
Abstract
Acute kidney injury (AKI) frequently complicates corona virus disease 2019 (COVID-19) and is associated with significant mortality. Kidney disease in COVID-19 is usually due to acute tubular injury, but a variety of glomerular processes, especially collapsing glomerulopathy, have been increasingly described. Until recently, proliferative glomerulonephritis with monoclonal immunoglobulin deposits (PGNMID) had not been reported in the setting of COVID-19. We present a case of dialysis-dependent AKI developing soon after symptomatic COVID-19 which, on kidney biopsy, was found to be due to PGNMID with IgG3 kappa deposits. As is typical of PGNMID, a search for evidence of extra-renal monoclonal immunoglobulin or clonal lymphocyte population was negative. However, the patient had a favorable response to anti-plasma cell therapy and was ultimately able to stop hemodialysis. Though monoclonal gammopathy of renal significance (MGRS) is usually not associated with infection, other cases of post-viral MGRS, including PGNMID, have been previously reported. PGNMID has recently been linked specifically to COVID-19, with this representing one of only four cases reported thus far. Though causality between the preceding viral infection and the subsequent glomerulonephritis cannot be proven in these reports, nephrologists should be aware that not all kidney disease occurring in the aftermath of COVID-19 is due to tubular injury or collapsing glomerulopathy. As such, kidney biopsy should be routinely considered in the setting of COVID-19-associated glomerular disease as findings may change management. In the case of COVID-19-associated PGNMID data to guide treatment are limited, but our report suggests that anti-plasma cell therapy may be effective.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute kidney injury (AKI) frequently complicates corona virus disease 2019 (COVID-19) and is associated with significant mortality [1,2,3]. Autopsy and biopsy studies have shown that kidney disease in the setting of COVID-19 is most commonly due to acute tubular injury [3,4,5,6,7,8], but multiple glomerular pathologies, especially collapsing glomerulopathy, have been reported [6,7,8,9,10]. Proliferative glomerulonephritis with monoclonal immunoglobulin deposits (PGNMID) in the setting of COVID-19 has only very recently been reported, having been found in four of 284 biopsies described in a recent case series [11]. We present one of these cases, providing the first detailed clinicopathologic report of COVID-19-associated PGNMID.
Case presentation
A 71-year-old woman with a history of asthma, sleep apnea, gastroesophageal reflux disease, and depression was admitted with 1 week of dyspnea, fever, nausea, vomiting, and diarrhea due to COVID-19. She was unvaccinated, as vaccination for COVID-19 was not yet available at the time. Apart from hypoxemia requiring oxygen at 2 L/min and hypoalbuminemia (1.7 g/dL), vital signs, physical examination, and laboratory data were normal, including a serum creatinine of 0.7–0.9 mg/dL (see Table 1). Body mass index was 26.8 kg/m2. Urinalysis was not obtained this first admission. Chest X-ray revealed patchy peripheral and basilar ground-glass opacities. She was treated with remdesivir and dexamethasone and discharged after 2 days on dexamethasone and oxygen. She was seen in telehealth follow-up with gradual resolution of her respiratory symptoms and oxygen requirement over the following weeks.
However, a month after discharge, she was readmitted to another hospital with generalized swelling and was found to have AKI with initial creatinine of 3.3 mg/dL, nephrotic-range proteinuria, hematuria, and culture-negative pyuria (see Table 1). Her kidney function subsequently worsened, with peak creatinine of 8.5 mg/dL, and she developed anuria. Hemodialysis was initiated. Serologies were negative apart from mildly decreased C3 with normal C4. Serum protein electrophoresis (SPEP) and 24-h urine protein electrophoresis (UPEP) with immunofixation were both negative for monoclonal proteins, and serum kappa-lambda ratio was normal. Renal ultrasound was normal with 11-cm kidneys bilaterally. Kidney biopsy was performed which revealed PGNMID with monoclonal IgG3-kappa deposits and mild (10–20%) interstitial fibrosis-tubular atrophy (IFTA). Abdominal fat pad biopsy was negative for amyloidosis. Bone marrow biopsy was negative for lymphocyte neoplasm by morphology, immunohistochemistry, and flow cytometry. Before discharge, she received treatment with one dose each of cyclophosphamide 600 mg by mouth (approximately 300 mg/m2), bortezomib 3 mg subcutaneous (approximately 1.5 mg/m2), and dexamethasone 40 mg by mouth (CyBorD). Of note, though her COVID-19 symptoms had long since resolved, at discharge she was still PCR-positive for SARS-CoV-2 by nasopharyngeal swab. She continued outpatient hemodialysis but was able to stop 1–2 weeks later after improvement in renal function. She was seen in clinic by a hematologist who opted to not prescribe further antineoplastic therapy. She did well for several weeks, with nadir creatinine of 2.1 mg/dL, though her proteinuria persisted, and she required ongoing loop diuretic therapy for management of her volume status.
