
Abstract
Cancer disease is developing all over the world mainly in developing countries. We should learn more about DNA–ligand interactions to design new drugs that target biological activities like transcription, replication and translation of particular genes. To understand the mechanism of action and design-specific DNA binders, the evaluation of DNA–ligand interactions is critical. Novel barbituric acid derivatives based on (benzyloxy)benzaldehydes were synthesized and evaluated as DNA-binding agents. Among products, molecular docking studies revealed that 4j and 4m have the best interactions with the ctDNA via the minor groove binding. These results were approved by the quantum mechanics calculations. The interaction profiles of the selected compound (4j and 4m) with DNA were evaluated by UV–Visible titration. UV–Visible titration data confirm this interaction. According to the molecular modeling results, the Structure–Activity relationships for all synthesized barbituric acid derivatives were proposed. It was observed that N,N-dimethyl barbituric acid/4-hydroxybenzaldehyde derivatives have better DNA interactions than barbituric acid/vanillin and barbituric acid/3-hydroxybenzaldehyde derivatives.
Similar content being viewed by others


Introduction
During the cell cycle, the DNA molecule loses its twisted state and its strands disintegrate. This event makes it possible to copy its genetic information [1].
Based on the difference in the rate of transcription of genetic information in normal and cancer cells, the effect of anticancer drugs on the DNA chain of cancer cells is greater than that of normal cells. This process is attractive to researchers and is the source of many anticancer drugs [2]. In recent years, many studies on the binding of organic compounds to DNA have been performed. Studying drug–DNA interaction mechanisms is crucial for designing the beneficial drugs that specifically target DNA [3]. Generally, drugs bind to the DNA through two types of covalent and non-covalent interactions [4].
Drug–DNA Covalent binding is irreversible, leading to complete inhibition of the cell division process and eventual cell death. Cyclophosphamide, Carmustine and Busulfan are examples of drugs that covalently bind to DNA. These drugs are often cytotoxic, non-selective, and have many side effects that can even cause new types of cancer in patients [5]. Consequently, drugs that act through non-covalent interactions with DNA are more attractive to researchers.
Non-covalent interactions cause some drugs to be located between adjacent nucleotide bases to form the DNA–ligand complex. Popular interactions that stabilize the DNA–ligand complex include van der Waals forces, hydrogen bonding, hydrophobic attractions, and charge-transfer forces. DNA-binding agents stop replication in the DNA chain of cancer cells and induce cell death [6].
Actinomycin, Doxorubicin, and Daunorubicin are examples of drugs that bind non-covalently to DNA [7].
One of the applications of spectrophotometric methods is to determine the constant binding of the drug to DNA. UV absorption spectroscopy is the simplest and most common method of studying DNA stability and its interactions with small molecules.
The easiest way to determine the interaction between DNA and drug is the comparison of maximum wavelength absorption (λmax) of free DNA with the λmax of drug-bound DNA. The observed changes in λmax are considered a parameter to evaluate the strength of the interaction between DNA and the drug [8]. Molecules such as Berenil [9] and Netropsin [10], which have a long linear structure, can become curved and enter the minor groove of DNA.
This study aimed to: (a) synthesize new long linear structure derivatives of barbituric acid, (b) in-silico study their interaction with DNA through docking and simulation of molecular dynamics, and finally (c) evaluate this interaction by UV spectroscopy technique.
Materials and methods
Materials and instrumental
Solvents and chemicals were purchased from Merck. ctDNA (Deoxyribonucleic acid sodium salt from calf thymus DNA) were purchased from Sigma.
Melting points are uncorrected and were measured on a Stuart SMP3 apparatus. IR spectra of products were recorded using the ALPHA II Compact FT-IR spectrometer on KBr disks. The NMR (1H and 13C) spectroscopy was performed using a Varian-INOVA 500 MHz and Bruker Ascend-400 MHz spectrometer. The mass spectra of products were recorded on an Agilent Technology (HP 5975C MSD) mass spectrometer operating at an ionization potential of 70 eV.
The UV–Vis absorption spectra were measured using a PerkinElmer apparatus with 1 cm quartz cuvette.
General procedure for the synthesis of (benzyloxy)benzaldehydes 3
In a 25-mL round-bottom flask, hydroxyl benzaldehyde 1a-c (1 mmol) and benzyl halides 2 (3 mmol) were dissolved in 3 mL DMF. Then 1 mmol potassium carbonate was added and the mixture was stirred at room temperature for 24 h. At the end of the reaction (followed by the TLC, n-hexane/EtOAc, 4:1), the reaction mixture was poured into the mixture of water–ice. The precipitates were separated, dried and used for the subsequent reaction without further purification.
General procedure for the synthesis of compounds 4a-r
In a 25-mL round-bottom flask, 1 mmol (benzyloxy)benzaldehydes 3 and 1 mmol barbituric acid or N,N-dimethyl barbituric acid were added to 2 mL glacial acetic acid and 2 mL ethanol. Then, the reaction mixture was stirred at room temperature for 24 h. After filtration, the precipitate was washed with water and crystallized in methanol. All products are yellow solid.
Physical and spectral data of products
4a: 5-(4-(benzyloxy)benzylidene)pyrimidine-2,4,6(1H,3H,5H)-trione [11]
Melting point: 270–275 °C.
1H-NMR (400 MHz, DMSO-d6) δ: 11.36 (s, 1H), 11.19 (s, 1H), 8.34–8.36 (d, J = 8.9 Hz, 2H), 8.25 (s, 1H), 7.48 (d, J = 7.0 Hz, 2H), 7.41 (t, J = 7.3 Hz, 2H), 7.35 (d, J = 14.4 Hz, 1H), 7.13 (s, 2H),, 5.24 (s, 2H). IR (KBr, cm−1) v: 3052.13, 2858.48, 1758.22, 1658.11, 1542.94.
4b: 5-(4-((2-fluorobenzyl)oxy)benzylidene)pyrimidine-2,4,6(1H,3H,5H)-trione
Melting point: 260–265 °C.
