
Abstract
Purpose of Review
To summarize experimental and clinical evidence on the association between tumor necrosis factor-α (TNF-α) and nonalcoholic fatty liver disease (NAFLD) and discuss potential treatment considerations.
Recent Findings
Experimental evidence suggests that TNF-α is a cytokine with a critical role in the pathogenesis of NAFLD. Although, the production of TNF-α may be an early event during the course of nonalcoholic fatty liver (NAFL), TNF-α may play a more substantial role in the pathogenesis of nonalcoholic steatohepatitis (NASH) and NAFLD-associated fibrosis. Moreover, TNF-α may potentiate hepatic insulin resistance, thus interconnecting inflammatory with metabolic signals and possibly contributing to the development of NAFLD-related comorbidities, including cardiovascular disease, hepatocellular carcinoma, and extra-hepatic malignancies. In clinical terms, TNF-α is probably associated with the severity of NAFLD; circulating TNF-α gradually increases from controls to patients with NAFL, and then, to patients with NASH. Given this potential association, various therapeutic interventions (obeticholic acid, peroxisome proliferator-activated receptors, sodium-glucose co-transporter 2 inhibitors, glucagon-like peptide-1 receptor agonists, probiotics, synbiotics, rifaximin, vitamin E, pentoxifylline, ursodeoxycholic acid, fibroblast growth factor-21, n-3 polyunsaturated fatty acids, statins, angiotensin receptor blockers) have been evaluated for their effect on TNF-α and NAFLD. Interestingly, anti-TNF biologics have shown favorable metabolic and hepatic effects, which may open a possible therapeutic window for the management of advanced NAFLD.
Summary
The potential key pathogenic role of TNF-α in NAFLD warrants further investigation and may have important diagnostic and therapeutic implications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Nonalcoholic fatty liver disease (NAFLD) is a highly prevalent disease affecting approximately 30% of the general population; the prevalence is much higher in certain populations, such as patients with type 2 diabetes mellitus (T2DM) and obesity, reaching 60% and 90%, respectively [1]. Although NAFLD has become the most common chronic liver disease worldwide, it remains largely underdiagnosed, whereas the absence of any approved pharmacological therapy results in increased health and socioeconomic burden [2]. NAFLD does not represent a single entity, but it encompasses distinct phenotypes: nonalcoholic fatty liver (NAFL, simple hepatic steatosis), nonalcoholic steatohepatitis (NASH), in which steatosis is combined with inflammation, degeneration, and various degrees of hepatic fibrosis, and finally, NAFLD-related cirrhosis, either compensated or decompensated, and hepatocellular carcinoma (HCC) in a subset of patients [3].
NASH and hepatic fibrosis are associated with increased risk of hepatic complications, including cirrhosis and HCC, as well as extra-hepatic complications, including cardiovascular disease (CVD), chronic kidney disease, and extrahepatic malignancies [3]. NASH is characterized by high concentrations of multiple cytokines, including tumor necrosis factor-α (TNF-α) and interleukin (IL)-6, which may play key roles in disease pathogenesis [4,5,6]. TNF-α has been proposed to orchestrate an inflammatory process that extends beyond the liver [6]; however, although TNF-α seems to participate in the pathogenesis of NASH, its exact role has not been fully elucidated and warrants further investigation.
This review, first, focuses on the possible pathogenic role of TNF-α in NAFLD; second, summarizes clinical evidence on the association between TNF-α and NAFLD; and third, discusses potential treatment considerations derived from this association.
TNF-α and Pathogenesis of NAFLD
TNF-α in NAFLD: Origin and Biological Functions
NAFLD is characterized by a state of chronic, low-grade hepatic and systemic inflammation affecting multiple organs beyond the liver [6]. TNF-α is regarded as a key cytokine in this process, critically implicated in the pathogenesis of NAFLD. In NAFLD, TNF-α is not only produced in the liver by the resident hepatic cells, Kupffer cells (KCs), but also by the immune cells infiltrating the liver in the presence of steatosis [7]. Dysfunctional visceral adipose tissue also contributes to the production and secretion of TNF-α, mainly produced by immune cells infiltrating the adipose tissue, when it is expanded (e.g., in obesity); TNF-α of extra-hepatic origin is delivered to the liver via systemic circulation, along with other cytokines and adipokines, which may also affect the development and progression of NAFLD [7].
Obesity may affect the liver, at least partly, through the secretion of adipokines (e.g., leptin, adiponectin, resistin, visfatin), which are produced mainly but not exclusively by the adipocytes, and cytokines (e.g., TNF-α, IL-1, IL-6), which are produced mainly by the immune cells infiltrating the adipose tissue when it expands; there is an ever continuing and dynamic interplay among various adipokines and cytokines, exhibiting synergistic or antagonistic action [6, 8]. As for example, leptin and resistin were shown to upregulate the expression of TNF-α in the liver, thus contributing to the onset of NAFLD and to its progression to NASH [9, 10]. In contrast, adiponectin normally downregulates TNF-α expression in the liver, but its levels are low in obesity, thus TNF-α production is enhanced, with subsequent effects in the liver [5]. The above considering, circulating TNF-α derives from various sources, including the adipose tissue, and may affect distant tissues, including the liver.
