagents were purchased from Sinopharm Chemical Reagent Co Ltd. (Beijing, China).
DNA analysis
Genomic DNA was extracted from peripheral blood or frozen muscle of the patients. First, all coding exons and exon–intron boundaries of ACTA1 and TPM3 were amplified by PCR and analyzed by Sanger sequencing. If no pathogenic mutation was identified in these two genes, other causative genes of congenital myopathies, including NM-related genes (NEB, CFL2, TPM2, TNNT1, KBTBD13, KLHL40, KLHL41, LMOD3, MYO18B, and MYPN), were evaluated by whole exon sequencing (WES). Exon capture was performed using the Illumina TruSeq Exon Enrichment Guide. Sequence reads were mapped to the human reference genome assembly (GRch37/hg19) using Integrative Genomics Viewer (IGV; Broad Institute, Cambridge, MA, USA). Variants potentially affecting protein structure or function, including nonsynonymous variants, frameshifts, or variants affecting splicing, were investigated. The variants showed a depth of more than 50 and a minor allele frequency of less than 0.01. We evaluated the impact of mutations using the SIFT/PROVEAN and Polyphen2 prediction system in the case of point mutations. The splice sites were evaluated with Human Splicing Finder. Sanger sequencing was performed to confirm the mutations found by WES. Because WES has clear limitations in reading the triplicate region of NEB, Sanger fill-in was performed for patients in whom a single heterozygous variant or no variant was identified. In addition, all patient samples were done CNV analysis by targeted array-CGH, as previously described [15].
Results
Clinical features
Details of the patients were shown in Table 1.
From January 1986 to October 2018, 5,429 suspected myopathy cases received muscle biopsies in our center. The proportion of NM cases in this group was 0.29% (16/5,429). In the 16 patients evaluated in this study, there were ten males (M) and six females (F), with a ratio M:F = 1:0.6. The patients’ age ranged from 6 to 49 years, with a mean age 26.7 ± 10.8 years. The disease course ranged from 3 to 35 years, with a mean disease course of 14.4 ± 11.8 years.
According to the classification of nemaline myopathy by European Neuromuscular Center (ENMC) in 2000, 16 patients in our study were divided into three groups: 8 cases belong to typical congenital subtype, 6 cases belong to childhood/juvenile onset subtype and 2 cases belong to adult onset subtype. Patients of typical congenital subtype presented hypotonia and chronic limb weakness during the neonatal period, with delayed motor milestone. The patients of childhood/juvenile onset subtype usually show lower limbs weakness without muscle atrophy as the initial manifestation. While with the progression of disease, lower limbs muscle atrophy will gradually appear. The course of the case with adult onset subtype progressed rapidly, the patient developed trunk and proximal extremities muscle atrophy in several years. Among all the patients, the creatine kinase was normal or slightly elevated, with an average of 103.9 ± 56.8 IU/L (range 32–235 IU/L). EMG results suggested myogenic damage and normal nerve conduction velocity.
Pathological features
As shown in Fig. 1, HE staining revealed variations in the muscle fiber size, with areas of small groups of atrophic muscle fibers. No muscle fiber necrosis and no inflammatory cell infiltration were discovered. Collection of eosinophilic rods assembled under the sarcolemma. Typical purple-colored rods were observed in the subsarcolemmal region of muscle fibers stained with MGT staining. NADH-TR staining could not show the rods, while the light stained area without oxidative enzyme activity usually means collections of rods. Electron microscopy showed that some myofibrillar dysplasia was manifested as myofilament rupture, irregular alignment and different myofibrils. The Z-line of myofibrils was small, irregular or disappeared. There are many uniform high electron density rods around the submucosa and the nucleus. Most rods were arranged equally with myofibrils, even a segment of myofibrils.