1H-NMR (499 MHz, DMSO-d6) δ: 11.30 (s, 1H), 11.17 (s, 1H), 8.34–8.36 (d, J = 8.5 Hz, 2H), 8.25 (s, 1H), 7.56–7.59 (t, J = 7.6 Hz, 1H), 7.42–7.46 (q, J = 9.98 Hz, 1H), 7.23–7.28 (q, J = 9.98 Hz, 2H), 7.14–7.16 (d, J = 8.7 Hz, 2H), 5.26 (s, 2H). 13C-NMR (125 MHz, DMSO-d6) δ: 164.34, 162.70, 162.61, 155.26, 150.67, 137.83, 131.38, 131.17, 131.24, 125.99, 125.07, 123.67, 123.56, 116.32, 116.08, 115.87, 115.00, 64.48. IR (KBr, cm−1) v: 3036.23, 2872.11, 1732.57, 1663.76, 1526.92, 1173.30. MS (m/z): 340.2.
4c: 5-(4-((3-fluorobenzyl)oxy)benzylidene)pyrimidine-2,4,6(1H,3H,5H)-trione
Melting point: 265–268 °C.
1H-NMR (499 MHz, DMSO-d6) δ: 11.30 (s, 1H), 11.17 (s, 1H), 8.34–8.36 (d, J = 9.98 Hz, 2H), 8.24 (s, 1H), 7.42–7.47 (q, J = 9.98 Hz, 1H), 7.29–7.31 (d, J = 9.98 Hz, 2H), 7.12–7.19 (m, 3H), 5.26 (s, 2H).13C-NMR (125 MHz, DMSO-d6) δ: 164.34, 163.64, 162.67, 161.69, 155.27, 150.67, 139.77, 137.83, 131.06, 125.95, 124.19, 116.28, 115.39, 115.23, 115.13, 114.99, 114.81, 69.22. IR (KBr, cm−1) v: 3200.91, 3069.84, 2857.37, 1743.91, 1671.79, 1529.76, 1176.17. MS (m/z): 340.2
4d: 5-(4-((4-fluorobenzyl)oxy)benzylidene)pyrimidine-2,4,6(1H,3H,5H)-trione
Melting point: 303–306 °C.
1H-NMR (499 MHz, DMSO-d6) δ: 11.29 (s, 1H), 11.17 (s, 1H), 8.36–8.38 (d, J = 9.98 Hz, 2H), 8.24 (s, 1H), 7.53–7.56 (t, J = 6.99 Hz, 2H), 7.23–7.27 (m, 2H), 7.14–7.16 (t, J = 9.98 Hz, 2H), 5.26 (s, 2H).13C-NMR (125 MHz, DMSO-d6) δ: 164.35, 163.35, 162.84, 162.62, 161.40, 155.32, 150.67, 137.87, 133.08, 130.73, 125.85, 116.18, 115.90, 115.73, 115.12, 114.99, 114.81, 69.39. IR (KBr, cm−1) v: 3190.58, 3057.91, 2867.82, 1732.10, 1665.50, 1500.81, 1172.83. MS (m/z): 340.2
4e: 5-(4-((4-chlorobenzyl)oxy)benzylidene)pyrimidine-2,4,6(1H,3H,5H)-trione
Melting point: 306–310 °C.
1H-NMR (499 MHz, DMSO-d6) δ: 11.29 (s, 1H), 11.17 (s, 1H), 8.34–8.35 (d, J = 8.5 Hz, 2H), 8.25 (s, 1H), 7.50–7.45 (q, J = 9.6 Hz, 4H), 7.13–7.11 (d, J = 8.6 Hz, 2H), 5.23 (s, 2H). 13C-NMR (125 MHz, DMSO-d6) δ: 164.34, 162.73, 162.61, 155.27, 150.66, 137.84, 135.89, 133.16, 130.17, 129.00, 125.92, 115.73, 116.25, 115.14, 69.24. IR (KBr, cm−1) v: 3189.47, 3064.58, 2863.98, 1730.62, 1670.69, 1529.14, 1170.74. MS (m/z): 356.2
4f: 5-(4-((4-bromobenzyl)oxy)benzylidene)pyrimidine-2,4,6(1H,3H,5H)-trione
Melting point: 305–309 °C.
1H-NMR (499 MHz, DMSO-d6) δ: 11.30 (s, 1H), 11.17 (s, 1H), 8.33–8.35 (d, J = 7.98 Hz, 2H), 8.25 (s, 1H), 7.59–7.57 (J = 7.98 Hz, 2H), 7.41–7.43 (J = 6.98 Hz, 2H), 7.10–7.12 (J = 9.48 Hz, 2H), 5.20 (s, 2H). 13C-NMR (125 MHz, DMSO-d6) δ: 164.34, 162.71, 162.61, 155.27, 150.66, 137.84, 136.30, 131.92, 130.45, 125.92, 121.70, 116.25, 115.14, 64.27. IR (KBr, cm−1) v: 3188.73, 3066.39, 1730.22, 1671.19, 1528.23, 1171.59. MS (m/z): 402.1
4g: 5-(4-((4-bromobenzyl)oxy)-3-methoxybenzylidene)pyrimidine-2,4,6(1H,3H,5H)-trione
Melting point: 293–266 °C.
1H-NMR (499 MHz, DMSO-d6) δ: 11.31 (s, 1H), 11.19 (s, 1H), 8.41 (s, 1H), 8.25 (s, 1H), 7.85–7.87 (d, J = 8.6 Hz, 2H), 7.85–7.87 (d, J = 8.6 Hz, 1H), 7.58–7.60 (d, J = 10.47 Hz, 2H), 7.58–7.60 (d, J = 10.47 Hz, 2H), 7.15–7.17 (d, J = 8.5 Hz, 1H), 5.22 (s, 2H), 3.82 (s, 3H). 13C-NMR (125 MHz, DMSO-d6) δ: 164.41, 162.80, 155.76, 152.79, 150.64, 148.47, 136.20, 131.93, 131.74, 130.52, 130.50, 126.09, 121.74. 117.58, 116.01, 112.87, 69.59, 55.97. IR (KBr, cm−1) v: 3308.66, 3195.03, 3083.67, 1728.09, 1680.49, 1551.47, 1267.86. MS (m/z): 432.1
4h: 5-(4-((4-chlorobenzyl)oxy)-3-methoxybenzylidene)pyrimidine-2,4,6(1H,3H,5H)-trione
Melting point: 285–290 °C.