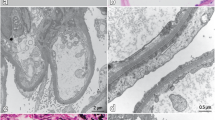
About 6 weeks after discharge, she presented again to our hospital with lower extremity edema, dyspnea, and nausea with persistent retching. Vital signs and examination, apart from 1 + edema, were normal. Admission laboratory data (see Table 1) revealed a creatinine of 6.7 mg/dL and serum albumin of 2.0 g/dL. Urine studies revealed ongoing hematuria and nephrotic-range proteinuria. Repeat SPEP, UPEP × 2 (on a spot collection followed by repeat 24 h collection), and serum kappa-lambda ratio were again negative. Hemodialysis was reinitiated with improvement in her nausea and edema. Repeat kidney biopsy was obtained (shown in Fig. 1) and the results were nearly identical to the first biopsy, again revealing PGNMID with minimal progression of chronicity (now with estimated 20% IFTA). After discussing the unclear risks and benefits, the patient opted to restart antineoplastic therapy with the goal of increasing the odds of being able to stop hemodialysis again. CyBorD therapy was reinitiated and continued after discharge. She ultimately was treated with cyclophosphamide 500 mg by mouth, bortezomib 2.8 mg subcutaneous, and dexamethasone 20 mg by mouth, each given every 1–2 weeks for four doses per cycle for six total cycles and 24 total doses. She again had a significant response, coming off hemodialysis after several weeks. Though she had some subsequent infectious complications, including a urinary tract infection and cellulitis at the site of her tunneled hemodialysis catheter, both were readily treated with antibiotics and catheter removal, and, overall, she tolerated the therapy well and was ultimately quite pleased to be able to stop dialysis again. At last check (about 4 months after restarting CyBorD), her serum creatinine had stabilized at < 2.0 mg/dL with improved but persistent proteinuria (2.5 g/g creatinine, down from peak of 16.4 g/g).

The second renal biopsy yielded 32 glomeruli for light microscopy, of which four were globally sclerosed and the rest marked by diffuse global endocapillary proliferation, including four glomeruli with cellular crescents [a hematoxylin and eosin stain (H&E), original magnification × 400; b periodic acid-Schiff (PAS) stain, original magnification × 400]. The tubulointerstitial compartment was notable for minimal interval progression of interstitial fibrosis-tubular atrophy, slightly increased from 10–20 to 20% (c trichrome stain, original magnification × 100), as well as edema, mononuclear cell infiltration, and red blood cell casts (d H&E, original magnification × 400). Immunofluorescence (IF) revealed granular glomerular staining of C3 (3 + , e original magnification × 400), IgG [not shown], and IgG-3 subclass (3 + , f original magnification × 400), and kappa (3 + , g original magnification × 400), but was negative for lambda (h original magnification × 400). Additional IF staining [not shown] of the glomeruli showed C1q (3 +), but was negative for IgM, IgA, lambda, and the other IgG subclasses, whereas staining for kappa and lambda light chains was similar throughout the tubulointerstitial compartment in proteinaceous casts and in protein droplets. Electron microscopy [not shown] revealed severe epithelial foot process effacement with normal glomerular basement membrane thickness; multiple electron-dense deposits in the mesangium, associated with increased mesangial matrix, and electron-dense deposits in the subendothelial space, but no deposits in the subepithelial space or tubular basement membranes; and no substructure of the deposits to suggest deposition of cryoglobulin, amyloid, or other fibrillar proteins
Discussion
PGNMID is a relatively novel subtype of monoclonal gammopathy of renal significance (MGRS) first described by Nasr et al. [12]. The pathogenesis of PGNMID involves deposition of clonal immunoglobulin, most commonly IgG3-kappa light chains, with subsequent complement fixation within the glomerular capillary wall triggering an inflammatory cascade that manifests in proliferative glomerulonephritis in typically a membranoproliferative and/or endocapillary pattern. IgG3 normally accounts for < 10% of circulating IgG but has been found to be the most nephritogenic of all IgG isoforms due to a higher molecular weight, cationic charge, and ability to fix complement [13].