Histochemical, Ultrastructural findings in the muscle biopsy samples from nemaline myopathy patients (Case 3). HE staining indicates that collection of rods of eosinophilic rods assemble under the sarcolemma of many muscle fiber membranes. (X400, a); MGT staining suggests that typical purple-colored rods are observed in the subsarcolemmal region of muscle fibers. (X400, b); NADH-TR staining shows areas deviod of oxidative enzyme activity in which rods exist. (X400, c); Electron microscopy show rod-like structure with high electron density among myofibrils. (X8000, d); The rod-like structure with high electron density among segments of myofibrils, and the surrounding myofibrillar structure is ruptured. (X15000, e Enlarged figure of d); Electron microscopy show that there are many rod-like structures with different electron densities near the nucleus (X10000, f)
Genetic analysis results
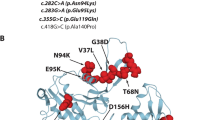
All 16 patients were underwent genetic analysis and nine patients (9/16 56.3%) were detected gene mutations. Among them, NEB gene mutation was discovered in six patients, KBTBD13 gene mutation was discovered in two patients and ACTA1 gene mutation was discovered in one patient. For the six patients harboring NEB gene mutation, we discovered the splice change c.21522 + 3A > G in five patients, which have been reported before [15]. And one nonsense change c.4417C > T and two splice changes c.23233-1G > T/c.5343 + 1G > A were newly discovered nucleotide changes according HGMD database for NEB gene in nemaline myopathy patients. For the two patients harboring KBTBD13 gene mutation, two missense mutations were discovered, the c.1170G > C (p.K390N) mutation was reported before [7] and the c.1169G > C (p.K390N) was novel mutation. For one patient harboring ACTA1 gene mutation, the missense mutation c.956 T > C (p.I319T) was a novel one which has not been reported before.
Discussion
Nemaline myopathy is one type of congenital myopathy, the cause of which is associated with multiple gene mutations. Our study systematically analyze the clinical and pathological characteristics of nemaline myopathy. And importantly, the genetic features of 16 cases of nemaline myopathy collected in our center were studies by WES, in which nine mutations were discovered, which broaden gene mutation spectrum of nemaline myopathy in China.
According to our data, nemaline myopathy is a rare neuromuscular disease, which accounts for 0.29% in all the 5429 patients of our myopathy database. The clinical presentation of NM patients is greatly variable, ranging from neonatal death to mild limb weakness. The muscle weakness of nemaline myopathy is usually generalized, with involvement of the neck flexors, the face and proximal muscles. Traditionally, proximal muscle weakness is characteristic in nemaline myopathy, while some patients suffer from distal muscle weakness, which suggests the heterogeneity of clinical manifestation and brings challenge for the diagnosis of nemaline myopathy. Case 7 and 11 in our study showed distal muscle weakness with NEB gene mutations, which is in accordance with the study by Kiiski, who reported distal nemaline/cap myopathy with a large deletion in the nebulin gene [16]. According to the ENMC classification, the 16 patients in our study were divided into three groups: typical congenital, childhood/juvenile onset, adult onset, and the proportion is 50%, 37.5%, 12.5% respectively, which is similar to the previous research [17]. There are two adult onset cases (1 and 16) with different disease progression. Case 1 in our study is sporadic late-onset nemaline myopathy (SLONM) subtype, which is usually immune-mediated and related to monoclonal gammopathy of undermined significance (MGUS) [18, 19]. He showed rapidly progressive disease course with limbs and trunk weakness [20]. Without approriate treatment, the prognosis of SLONM is unfavorable and the patients might suffer from progressive respiratory failure. Previous studies have suggested that early monitoring and appropriate treatment of respiratory function for NM patients could effectively improve their quality of life and prolong survival. Case 16 had the age of onset at 25 years old, while he had family history and harbored NEB gene mutation. We assume that the reason for his onset so late is due to the chronic progression of the disease, and he did not show symptoms until the age of 25. With the deepening understanding of the NM, in 2019, Sewry and his colleagues proposed a new classification for memaline myopathies which gave a strong genotype–phenotype correlation, which is a great progression with the development of gene detection technique [21]. According to the new classification, cases in our study could be divided into three groups: congenital nemaline myopathy (Case 3, 7, 8, 11, 13, 14), mild (childhood or juvenile onset) nemaline myopathy (Case 16) and Childhood-onset nemaline myopathy with slowness of movements and core-rod histology (Case 5, 10).