1H-NMR (499 MHz, DMSO-d6) δ: 11.30 (s, 1H), 11.18 (s, 1H), 8.40 (s, 1H), 8.24 (s, 1H), 7.85–7.87 (d, J = 8.48 Hz, 1H), 7.47 (s, 4H), 7.16–7.18 (d, J = 8.4, 1H), 5.23 (s, 2H), 3.81 (s, 3H). 13C-NMR (125 MHz, DMSO-d6) δ: 164.41, 162.81, 155.75, 152.81, 150.64, 148.48, 135.87, 133.19, 131.74, 130.24, 129.01, 126.09, 117.59, 116.01, 112.87, 69.55, 55.97. IR (KBr, cm−1) v: 3307.14, 3079.84, 1728.19, 1680.62, 1551.86. MS (m/z): 386.1
4i: 5-(4-((2-fluorobenzyl)oxy)benzylidene)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione
Melting point: 197–201 °C.
1H-NMR (499 MHz, DMSO-d6) δ: 8.31–8.32 (d, J = 6.48 Hz, 3H), 7.57–7.60 (t, J = 8.483 Hz, 1H), 7.42–7.45 (m, 1H), 7.23–7.29 (q, J = 8.98 Hz, 2H), 7.15–7.17 (d, J = 7.98 Hz, 2H), 5.27 (s, 2H), 3.20–3.22 (d, 6H). 13C-NMR (125 MHz, DMSO-d6) δ: 163.04, 162.73, 161.23, 159.98, 156.08, 151.58, 137.69, 131.41, 131.25, 126.01, 125.10, 123.66, 115.88, 114.99, 115.87, 64.51, 29.10, 28.40. IR (KBr, cm−1) v: 3078.72, 1728.11, 1664.12, 1572.83. MS (m/z): 384.09.
4j: 5-(4-((3-fluorobenzyl)oxy)benzylidene)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione
Melting point: 187–188 °C.
1H-NMR (499 MHz, DMSO-d6) δ: 8.30–8.32 (d, J = 8.6 Hz, 3H), 7.43–7.47 (q, J = 6.98 Hz, 1H), 7.30–7.32 (d, J = 7.98 Hz, 2H), 7.12–7.19 (m, 3H), 5.25 (s, 2H), 3.19–3.21 (d, J = 10.7 Hz, 6H,). 13C-NMR (125 MHz, DMSO-d6) δ: 163.64, 163.03, 162.69, 161.23, 156.08, 151.57, 139.77, 137.68, 131.09, 125.98, 124.21, 116.10, 115.41, 115.24, 115.12, 115.0, 114.83, 69.23, 28.09, 28.48. IR (KBr, cm−1) v: 3128.99, 2948.61, 1661.07, 1574.82, 1258.26. MS (m/z): 368.2
4k: 5-(4-((4-fluorobenzyl)oxy)benzylidene)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione
Melting point: 197–198 °C.
1H-NMR (499 MHz, DMSO-d6) δ: 8.30–8.32 (d, J = 8.5 Hz, 3H), 7.51–7.54 (t, J = 6.98 Hz, 2H), 7.21–7.23 (t, J = 9.98 Hz, 2H), 7.11–7.14 (d, J = 9.48 Hz, 2H), 5.21 (s, 2H), 3.19–3.21 (d, J = 10.8 Hz, 6H). 13C-NMR (125 MHz, DMSO-d6) δ: 156.14, 137.75, 130.73, 130.66, 115.91, 115.74, 115.10, 69.41, 28.47. IR (KBr, cm−1) v: 3056.78, 2960.43, 1727.56, 1667.53, 1544.43. MS (m/z): 368.2
4m: 5-(4-((4-bromobenzyl)oxy)benzylidene)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione
Melting point: 213–215 °C.
1H-NMR (500 MHz, DMSO-d6) δ: 8.30–8.32 (d, J = 8.1 Hz, 3H), 7.59–7.61 (d, J = 8.48 Hz, 2H), 7.42–7.44 (d, J = 6.98 Hz, 2H), 7.11–7.13 (d, J = 8.48 Hz, 2H), 5.22 (s, 2H), 3.19–3.22 (d, J = 10.98, 6H). 13C-NMR (125 MHz, DMSO-d6) δ: 163.05, 162.72, 161.24, 156.07, 151.58, 137.68, 136.31, 131.93, 130.46, 125.95, 121.71, 116.10, 115.13, 69.28, 29.10, 28.49. IR (KBr, cm−1) v: 3066.97, 2951.41, 1726.68, 1669.08, 1541.07, 1181.79. MS (m/z): 430.1
4n: 5-(4-((4-bromobenzyl)oxy)-3-methoxybenzylidene)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione
Melting point: 215–218 °C.
1H-NMR (500 MHz, DMSO-d6) δ: 8.28 (s, 1H), 8.32 (s, 1H), 7.87–7.99 (d, J = 8.48 Hz, 1H), 7.60–7.62 (d, J = 8.98 Hz, 2H), 7.42–7.43 (d, J = 8.48 Hz, 2H), 7.17–7.18 (d, J = 9.48, Hz, 1H,), 5.22 (s, 2H), 3.83 (s, 3H), 3.21–3.23 (s, 6H). IR (KBr, cm−1) v: 3114.82, 2952.15, 1725.74, 1663.51, 1547.78. MS (m/z): 460.2
4o: 5-(4-((4-chlorobenzyl)oxy)-3-methoxybenzylidene)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione
Melting point: 224–227 °C.