In most forms of MGRS a clonal immunoglobulin and/or clonal lymphocyte population can be detected on SPEP, serum kappa-lambda ratio, UPEP, bone marrow biopsy, and/or peripheral blood flow cytometry. However, as in this case, PGNMID often develops in the absence of detectable serum or urine monoclonal immunoglobulin or monoclonal cell line, which significantly complicates diagnosis and management. Specifically, a monoclonal gammopathy is detected in only 20–40% of PGNMID cases, with only a subset of these having a detectable lymphocyte clone [12,13,14,15].
COVID-19-associated AKI is most commonly due to acute tubular injury and is associated with high mortality [1,2,3,4,5,6,7]. Kidney disease in COVID-19 appears to be a result of both indirect and direct effects of SARS-CoV-2 infection [16]. Many cases appear to be attributable to indirect ischemic injury and/or toxic injury from nephrotoxins or rhabdomyolysis [3]. However, the receptors for cellular viral entry, including angiotensin converting enzyme-2 (ACE2), are highly expressed in the proximal tubule [17, 18], and emerging data suggest that SARS-CoV-2 may directly infect proximal tubular cells [16]. Furthermore, ACE2 is also expressed by glomerular podocytes [18], which may explain why proteinuria and hematuria appear to be relatively prominent in COVID-19-associated AKI [1, 3] and why COVID-19 is linked to a variety of glomerular pathologies. Most common of these is collapsing glomerulopathy, which appears to have a predilection for patients expressing APOL1 risk alleles [6, 7, 9,10,11]. Other glomerular lesions reported in multiple patients include membranous glomerulopathy [7, 10, 11] and thrombotic microangiopathy [6, 8, 11]. Nephritic or crescentic lesions in the setting of COVID-19 appear to be less common, though there have been limited reports of pauci-immune glomerulonephritis [6, 11], infection-associated glomerulonephritis [8, 11], lupus nephritis [10, 11], anti-GBM nephritis [10, 11], and IgA nephropathy [7, 11].
Our patient developed rapidly progressive glomerulonephritis 4–6 weeks after symptomatic COVID-19. Interestingly, though her respiratory viral symptoms had resolved 1–2 weeks before she developed glomerulonephritis, she had detectable nasopharyngeal SARS-CoV-2 RNA 1 month after her first kidney biopsy. Though the temporal relationship between the SARS-CoV-2 infection and glomerulonephritis is suggestive, causality in this case or other reported cases [11] of COVID-19-associated PGNMID cannot be proven. Indeed, MGRS and PGNMID usually are not post-infectious processes. However, MGRS and specifically PGNMID have previously been rarely reported to occur in the context of viral infection. Most similar to this case, Fujita et al. [19] described two patients who developed PGNMID with IgG3-kappa after parvovirus B-19 infection. Others have published single case reports of type I cryoglobulinemic glomerulonephritis with monoclonal IgG1-kappa in the setting of hepatitis C infection [20] and light chain cast nephropathy with monoclonal IgG-kappa (IgG subclass not reported) triggered by hepatitis E [21].
The usual presentation of PGNMID includes AKI, nephrotic syndrome, and/or nephritic syndrome. As seen in this case, a varying degree of hypocomplementemia is found in a substantial minority of patients [12, 13]. The prognosis of PGNMID is variable and, as with many glomerular diseases, appears to depend primarily on the extent of glomerulosclerosis and IFTA detected on biopsy. Over 30 months, roughly one-third of cases will recover, one-third will stabilize, and 20% will progress to end-stage kidney disease [13].