With great clinical heterogeneity of nemaline myopathy, the diagnosis of NM is based on the appearance of dark red staining inclusions with the MGT stainning in muscle pathology [2]. While HE stained sections of skeletal muscle from patients with NM can appear normal, or exhibit some fiber size variation. By electron microscopy, rods are electron dense ovoid structures which are often parallel to the long axis of the sarcomere or near the Z disc, measuring 1–7 µm in length and 0.3–2 µm in width. Luther et al. [22] have demonstrated that rods have a lattice structure similar to Z-disks on electron microscope and the major constituent of rods is α-actinin, which is exactly the component of Z disc.
As congenital myopathy, nemaline myopathy is mainly caused by gene mutations which is related to different components of striated muscle. With the development of gene sequencing technology, 13 genes have been discovered to be associated to the onset of nemaline myopathy. Among these, NEB gene mutations are responsible for 50% of the cases of NM [23]. NEB gene mutations are seen most commonly in autosomal recessive NM cases and these patients present as typical congenital NM [24]. NEB gene is a large one which contains 183 exons in a 249 kb genomic region. It encodes a 600 to 900 kD protein nebuline which is one component of thin filaments within the sarcomeres of skeletal muscle and function as molecular ruler in the regulation of muscle contraction, Z-disc formation, myofibril organization and assembly [25]. Kiss and colleagues discovered shorter thin filament as well as impaired tropomyosin and troponin movement in nebulin-knockout mice, which indicates that nebulin participate the thin filament activation and cross-bridge recruitment [26]. In our study, 67% patients harbor NEB gene mutations. And all the patients with NEB gene mutations showed relatively benign clinical course with slow clinical progression. Splicing change c.21522 + 3A > G has been reported before and the research reported only one case [15]. While in our research, 5 out 6 unrelated nemaline myopathy patients with NEB mutations harbored this mutational site, which reminded us that this mutational site might be a mutational hotspot for nemaline myopathy patients in China. And there were three NEB mutations firstly discovered in our study including one nonsense change c.4417C > T and two splicing change c.23233-1G > T/c.5343 + 1G > A which give rise to truncated nebulin without normal function and results in reduced cross-bridge interaction and impaired muscle force.
Except NEB gene mutation, ACTA1 gene mutation is the most common one that cause nemaline myopathy. The ACTA1 gene encodes skeletal muscle α-actin, the principal actin isoform in adult skeletal muscle, which is highly conserved and forms the core of the thin filament of the sarcomere. Sarcomere contractility of the patients with ACTA1 mutations reduces due to the decreased myosin heads binding to actin, which is mainly composed of α-actin encoded by ACTA1 gene [27]. The clinical manifestations of nemaline myopathy caused by ACTA1 gene mutations is often severe, with the neonate presenting as a floppy infant, sometimes with failure to establish respiration and spontaneous movements. More rarely, mutations in ACTA1 may cause the intermediate, mild, or typical forms of nemaline myopathy [28]. The clinical subtype of the case in our research is typical congenital, which is a rare one. The missense change c.956 T > C in highly conserved ACTA1 gene causes disrupted protein structure and finally impaired muscle contraction.
KBTBD13, unlike other mutant genes encoding the thin filament component, which encodes Kelch-like proteins associated with thin filament regulation. The protein it encodes has an N-terminal BTB/POZ domain, followed by a central α-helical linker region and a C-terminal Kelch repeat domain that contains five repeats and is predicted to form a β-propeller structure. Mutations in KBTBD13 gene were found to cause dominantly inherited nemaline myopathy, which features onset of weakness in early childhood and the presence of both nemaline bodies and core-like formations in the muscle fibers. Cullin-3-RING ubiquitin ligase mediates the proteostasis in skeletal muscle and acts as critical regulator in the muscle development and function. As the E3 ubiquitin ligase Cullin-3 substrate adaptors, BTB/Kelch domain proteins encoded by KBTBD13 participate in the degradation of non-muscle α-actinins (ACTN1) during myogenesis [29, 30]. The newly discovered missense change c.1169A > C in KBTBD13 gene is located in the highly conserved fifth repeats of Kelch domain, is predicted to damage the corresponding strands of the molecular β-propeller blades of KBTBD13 protein. Then the accumulation of ACTN1 in muscle fibers will interfere the development, function and maintenance of skeletal muscle and finally give rise to reduced muscle force.