1H-NMR (500 MHz, DMSO-d6) δ: 8.28–8.31 (d, J = 14.4 Hz, 2H), 7.87–7.89 (d, J = 8.48 Hz, 1H), 7.48 (s, 4H), 7.17–7.18 (d, J = 7.98 Hz, 1H), 5.23 (s, 2H), 3.82 (s, 3H), 3.21–3.23 (d, J = 6.0 Hz, 6H). IR (KBr, cm−1) v: 3115.91, 2956.35, 1726.33, 1665.14, 1547.50. MS (m/z): 414.2
4p: 5-(3-((3-fluorobenzyl)oxy)benzylidene)pyrimidine-2,4,6(1H,3H,5H)-trione
Melting point: 225–228 °C.
1H-NMR (500 MHz, DMSO-d6) δ: 11.39 (s, 1H), 11.24 (s, 1H), 8.24 (s, 1H), 7.90 (s, 1H), 7.63–7.64 (d, J = 7.98 Hz, 1H), 7.37–7.46 (m, 2H), 7.28–7.30 (m, 2H), 7.14–7.20 (m, 2H), 5.16 (s, 2H). 13C-NMR (125 MHz, DMSO-d6) δ: 163.84, 163.64, 162.07, 158.01, 154.80, 150.64, 140.25, 134.40, 131.02, 129.66, 126.82, 124.09, 119.87, 119.53, 119.20, 115.23, 114.87, 68.99. IR (KBr, cm−1) v: 3230.76, 3095.48), 1754.88, 1673.28, 1571.44, 1235.13. MS (m/z): 340.09.
4q: 5-(3-((4-fluorobenzyl)oxy)benzylidene)pyrimidine-2,4,6(1H,3H,5H)-trione
Melting point: 260–265 °C.
1H-NMR (500 MHz, DMSO-d6) δ: 11.39 (s, 1H), 11.24 (s, 1H), 8.24 (s, 1H), 7.90 (s, 1H), 7.62–7.63 (d, J = 7.98 Hz, 2H), 7.49–7.52 (t, J = 5.99 Hz, 2H), 7.37–7.40 (m, 1H), 7.17–7.23 (m, 3H), 5.11 (s, 2H). 13C-NMR (125 MHz, DMSO-d6) δ: 163.85, 163.26, 162.08, 161.32, 158.12, 154.86, 150.65, 134.37, 133.50, 130.57, 129.64, 126.77, 119.82, 119.56, 119.15, 115.85, 115.68, 69.14. IR (KBr, cm−1) v: 3230.91, 3100.15, 1756.02, 1673.16, 1575.09, 1237.59. MS (m/z): 340.09.
4r: 5-(3-((4-chlorobenzyl)oxy)benzylidene)pyrimidine-2,4,6(1H,3H,5H)-trione
Melting point: 273–274 °C.
1H-NMR (500 MHz, DMSO-d6) δ: 11.39 (s, 1H), 11.24 (s, 1H), 8.24 (s, 1H), 7.89 (s, 1H), 7.62–7.63 (d, J = 7.48 Hz, 1H), 7.43–7.49 (q, J = 7.98 Hz, 4H), 7.37–7.40 (t, J = 8.48 Hz, 1H), 7.17–7.19 (d, J = 8.48 Hz, 1H), 5.13 (s, 2H), 5.11 (s, 2H, Hb). 13C-NMR (125 MHz, DMSO-d6) δ: 163.85, 162.07, 158.04, 154.84, 150.65, 136.31, 134.38, 132.97, 130.04, 129.65, 128.94, 126.84, 119.82, 119.54, 119.18, 69.01. IR (KBr, cm−1) v: 3228.24, 3102.01, 1755.44, 1673.59, 1574.90, 1218.99. MS (m/z): 356.2
Molecular modeling
To optimize the three-dimensional structure of the studied ligands, ab initio calculations were performed using the ORCA quantum chemistry package [12]. The BP86/Def2-TZVP and MP2/Def2-TZVPP were utilized for optimization and energy calculation, respectively. Force field compatible partial atomic charges were calculated by the AM1-BCC method with the Antechamber package [13]. We used Molekel to visualize the molecular orbital structures [Ugo Varetto, Molekel 5.4]. The Matplotlib was used to draw the UV absorption spectra (https://matplotlib.org).
Molecular docking studies
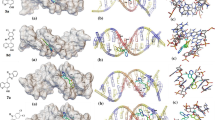
Molecular docking simulations were carried out using AutoDock4 software with default settings to gain a molecular view of the interaction of ligands and DNA duplex. The docked poses for the two of the best-scored hybrids 4j and 4m, shown in Fig. 1, depict its binding along the minor groove of DNA. Crystallographic structures of two DNA strands 1CGC (CG-rich), and 1DNE (AT-rich), were obtained from the RCSB database (http://www.rcsb.org/pdb). The molecular docking simulations were performed based on the Lamarckian genetic algorithm of the AutoDock 4.2. The 60 × 60 × 60 Å cubic lattice box was centered on the DNA strand with the lattice distance of 0.375 Å by using AutoGrid [12]. Docking parameters were implemented as the maximum number of energy evaluations 2,500,000, population size 150, gene mutation rate 0.02, crossover rate 0.8, maximum generation 27,000, the genetic algorithm runs 200. The cluster analysis of similar conformations was accomplished with the RMSD (maximum root mean square deviation) of 2.0 Å.

Interaction profiles of 4j and 4m with the 1CGC strand and 1DNE strands
MD simulation studies
Molecular dynamics (MD) simulation was performed by the AMBER99SB-ILDN force field accessible in the GROMACS 4.6.5 software package [14]. The predicted DNA–ligand complex was placed in the center of a dodecahedron box filled with TIP3P water molecules [15]. To neutralize the net charge of the system, enough Mg2+ ions were added. After each update, the LINCS algorithm was used to reset all covalent bonds to their default lengths [16]. For both long-range electrostatic and van der Waals interactions, the Particle Mesh Ewald (PME) method with a cutoff length of 14 Å and cubic interpolation was used [17].