There are currently no evidence-based treatment guidelines for PGNMID due to a paucity of data available for this rare disease. If a monoclonal cell line or immunoglobulin is detected, treatment can be tailored appropriately. There is less consensus on how to approach therapy in the majority of cases of PGNMID which lack a detectable clonal immunoglobulin or lymphocyte population. However, based on a few recently published case series, empiric anti-plasma cell or anti-lymphocyte therapy appears to be associated with beneficial results [14, 15]. While patients with modest proteinuria and renal dysfunction may be monitored with antiproteinuric therapy and supportive care alone, most experts recommend, barring advanced fibrosis on biopsy, antineoplastic therapy in patients with significant proteinuria (e.g., > 1 g/day) or significant kidney dysfunction. Indeed, though her long-term renal prognosis remains uncertain, our patient twice had favorable responses to antineoplastic therapy, with substantial improvement in renal function and proteinuria and significant dialysis-free periods.
In conclusion, PGNMID is a rare form of MGRS that, in contrast to most types of MGRS, is usually associated with no detectable extra-renal monoclonal immunoglobulin or monoclonal cell line. Though the most common renal lesions in COVID-19 are acute tubular injury and collapsing glomerulopathy, multiple other glomerular processes have been reported. Though typically not a post-infectious process, four cases of post-viral MGRS had been previously published before PGNMID was linked in recent reports to COVID-19. Though data to guide the treatment of PGNMID in any context are limited, our report suggests that anti-plasma cell therapy can improve the course of COVID-19-associated PGNMID.
References
Cheng Y, Luo R, Wang K, Zhang M, Wang Z, Dong L, et al. Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int. 2020;97(5):829–38. https://doi.org/10.1016/j.kint.2020.03.005.
Ng JH, Hirsch JS, Hazzan A, Wanchoo R, Shah HH, Malieckal DA, et al. Outcomes among patients hospitalized with COVID-19 and acute kidney injury. Am J Kidney Dis. 2021;77(2):204-15 e1. https://doi.org/10.1053/j.ajkd.2020.09.002.
Mohamed MMB, Lukitsch I, Torres-Ortiz AE, Walker JB, Varghese V, Hernandez-Arroyo CF, et al. Acute kidney injury associated with coronavirus disease 2019 in urban New Orleans. Kidney360. 2020;1(7):614–22. https://doi.org/10.34067/kid.0002652020.
Su H, Yang M, Wan C, Yi LX, Tang F, Zhu HY, et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020;98(1):219–27. https://doi.org/10.1016/j.kint.2020.04.003.
Golmai P, Larsen CP, DeVita MV, Wahl SJ, Weins A, Rennke HG, et al. Histopathologic and ultrastructural findings in postmortem kidney biopsy material in 12 patients with AKI and COVID-19. J Am Soc Nephrol. 2020;31(9):1944–7. https://doi.org/10.1681/ASN.2020050683.
Sharma P, Uppal NN, Wanchoo R, Shah HH, Yang Y, Parikh R, et al. COVID-19-associated kidney injury: a case series of kidney biopsy findings. J Am Soc Nephrol. 2020;31(9):1948–58. https://doi.org/10.1681/ASN.2020050699.
Nasr SH, Alexander MP, Cornell LD, Herrera LH, Fidler ME, Said SM, et al. Kidney biopsy findings in patients with COVID-19, kidney injury, and proteinuria. Am J Kidney Dis. 2021;77(3):465–8. https://doi.org/10.1053/j.ajkd.2020.11.002.
Akilesh S, Nast CC, Yamashita M, Henriksen K, Charu V, Troxell ML, et al. Multicenter clinicopathologic correlation of kidney biopsies performed in COVID-19 patients presenting with acute kidney injury or proteinuria. Am J Kidney Dis. 2021;77(1):82-93 e1. https://doi.org/10.1053/j.ajkd.2020.10.001.
Wu H, Larsen CP, Hernandez-Arroyo CF, Mohamed MMB, Caza T, Sharshir M, et al. AKI and collapsing glomerulopathy associated with COVID-19 and APOL 1 high-risk genotype. J Am Soc Nephrol. 2020;31(8):1688–95. https://doi.org/10.1681/ASN.2020050558.
Kudose S, Batal I, Santoriello D, Xu K, Barasch J, Peleg Y, et al. Kidney biopsy findings in patients with COVID-19. J Am Soc Nephrol. 2020;31(9):1959–68. https://doi.org/10.1681/ASN.2020060802.