It is worth noting that nemaline myopathy cases with KBTBD13 gene mutations are rarely reported, while the muscle pathology shows characteristic rods and cores lesions [7, 31]. The characteristic pathological features suggest genotype–phenotype correlation, which has certain hints for genetic detection. In fact, it should be noted that not every patient’s pathological manifestations are typical rods and cores, which may manifest as an unevenness of stain resembling core-like areas and need to be confirmed by electron microscopy.
Conclusion
In our study, we systemically reviewed the clinical, pathological and genetic characteristics of nemaline myopathy patients in China. The NEB, KBTBD13, and ACTA1 gene mutations were discovered in 9 out of 16 patients in our database. Mutational hotspot c.21522 + 3A > G in NEB gene was discovered in nemaline myopathy patients in China and three mutational sites in NEB gene were reported which haven't been discovered before. The clinical and pathological characteristics of nemaline myopathy patients in China was systematically reviewed and gene mutation spectrum was broadened. Unfortunately, seven patients in our study who underwent genetic analysis had no mutation been discovered in known mutational genes related to nemaline myopathy. This reminded us that there might be undiscovered mutational genes in the cases and nemaline myopathy is a disease with great genetic heterogeneity. With the development of genetic sequencing technology, more and more mutational genes may be discovered and the pathogenesis of nemaline myopathy may be illustrated clearly.
References
Gilbreath HR, Castro D, Iannaccone ST (2014) Congenital myopathies and muscular dystrophies. Neurol Clin 32(3):689–703. https://doi.org/10.1016/j.ncl.2014.04.006
Mah JK, Joseph JT (2016) An overview of congenital myopathies. Continuum (Minneap Minn) 22(6):1932–1953. https://doi.org/10.1212/CON.0000000000000404
Wallgren-Pettersson C, Laing NG (2000) Report of the 70th ENMC International Workshop: nemaline myopathy, 11–13 June 1999, Naarden. Netherlands Neuromuscul Disord 10(4–5):299–306
Nowak KJ, Wattanasirichaigoon D, Goebel HH, Wilce M, Pelin K, Donner K, Jacob RL, Hubner C, Oexle K, Anderson JR, Verity CM, North KN, Iannaccone ST, Muller CR, Nurnberg P, Muntoni F, Sewry C, Hughes I, Sutphen R, Lacson AG, Swoboda KJ, Vigneron J, Wallgren-Pettersson C, Beggs AH, Laing NG (1999) Mutations in the skeletal muscle alpha-actin gene in patients with actin myopathy and nemaline myopathy. Nat Genet 23(2):208–212. https://doi.org/10.1038/13837
Pelin K, Hilpela P, Donner K, Sewry C, Akkari PA, Wilton SD, Wattanasirichaigoon D, Bang ML, Centner T, Hanefeld F, Odent S, Fardeau M, Urtizberea JA, Muntoni F, Dubowitz V, Beggs AH, Laing NG, Labeit S, de la Chapelle A, Wallgren-Pettersson C (1999) Mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Proc Natl Acad Sci USA 96(5):2305–2310
Wattanasirichaigoon D, Swoboda KJ, Takada F, Tong HQ, Lip V, Iannaccone ST, Wallgren-Pettersson C, Laing NG, Beggs AH (2002) Mutations of the slow muscle alpha-tropomyosin gene, TPM3, are a rare cause of nemaline myopathy. Neurology 59(4):613–617