Energy optimization was performed by 5000 steps of steepest descent and 2500 steps of the conjugated gradient method. After the initial optimization, in the NVT condition (constant number, volume and temperature), while the heavy atoms of ligand and receptor were fixed, the temperature rose from absolute zero (0 K) to 300 K in 300 picoseconds (ps) and remained at this temperature for 200 ps to reach to equilibrium. As the separate temperature-coupling groups, the DNA-ligand and ions-solvent were treated using a modified Berendsen thermostat (V-rescale) [18].
After stabilization of temperature, the MD simulations were performed for 1000 ps in NPT condition (constant number, pressure and temperature), at 1 atm pressure using similar parameters together with isotropic pressure coupling [19]. When the system reached equilibrium, all limitations were removed and the 50 ns MD simulations were accomplished at the NPT ensemble.
UV–Visible spectral studies
Preparation of stock solutions
The stock solution of ctDNA was prepared by dissolving 1 mg of ctDNA in 5 mL of 0.1 M phosphate buffer (K2HPO4/KH2PO4) at pH 7.4 with frequent stirring at room temperature (30 °C) for 24 h. The concentration of ctDNA was calculated by UV absorption spectrophotometry using the molar absorption coefficient (ε260 = 6600 cm−1mol1−, as the molarity of phosphate groups) at 260 nm [20]. The adsorption ratio of ctDNA was measured at 260 and 280 nm to evaluate the protein content rate of the ctDNA solution. The A260/A280 ratio was calculated to be 1.9 (A260/A280 > 1.8) showed that the ctDNA sample was appropriately free of protein content [21]. The stock solutions of the barbituric acid derivatives were prepared in DMSO with a concentration of 10 mM. Serial dilution of stock solution was carried out using 0.1 M phosphate buffer (pH = 7.4).
Spectroscopy measurements
In docking calculations, the best results were related to the 4j and 4m. Therefore, these products were considered for UV spectrophotometric studies. UV–Visible spectroscopic titrations were done by increasing the concentration of ctDNA (0–200 μl) versus the constant concentration of the ligands. Through titration, an equal amount of ctDNA was added to the barbituric acid derivatives and blank to remove its absorption spectrum. The binding constant (Ka) can be estimated based on the Benesi and Hildebrand equation [22]:
In this equation, A0 indicates the adsorption of barbituric acid derivatives in the absence of ctDNA, A indicates the adsorption of barbituric acid derivatives in the presence of ctDNA, Ka is the binding constant, εG and εG-H are the adsorption coefficients of free ligand and its complex with DNA, respectively [23].
Results and discussion
Chemistry
The synthetic routes for the preparation of barbituric acid derivatives (4a-r) were depicted in Scheme 1. For this purpose, various (benzyloxy)benzaldehydes (3) were obtained by the reaction of hydroxyl benzaldehydes (1a-c) and benzyl halides (2) in the presence of K2CO3 at room temperature in DMF. The Knoevenagel reaction between (benzyloxy)benzaldehydes (3) with barbituric acid/N,N-dimethyl barbituric acid was carried out using NH4HPO4 as the base in 2 mL glacial acetic acid and 2 mL ethanol at room temperature to obtain the desired products 4a-r in moderate to good yields. The structure of all new barbituric acid derivatives were confirmed by IR, 1H, 13C NMR, and mass spectroscopy.

Synthesis of barbituric acid derivatives (4a-r)
Barbituric acid products derived from 4-hydroxy benzaldehyde or vanillin are shown in Table 1 and the products derived from 3-hydroxy benzaldehyde are shown in Table 2.
In-Silico ADME prediction
To predict the drug-likeness properties of the compounds, the molecular weight factors, the number of hydrogen bonding donors, the number of hydrogen bonding acceptors, the lipophilicity index, and the number of rotating bonds of a compound must be calculated according to Lipinski's rule of five [24].
Physiochemical properties have significant effects on the behavior of compounds within a living system. Hence, it is vital to predict ADME properties through hit identification. We used 2D structures to predict the ADME data of products. Results of estimated parameters indicated that all barbituric acid derivatives (4a-r) met the criteria for drug-likeness and followed Lipinski’s law (Table 2S).
Quantum mechanics studies
Frontier molecular orbitals are one of the essential parameters for determining the biological activity of the molecules. The HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) can use to determine the stability, reactivity, and biological activity of the molecules. Table 3 shows the results of quantum mechanics calculations for 4j and 4m using the MP2/Def2-TZVPP||PB86/Def2-TZVPMP2 method.
The electrons fill the molecular orbitals from the lowest energy level orbitals up to the HOMO. HOMO energy is equal to ionization potential (I = -EHOMO). The higher HOMO energy equals the lower ionization potential of a molecule, leading to the increase of the tendency of that molecule to lose an electron and participate in charge-transfer interactions. The LUMO electron density contains the parts of a molecule with a strong tendency to accept the electron. The electron affinity (A = −ELUMO) is described as the amount of energy released by electron absorption. The greater the electron affinity of a molecule means the greater tendency to capture the electron [21].
The amounts of ionization potentials (I = −EHOMO) and electron affinity (A = −ELUMO) in the compounds 4j and 4m were almost equal, so they have a similar tendency to give electrons and participate in charge-transfer interactions.
I and A values of the Pyriproxyfen [25] are calculated and written in Table 2 compared with compounds 4j and 4m. 4j and 4m had lower I and A values than Pyriproxyfen. So, the charge-transfer interaction, as well as the electron affinity of Pyriproxyfen is more than 4j and 4m.