May RM, Cassol C, Hannoudi A, Larsen CP, Lerma E, Haun RS, et al. A multi-center retrospective cohort study defines the spectrum of kidney pathology in Coronavirus 2019 Disease (COVID-19). Kidney Int. 2019. https://doi.org/10.1016/j.kint.2021.07.015.
Nasr SH, Markowitz GS, Stokes MB, Seshan SV, Valderrama E, Appel GB, et al. Proliferative glomerulonephritis with monoclonal IgG deposits: a distinct entity mimicking immune-complex glomerulonephritis. Kidney Int. 2004;65(1):85–96. https://doi.org/10.1111/j.1523-1755.2004.00365.x.
Nasr SH, Satoskar A, Markowitz GS, Valeri AM, Appel GB, Stokes MB, et al. Proliferative glomerulonephritis with monoclonal IgG deposits. J Am Soc Nephrol. 2009;20(9):2055–64. https://doi.org/10.1681/ASN.2009010110.
Gumber R, Cohen JB, Palmer MB, Kobrin SM, Vogl DT, Wasserstein AG, et al. A clone-directed approach may improve diagnosis and treatment of proliferative glomerulonephritis with monoclonal immunoglobulin deposits. Kidney Int. 2018;94(1):199–205. https://doi.org/10.1016/j.kint.2018.02.020.
Kousios A, Duncan N, Tam FWK, Chaidos A, Cook HT, Roufosse C, et al. Proliferative glomerulonephritis with monoclonal Ig deposits (PGNMID): diagnostic and treatment challenges for the nephrologist! Kidney Int. 2019;95(2):467–8. https://doi.org/10.1016/j.kint.2018.10.016.
Khan S, Chen L, Yang CR, Raghuram V, Khundmiri SJ, Knepper MA. Does SARS-CoV-2 infect the kidney? J Am Soc Nephrol. 2020;31(12):2746–8. https://doi.org/10.1681/ASN.2020081229.
Zhang YM, Zhang H. Genetic roadmap for kidney involvement of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. Clin J Am Soc Nephrol. 2020;15(7):1044–6. https://doi.org/10.2215/CJN.04370420.
Pan XW, Xu D, Zhang H, Zhou W, Wang LH, Cui XG. Identification of a potential mechanism of acute kidney injury during the COVID-19 outbreak: a study based on single-cell transcriptome analysis. Intensive Care Med. 2020;46(6):1114–6. https://doi.org/10.1007/s00134-020-06026-1.
Fujita E, Shimizu A, Kaneko T, Masuda Y, Ishihara C, Mii A, et al. Proliferative glomerulonephritis with monoclonal immunoglobulin G3kappa deposits in association with parvovirus B19 infection. Hum Pathol. 2012;43(12):2326–33. https://doi.org/10.1016/j.humpath.2012.04.004.
Hemminger J, Kandarpa M, Tsai A, Nadasdy T. Proliferative glomerulonephritis with monoclonal IgG1kappa deposits in a hepatitis C virus-positive patient. Am J Kidney Dis. 2016;67(4):703–8. https://doi.org/10.1053/j.ajkd.2015.08.032.
Agrawal P, Kumar V, Kumar A, Sachdeva MUS, Malhotra P, Nada R. Monoclonal gammopathy of renal significance triggered by viral E hepatitis. Indian J Nephrol. 2019;29(1):50–2. https://doi.org/10.4103/ijn.IJN_417_17.
Funding
None.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no relevant financial disclosures or other conflicts of interest.
Ethical approval
Not Applicable. (Per University of New Mexico IRB Policy, retrospective case reports/series with ≤ 3 subjects do not require IRB approval).
Informed consent
Written informed consent was obtained from the patient for publication of this case report and any accompanying images.
Prior publication
This case report was presented as an abstract for American Society of Nephrology Kidney Week 2021. As referenced in the manuscript, the biopsy in this case was included in a recently published biopsy series co-authored by Dr. Messias. This case report was independently drafted by the other authors who had no role in the biopsy series but later invited Dr. Messias to be a coauthor for the pathology component of this case report.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Shieh, M., Giannini, J.A., Combs, S.A. et al. Proliferative glomerulonephritis with monoclonal immunoglobulin deposits triggered by COVID-19: a case report. CEN Case Rep 11, 380–385 (2022). https://doi.org/10.1007/s13730-022-00687-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13730-022-00687-1