Sambuughin N, Yau KS, Olive M, Duff RM, Bayarsaikhan M, Lu S, Gonzalez-Mera L, Sivadorai P, Nowak KJ, Ravenscroft G, Mastaglia FL, North KN, Ilkovski B, Kremer H, Lammens M, van Engelen BG, Fabian V, Lamont P, Davis MR, Laing NG, Goldfarb LG (2010) Dominant mutations in KBTBD13, a member of the BTB/Kelch family, cause nemaline myopathy with cores. Am J Hum Genet 87(6):842–847. https://doi.org/10.1016/j.ajhg.2010.10.020
Gupta VA, Ravenscroft G, Shaheen R, Todd EJ, Swanson LC, Shiina M, Ogata K, Hsu C, Clarke NF, Darras BT, Farrar MA, Hashem A, Manton ND, Muntoni F, North KN, Sandaradura SA, Nishino I, Hayashi YK, Sewry CA, Thompson EM, Yau KS, Brownstein CA, Yu TW, Allcock RJ, Davis MR, Wallgren-Pettersson C, Matsumoto N, Alkuraya FS, Laing NG, Beggs AH (2013) Identification of KLHL41 mutations implicates BTB-kelch-mediated ubiquitination as an alternate pathway to myofibrillar disruption in nemaline myopathy. Am J Hum Genet 93(6):1108–1117. https://doi.org/10.1016/j.ajhg.2013.10.020
Ravenscroft G, Miyatake S, Lehtokari VL, Todd EJ, Vornanen P, Yau KS, Hayashi YK, Miyake N, Tsurusaki Y, Do H, Saitsu H, Osaka H, Yamashita S, Ohya T, Sakamoto Y, Koshimizu E, Imamura S, Yamashita M, Ogata K, Shiina M, Bryson-Richardson RJ, Vaz R, Ceyhan O, Brownstein CA, Swanson LC, Monnot S, Romero NB, Amthor H, Kresoje N, Sivadorai P, Kiraly-Borri C, Haliloglu G, Talim B, Orhan D, Kale G, Charles AK, Fabian VA, Davis MR, Lammens M, Sewry CA, Manzur A, Muntoni F, Clarke NF, North KN, Bertini E, Nevo Y, Willichowski E, Silberg IE, Topaloglu H, Beggs AH, Allcock RJ, Nishino I, Wallgren-Pettersson C, Matsumoto N, Laing NG (2013) Mutations in KLHL40 are a frequent cause of severe autosomal-recessive nemaline myopathy. Am J Hum Genet 93(1):6–18. https://doi.org/10.1016/j.ajhg.2013.05.004
Yuen M, Sandaradura SA, Dowling JJ, Kostyukova AS, Moroz N, Quinlan KG, Lehtokari VL, Ravenscroft G, Todd EJ, Ceyhan-Birsoy O, Gokhin DS, Maluenda J, Lek M, Nolent F, Pappas CT, Novak SM, D’Amico A, Malfatti E, Thomas BP, Gabriel SB, Gupta N, Daly MJ, Ilkovski B, Houweling PJ, Davidson AE, Swanson LC, Brownstein CA, Gupta VA, Medne L, Shannon P, Martin N, Bick DP, Flisberg A, Holmberg E, Van den Bergh P, Lapunzina P, Waddell LB, Sloboda DD, Bertini E, Chitayat D, Telfer WR, Laquerriere A, Gregorio CC, Ottenheijm CA, Bonnemann CG, Pelin K, Beggs AH, Hayashi YK, Romero NB, Laing NG, Nishino I, Wallgren-Pettersson C, Melki J, Fowler VM, MacArthur DG, North KN, Clarke NF (2014) Leiomodin-3 dysfunction results in thin filament disorganization and nemaline myopathy. J Clin Invest 124(11):4693–4708. https://doi.org/10.1172/JCI75199
Malfatti E, Bohm J, Lacene E, Beuvin M, Romero NB, Laporte J (2015) A Premature stop codon in MYO18B is associated with severe nemaline myopathy with cardiomyopathy. J Neuromuscul Dis 2(3):219–227. https://doi.org/10.3233/JND-150085
Miyatake S, Mitsuhashi S, Hayashi YK, Purevjav E, Nishikawa A, Koshimizu E, Suzuki M, Yatabe K, Tanaka Y, Ogata K, Kuru S, Shiina M, Tsurusaki Y, Nakashima M, Mizuguchi T, Miyake N, Saitsu H, Ogata K, Kawai M, Towbin J, Nonaka I, Nishino I, Matsumoto N (2017) Biallelic mutations in MYPN, encoding myopalladin, are associated with childhood-onset, slowly progressive nemaline myopathy. Am J Hum Genet 100(1):169–178. https://doi.org/10.1016/j.ajhg.2016.11.017