Molecular docking studies
To evaluate the molecular mechanism of DNA interaction in atomic details, molecular docking was performed to predict the best possible binding mode between two DNA strands for the new synthesized barbituric acid derivatives (4a-r). The binding affinities of the 17 molecules of barbituric acid derivatives (4a-r) with two DNA strands were investigated.
We considered the docking of recognized small molecules with two DNA chains, 1CGC and 1DNE. The affinity of small molecules to the CG sequence can evaluate by using 1CGC. 1CGC is a decamer d(CCGGCGCCGG) that contains the repeated CG sequence. Also, to assess the affinity of small molecules to AT sequence, we used 1DNE. 1DNE is a dodecamer d(CGCGATATCGCG) with two repeats of AT at the central section of the strand.
Table 3S shows the results of molecular docking of DNA interaction of the barbituric acid derivatives.
The free energy (ΔG) of the ligand–receptor binding indicates the amount of ligand tending to bind to the receptor. The docking process can calculate this parameter [26].
According to the free energy values of DNA-binding (Table 3S), the strongest DNA-bindings are related to the 4j and 4m derivatives. For these compounds, the free energy value for the CG-rich chain is −8.80 kcal/mol and for the AT-rich chain is -8.1 kcal/mol. These values for Pyriproxyfen are −7.40 kcal/mol for the CG-rich chain and -6.7 kcal/mol for the AT-rich chain. Figure 1 shows that compounds 4j and 4m bind along the minor groove of DNA.
Table 4 shows the graphs of DNA-interaction of the 4j and 4m with the CG-rich strand (1CGC) and AT-rich strand (1DNE).
In the CG-rich strand, the O3 atom of 4j formed a hydrogen bond with the lengths of 3.12 and 2.82 Å with the H2 atom of the guanine rings DG16 and DG6, respectively. Also, 4j had vdW and dipole–dipole interactions with DC7, DC15, DC8, DC5, DC17, DC18, DG4 and DG19 nucleotides. In the AT-rich strand, 4j interacted with DT6, DT20, DA7, DA19, DT8, DT18 and DC9, DA17 nucleotides via vdW and dipole–dipole interactions.
In the CG-rich strand of the 4m, the O3 atom formed a hydrogen bond with the H2 atom of the guanine ring (DG6) with a length of 2.80 Å. Also, 4m had vdW and dipole–dipole interactions with DC15, DC17, DC5, DG4, DG19, DC18, DC7 and DC8 nucleotides. Also, in the AT-rich strand, 4m interacted with DC21, DA5, DT6, DA7, DA19, DT18, DT8 and DC9 nucleotides via vdW and dipole–dipole interactions.
Ligand efficiency (LE) is the free energy binding calculated by docking per heavy atom of the ligand structure. It indicates the ability of the functional group (based on molecular weight) to interact with the receptor [27]. This parameter was calculated by the following equation:
Table 5 compared the LE values of 4j and 4m with the Pyriproxyfen as a DNA-binding model.
The LE calculated for the 1CGC, for derivatives 4j and 4m, was 0.35 and 0.32, respectively, and 0.3 for the 1DNE chain. Therefore, these two compounds are optimal in terms of this parameter. The LE values for Pyriproxyfen were 0.3 for 1CGC and 0.27 for 1DNE, which were lower than 4j and 4m. These results indicate that in interaction with DNA, the role of atoms in compounds 4j and 4m is greater than that of Pyriproxyfen, which suggests the effectiveness of the design of this group of compounds.
Considering the docking error in estimating the ΔG of binding (± 2.0 kcal mol−1), it is not reliable to predict the binding affinity for DNA strands only by docking results. Hence, in the next phase, we used docking results as the starting data to perform molecular dynamics simulation (MD) to get more plausible results.
Molecular dynamics simulation
To achieve a more realistic model of ligand–DNA interaction, 50 ns MD simulations were accomplished on the predicted docking model (for 4j and 4m derivatives). To analyze the system's stability during 50 ns of MD simulation, the average total energy and temperature of systems were calculated. The standard deviation (SD) and relative standard deviation (RSD) were calculated and used to evaluate the stability of the systems. The coefficient of variation was calculated by the RSD equation [22]:
In all systems, the rate of fluctuation (RSD) of energy was less than 0.40% and the temperature was less than 1.00%. The low value of RSD shows that the systems were stable and the principle of total energy conservation was achieved in these systems.
The location of the atoms in each conformer was studied to evaluate the stability of the complexes during the 50 ns MD simulation. In this regard, the RMSD matrixes of 4j or 4m and DNA strands were calculated to display the changes in the conformation of each structure (Tables 11S and 12S). The RMSD matrix shows conformational changes at any moment compared to all conformers that have been seen through the MD simulation. The observed subsquares in the diameter of the RMSD matrix square show the conformational change. Hence, inside each square, the conformation of the complex was stable. The blue to red color spectrum indicates changes in the RMSD matrix from 0 to more than 0.5 nm, respectively. Tables 11S and 12S show the RMSD matrix of the DNA strands in 1CGC and 1DNE in complex with 4j and 4 m. In the 4j complexes with 1CGC and 1DNE, the changes in the RMSD matrix of the 1DNE chain were more significant than those of the 1CGC strand. 4j showed a stable binding to the DNA strand during the MD simulation. After the rotation of 4j in the minor groove of the DNA, the plate of 1,3-dimethyl barbiturate ring is placed almost perpendicular to the axis of the DNA strand. During simulation, 4j established 1.16 ± 0.48 hydrogen bonds with the 1CGC strand and had nearly no hydrogen bonds with the 1DNE strand. The 4j-DNA-binding energies indicate that this compound is more likely to bind to 1CGC than 1DNE.
4m formed stable complexes with 1CGC and 1DNE strands. 4m established 0.56 ± 0.53 hydrogen bonds with the CG-rich strand and had no hydrogen bonds with the AT-rich strand.