Sandaradura SA, Bournazos A, Mallawaarachchi A, Cummings BB, Waddell LB, Jones KJ, Troedson C, Sudarsanam A, Nash BM, Peters GB, Algar EM, MacArthur DG, North KN, Brammah S, Charlton A, Laing NG, Wilson MJ, Davis MR, Cooper ST (2018) Nemaline myopathy and distal arthrogryposis associated with an autosomal recessive TNNT3 splice variant. Hum Mutat 39(3):383–388. https://doi.org/10.1002/humu.23385
Yin X, Pu CQ, Wang Q, Liu JX, Mao YL (2014) Clinical and pathological features of patients with nemaline myopathy. Mol Med Rep 10(1):175–182. https://doi.org/10.3892/mmr.2014.2184
Lee JM, Lim JG, Shin JH, Park YE, Kim DS (2017) Clinical and genetic diversity of nemaline myopathy from a single neuromuscular center in Korea. J Neurol Sci 383:61–68. https://doi.org/10.1016/j.jns.2017.10.020
Kiiski KJ, Lehtokari VL, Vihola AK, Laitila JM, Huovinen S, Sagath LJ, Evila AE, Paetau AE, Sewry CA, Hackman PB, Pelin KB, Wallgren-Pettersson C, Udd B (2019) Dominantly inherited distal nemaline/cap myopathy caused by a large deletion in the nebulin gene. Neuromuscul Disord 29(2):97–107. https://doi.org/10.1016/j.nmd.2018.12.007
Ryan MM, Schnell C, Strickland CD, Shield LK, Morgan G, Iannaccone ST, Laing NG, Beggs AH, North KN (2001) Nemaline myopathy: a clinical study of 143 cases. Ann Neurol 50(3):312–320
Uruha A, Benveniste O (2017) Sporadic late-onset nemaline myopathy with monoclonal gammopathy of undetermined significance. Curr Opin Neurol 30(5):457–463. https://doi.org/10.1097/WCO.0000000000000477
Monforte M, Primiano G, Silvestri G, Mirabella M, Luigetti M, Cuccagna C, Ricci E, Servidei S, Tasca G (2018) Sporadic late-onset nemaline myopathy: clinical, pathology and imaging findings in a single center cohort. J Neurol 265(3):542–551. https://doi.org/10.1007/s00415-018-8741-y
Schnitzler LJ, Schreckenbach T, Nadaj-Pakleza A, Stenzel W, Rushing EJ, Van Damme P, Ferbert A, Petri S, Hartmann C, Bornemann A, Meisel A, Petersen JA, Tousseyn T, Thal DR, Reimann J, De Jonghe P, Martin JJ, Van den Bergh PY, Schulz JB, Weis J, Claeys KG (2017) Sporadic late-onset nemaline myopathy: clinico-pathological characteristics and review of 76 cases. Orphanet J Rare Dis 12(1):86. https://doi.org/10.1186/s13023-017-0640-2
Sewry CA, Laitila JM, Wallgren-Pettersson C (2019) Nemaline myopathies: a current view. J Muscle Res Cell Motil 40(2):111–126. https://doi.org/10.1007/s10974-019-09519-9
Luther PK, Squire JM (2002) Muscle Z-band ultrastructure: titin Z-repeats and Z-band periodicities do not match. J Mol Biol 319(5):1157–1164. https://doi.org/10.1016/S0022-2836(02)00372-8
Ottenheijm CA, Hooijman P, DeChene ET, Stienen GJ, Beggs AH, Granzier H (2010) Altered myofilament function depresses force generation in patients with nebulin-based nemaline myopathy (NEM2). J Struct Biol 170(2):334–343. https://doi.org/10.1016/j.jsb.2009.11.013