Although the energy released to bind the 4m compound to the 1CGC strand is a smaller amount than 1DNE, given its stability in binding to both DNA strands, its selectivity cannot be stated [28]. The results of the molecular dynamics simulation data are shown in Fig. 2. A comparison of the docking data (Fig. 1) and the MDD data (Fig. 2) shows that although the system has undergone many changes during the simulation, the designed ligands are well placed inside the minor groove. Examination of the behavior of the 4j and 4m showed that they tend to be located between nucleotide bases, although this process was not observed in the simulation time.

Interaction profiles of 4j and 4m with the 1CGC strand and 1DNE strands during MD
To understand the dynamic stability of DNA–ligand complexes, the radius of gyration (Rg) values are calculated [29]. As seen in Table 6, the changes of Rg were not statistically significant, and the DNA–ligand complexes have good stability MD.
UV–Visible spectral studies
To confirm the results achieved from MD simulation, the interactions of the 4j and 4m with DNA strands were investigated using the spectrophotometric method.
The UV spectra of titrated compounds with different concentrations of ctDNA are indicated in Fig. 3.

Spectrophotometric titration of the 4j (left) and 4m (right) with different concentrations of ctDNA
The binding constants of 4j and 4m (Ka) to the DNA strand were measured, and its values are listed in Table 7.
To in vitro investigate the interaction of the 4j and 4m with ctDNA, we measured the protein content of ctDNA to increase the accuracy of the titration analysis. For this purpose, the ctDNA adsorption ratio was measured at 260 and 280 nm. The A260/A280 ratio was calculated to be 1.9. It indicates the absence of protein in the ctDNA sample. Therefore, the obtained Kas indicate the interaction between 4j and 4m and ctDNA.
The absorption spectra of 4j and 4m had a peak at 263 nm and 259 nm, respectively (Fig. 2). The constant values of binding to the DNA strand (Ka) were determined by drawing A0/A-A0 versus 1/[ctDNA] and calculating the intercept ratio to the line slope.
Based on the results, the Ka of the 4m was higher than 4j. Therefore, it performs stronger interactions with ctDNA than 4j.
Changes in the UV spectrum of compound 4m during titration had a redshift (bathochromic) displacement. In the spectrophotometric titration study of a ligand with ctDNA, redshift indicates intercalative interactions [8]. So, this compound tends to locate between the nucleotide bases of the ctDNA.
The structure activity relationships (SARs) survey
It can be concluded from the molecular docking data of 17 synthesized products that: Compounds derived from 4-(benzyloxy)benzaldehyde perform a better interaction with DNA than 4-(benzyloxy)-3-methoxybenzaldehyde and 3-(benzyloxy)benzaldehyde. Also, products containing N,N-dimethyl barbituric acid performed better than barbituric acid.
Conclusion
In conclusion, efficient access to the synthesis of novel barbituric acid derivatives with promising DNA interacting properties has been accomplished. Molecular docking studies revealed that among all products, 4j and 4m are the best interacting agents and are capable of interacting with the ctDNA through the minor groove binding. Based on the flexible shape, they match well with the topology of DNA strands. Quantum mechanics calculations predicted similar trends. The interactions of 4j and 4m with DNA were studied by UV–Vis spectroscopy. The data obtained from spectrophotometric measurements confirm this interaction. Based on the molecular modeling results, the Structure Activity Relationships (SARs) for these new barbituric acid derivatives were proposed and it was observed that the derivative of N,N-dimethyl barbituric acid/4-hydroxybenzaldehyde have better DNA-interactions than barbituric acid/vanillin and barbituric acid/3-hydroxybenzaldehyde derivatives.
Abbreviations
- ADT:
-
AutoDock tools
- ADME:
-
Adsorption distribution metabolism excretion
- 1CGC:
-
CG-riched DNA strand
- ctDNA:
-
Deoxyribonucleic acid sodium salt from calf thymus DNA
- 1DNE:
-
A-riched DNA strand
- HOMO:
-
High occupied molecular orbital
- LE:
-
Ligand efficiency
- LUMO:
-
Low unoccupied molecular orbital
- MD:
-
Molecular dynamic
- RMSD-lig:
-
Root mean square deviation-ligand
- RMSD-bb:
-
Root mean square deviation-backbone
- RO5:
-
Lipinski’s rule of five known as simply the Rule of Five
- RSD:
-
Relative standard deviation
References
D.A. Guertin, D.M. Sabatini, in: J. Mendelsohn, P.M. Howley, M.A. Israel, J.W. Gray, C.B. Thompson, (Eds.), The Molecular Basis of Cancer (Third Edition), W.B. Saunders, Philadelphia, 2008.
F. Sadoughi, P.M. Dana, Z. Asemi, B. Yousefi, DNA damage response and repair in osteosarcoma: defects, regulation and therapeutic implications. DNA Repair 102, 103105 (2021). https://doi.org/10.1016/j.dnarep.2021.103105
Y.J. Luo, B.L. Wang, S.B. Kou, Z.Y. Lin, K.L. Zhou, Y.Y. Lou, J.H. Shi, Assessment on the binding characteristics of dasatinib, a tyrosine kinase inhibitor to calf thymus DNA: insights from multi-spectroscopic methodologies and molecular docking as well as DFT calculation. J. Biomol. Struct. Dyn. 38(14), 4210–4220 (2020). https://doi.org/10.1080/07391102.2019.1676824
A. Mukherjee, W.D. Sasikala, Drug–DNA intercalation: From discovery to the molecular mechanism. Adv. Protein Chem. Struct. Biol. 92, 1–62 (2013). https://doi.org/10.1016/B978-0-12-411636-8.00001-8
M.J. Lind, Principles of systemic anticancer therapy. Medicine 48(2), 90–96 (2020). https://doi.org/10.1016/j.mpmed.2019.11.005
J. Sheng, J. Gan, Z. Huang, Structure-based DNA-targeting strategies with small molecule ligands for drug discovery. Med. Res. Rev. 33(5), 1119–1173 (2013)
K.K. Kwok. E.C. Vincent, J.N. Gibson, in: F.J. Dowd, B.S. Johnson, A.J. Mariotti, (Eds.), Pharmacology and Therapeutics for Dentistry (Seventh Edition), Mosby 2017.