Lehtokari VL, Kiiski K, Sandaradura SA, Laporte J, Repo P, Frey JA, Donner K, Marttila M, Saunders C, Barth PG, den Dunnen JT, Beggs AH, Clarke NF, North KN, Laing NG, Romero NB, Winder TL, Pelin K, Wallgren-Pettersson C (2014) Mutation update: the spectra of nebulin variants and associated myopathies. Hum Mutat 35(12):1418–1426. https://doi.org/10.1002/humu.22693
Donner K, Sandbacka M, Lehtokari VL, Wallgren-Pettersson C, Pelin K (2004) Complete genomic structure of the human nebulin gene and identification of alternatively spliced transcripts. Eur J Hum Genet 12(9):744–751. https://doi.org/10.1038/sj.ejhg.5201242
Kiss B, Lee EJ, Ma W, Li FW, Tonino P, Mijailovich SM, Irving TC, Granzier HL (2018) Nebulin stiffens the thin filament and augments cross-bridge interaction in skeletal muscle. Proc Natl Acad Sci USA 115(41):10369–10374. https://doi.org/10.1073/pnas.1804726115
Joureau B, de Winter JM, Conijn S, Bogaards SJP, Kovacevic I, Kalganov A, Persson M, Lindqvist J, Stienen GJM, Irving TC, Ma W, Yuen M, Clarke NF, Rassier DE, Malfatti E, Romero NB, Beggs AH, Ottenheijm CAC (2018) Dysfunctional sarcomere contractility contributes to muscle weakness in ACTA1-related nemaline myopathy (NEM3). Ann Neurol 83(2):269–282. https://doi.org/10.1002/ana.25144
Moreno CAM, Abath Neto O, Donkervoort S, Hu Y, Reed UC, Oliveira ASB, Bonnemann C, Zanoteli E (2017) Clinical and histologic findings in ACTA1-related nemaline myopathy: case series and review of the literature. Pediatr Neurol 75:11–16. https://doi.org/10.1016/j.pediatrneurol.2017.04.002
Papizan JB, Vidal AH, Bezprozvannaya S, Bassel-Duby R, Olson EN (2018) Cullin-3-RING ubiquitin ligase activity is required for striated muscle function in mice. J Biol Chem 293(23):8802–8811. https://doi.org/10.1074/jbc.RA118.002104
Blondelle J, Tallapaka K, Seto JT, Ghassemian M, Clark M, Laitila JM, Bournazos A, Singer JD, Lange S (2019) Cullin-3 dependent deregulation of ACTN1 represents a new pathogenic mechanism in nemaline myopathy. JCI Insight. https://doi.org/10.1172/jci.insight.125665
Kang ZX, Wei XJ, Miao J, Gao YL, Wang ZY, Yu XF (2020) A family with nemaline myopathy type 6 caused by hseterozygous mutation (c1222C>T) in the KBTBD13 gene in China: a case report. Neuropathology 40(1):104–108. https://doi.org/10.1111/neup.12610
Acknowledgement
The authors would like to thank the nemaline myopathy patients for their participation in the study.
Funding
This research was supported by National Natural Science Foundation for Young Scientists of China (81701232, 81601086), The Natural Science Foundation of Shanxi Province (CN) (201601D101096).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Chinese PLA General hospital research committee and with the 1964 Helsinki declaration and its later amendments of comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants inlcuded in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yin, X., Pu, C., Wang, Z. et al. Clinico-pathological features and mutational spectrum of 16 nemaline myopathy patients from a Chinese neuromuscular center. Acta Neurol Belg 122, 631–639 (2022). https://doi.org/10.1007/s13760-020-01542-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13760-020-01542-9