M. Sirajuddin, S. Ali, A. Badshah, Drug–DNA interactions and their study by UV–Visible, fluorescence spectroscopies and cyclic voltametry. J. Photochem. Photobiol. B Biol. 124, 1–19 (2013). https://doi.org/10.1016/j.jphotobiol.2013.03.013
F. Zsila, Glycosaminoglycans are potential pharmacological targets for classic DNA minor groove binder drugs berenil and pentamidine. Phys. Chem. Chem. Phys. 17(38), 24560–24565 (2015). https://doi.org/10.1039/C5CP03153B
G.T. Nguyen, W.Y. Leung, T.N. Tran, H. Wang, V. Murray, W.A. Donald, Mechanism for the binding of netropsin to hairpin DNA revealed using nanoscale ion emitters in native mass spectrometry. Anal. Chem. 92(1), 1130–1137 (2019). https://doi.org/10.1021/acs.analchem.9b04209
S. Sundriyal, B. Viswanad, P. Ramarao, A.K. Chakraborti, P.V. Bharatam, New PPARγ ligands based on barbituric acid: virtual screening, synthesis and receptor binding studies. Bioorg. Med. Chem. Lett. 18(18), 4959–4962 (2008). https://doi.org/10.1016/j.bmcl.2008.08.028
F. Neese, The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2(1), 73–78 (2012). https://doi.org/10.1002/wcms.81
A. Jakalian, D.B. Jack, C.I. Bayly, Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 23(16), 1623–1641 (2002). https://doi.org/10.1002/jcc.10128
K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J.L. Klepeis, R.O. Dror, D.E. Shaw, Improved side-chain torsion potentials for the Amber ff99SB protein force field. Prot. Struct. Funct. Bioinform. 78(8), 1950–1958 (2010). https://doi.org/10.1002/prot.22711
K. Toukan, A. Rahman, Molecular-dynamics study of atomic motions in water. Phys. Rev. B. 31(5), 2643 (1985). https://doi.org/10.1103/PhysRevB.31.2643
B. Hess, H. Bekker, H.J. Berendsen, J.G. Fraaije, LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18(12), 1463–1472 (1997). https://doi.org/10.1002/(SICI)1096-987X(199709)18:12%3c1463::AID-JCC4%3e3.0.CO;2-H
T. Darden, D. York, L. Pedersen, Particle mesh Ewald: An N⋅ log (N) method for Ewald sums in large systems. J. Chem. Phys. 98(12), 10089–10092 (1993). https://doi.org/10.1063/1.464397
H.J. Berendsen, J.V. Postma, W.F. Van Gunsteren, A.R.H.J. DiNola, J.R. Haak, Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81(8), 3684–3690 (1984). https://doi.org/10.1063/1.448118
M. Parrinello, A. Rahman, Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52(12), 7182–7190 (1981). https://doi.org/10.1063/1.328693
M.E. Reichmann, S.A. Rice, C.A. Thomas, P. Doty, A further examination of the molecular weight and size of desoxypentose nucleic acid. J. Am. Chem. Soc. 76(11), 3047–3053 (1954)
J. Marmur, A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J. Mol. Biol. 3, 208–218 (1961). https://doi.org/10.1016/S0022-2836(61)80047-8
M. Khazaei, D. Dastan, A. Ebadi, Binding of Foeniculum vulgare essential oil and its major compounds to double-stranded DNA: In silico and in vitro studies. Food Biosci. 41, 100972 (2021). https://doi.org/10.1016/j.fbio.2021.100972
J.J. Stephanos, Drug-protein interactions: two-site binding of heterocyclic ligands to a monomeric hemoglobin. J. Inorg. Biochem. 62(3), 155–169 (1996). https://doi.org/10.1016/0162-0134(95)00144-1
C.A. Lipinski, F. Lombardo, B.W. Dominy, P.J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 23(1–3), 3–25 (1997). https://doi.org/10.1016/S0169-409X(96)00423-1
F. Ahmadi, N. Jamali, R. Moradian, B. Astinchap, Binding studies of pyriproxyfen to DNA by multispectroscopic atomic force microscopy and molecular modeling methods. DNA Cell Biol. 31(2), 259–268 (2012). https://doi.org/10.1089/dna.2011.1303
N. Shahabadi, S. Zendehcheshm, Evaluation of ct-DNA and HSA binding propensity of antibacterial drug chloroxine: multi-spectroscopic analysis, atomic force microscopy and docking simulation. Spectrochim. Acta A Mol. Biomol. Spectrosc. 230, 118042 (2020). https://doi.org/10.1016/j.saa.2020.118042
A.L. Hopkins, G.M. Keserü, P.D. Leeson, D.C. Rees, C.H. Reynolds, The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 13(2), 105–121 (2014). https://doi.org/10.1038/nrd4163
A. Pandey, R. Mishra, A. Yadav, Understanding interactions of DNA minor groove binders using advanced computational techniques. Int. J. Anal. Exp. Modal Anal. 12, 1300–1315 (2020)
A.K. Chaubey, K.D. Dubey, R.P. Ojha, MD simulation of LNA-modified human telomeric G-quadruplexes: a free energy calculation. Med. Chem. Res. 24(2), 753–763 (2015). https://doi.org/10.1007/s00044-014-1182-y
Acknowledgements
We thank the Research and Technology Vice-chancellor of Hamadan University of Medical Sciences for partial financial support.
Funding
This study has been adapted from a Pharm. D. thesis at Hamadan University of Medical Sciences (Grant No. 9807235421).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors declare no conflicting financial interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ebadi, A., Najafi, Z., Pakdel-yeganeh, H. et al. Design, synthesis, molecular modeling and DNA-binding studies of new barbituric acid derivatives. J IRAN CHEM SOC 19, 3887–3898 (2022). https://doi.org/10.1007/s13738-022-02576-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-022-02576-x