In the liver, KCs respond to two main types of stimuli: the intrahepatic danger-associated molecular patterns (DAMPs), which are released by the lipid-infiltrated and damaged hepatocytes, and the gut-derived bacterial antigens, also known as pathogen-associated molecular patterns (PAMPs), which translocate from the intestine to the liver due to an impaired intestinal epithelial barrier [11]. Both DAMPs and PAMPs bind Toll-like receptors (TLRs) on the surface of KCs and activate the intracellular pathway of nuclear factor-kappa B (NF-κB), which is the key signaling pathway for the transcription of TNF-α, as well as other cytokines and chemokines [12]. Notably, hepatocytes that are stressed by fat accumulation (lipotoxicity) also contribute to TNF-α production via the interaction between DAMPs/PAMPs and TLRs, albeit to a lesser extent [11].
Once released, TNF-α manifests its biological functions by binding its cognate receptors, TNF receptor 1 (TNFR1) and TNF receptor 2 (TNFR2), which are expressed on the cell membrane [13]. Binding either TNFR1, which is ubiquitously expressed, or TNFR2, whose expression is restricted to immune and endothelial cells, TNF-α initiates two important downstream signaling pathways, i.e., the c-Jun N-terminal kinase (JNK) and NF-κB pathways [13]. Through these intracellular pathways, TNF-α induces the transcription of target genes involved in inflammation, cell proliferation, and survival [14]. Besides these cellular responses, TNF-α also induces cell death, which is only mediated by TNFR1 but not TNFR2. TNFR1 contains a death domain, which enables the assembly of a death-inducing protein complex that sufficiently mediates cell death [14].
Contribution of TNF-α to NAFL, NASH, and NAFLD-Related Fibrosis
Although the production of TNF-α may be an early event in NAFL, potentially enhancing the severity of hepatic steatosis, NAFL is strongly associated with insulin resistance (IR), which is considered a major pathogenic driver of lipid accumulation in the liver [7]. As mentioned above, lipotoxicity is a main trigger for the production of TNF-α in the hepatocytes [15]; nevertheless, TNF-α may also influence the development of NAFL by modulating hepatic lipid metabolism. Indeed, transgenic mouse models of NAFLD lacking TNF-α receptors (TNFR − / −) were protected from severe hepatic fat accumulation compared with those with intact TNF-α signaling [16,17,18]. Moreover, treatment of NAFLD mice with anti-TNF antibody [19] or selective anti-TNFR1 antibody [20] attenuated hepatic steatosis. Of note, TNF-α was proposed as a potential positive regulator of the mammalian target of rapamycin (mTOR) complex-1 pathway in the hepatocytes, thus inducing the expression of the sterol regulatory element-binding protein (SREBP)-1c, a key transcription factor of de novo lipogenesis; it is highlighted that insulin is also a major regulator of the mTOR pathway [20]. An effective mTOR pathway is essential for both the expression and activation of SREBP-1c and subsequent upregulation of target lipogenic enzymes, which facilitates the intra-hepatic conversion of carbohydrates to free fatty acids, thus increasing lipid accumulation in the hepatocytes.
Apart from its possible role in the pathogenesis of NAFL, TNF-α may play a more substantial role in the pathogenesis of NASH and NAFLD-associated fibrosis [4]. In NASH, TNF-α provokes the release of a variety of pro-inflammatory mediators, including, but not limited to IL-1β, IL-6, IL-18, monocyte chemoattractant protein (MCP)-1, C–C motif chemokine 5 (CCL5), resulting in a massive activation of the immune response [21]. In addition, TNF-α induces the expression of adhesion molecules (e.g., intracellular adhesion molecule-1 (ICAM-1)) on the surface of the endothelial cells of liver vessels, which facilitates the migration of monocytes, neutrophils, and lymphocytes from the bloodstream into the liver, which, in turn, may produce further TNF-α, thus creating a vicious cycle [21]. As a result, TNF-α fuels a prolonged and sustained liver inflammation through the activation of inflammatory mediators, as well as migration and activation of immune cells.
Hepatocellular death, another important feature distinguishing NASH from NAFL, is reciprocally connected with inflammation, as both processes may positively regulate each other [22]. In the hepatocytes, TNF-α promotes two separate death mechanisms: apoptosis and necroptosis, regulated mainly through TNFR1 signaling [13]. Apoptosis is a caspase-dependent programmed death pathway, whereas necroptosis is another form of programmed cell death that is caspase-independent and is mediated by the receptor-interacting protein (RIP)1, RIP3, and mixed lineage kinase domain-like protein (MLKL) [13]. While apoptosis is considered silent and immunologically inert, necroptosis activates the inflammatory cascade, thus the immune response [23]. However, TNF-α/TNFR interaction has been regarded as a less potent inducer of cell death compared with other mediators of death signals, such as Fas ligand (FasL) and TNF-related apoptosis-inducing ligand (TRAIL), because its pro-apoptotic effect may be counteracted by its cytoprotective effect and its involvement in cell survival mediated by NF-κB [24]. Thus, based on this possibly dual facet of TNF-α, also observed in other cytokines and adipokines [25], we could speculate that the upregulation of TNF-α in NAFL and NASH may primarily target to a cytoprotective effect, when lipids are increasingly accumulated into the hepatocytes; however, if this hepatoprotective effect fails, TNF-α induces the cell death of the affected hepatocytes. Of course, this hypothesis remains to be shown.
TNF-α is also involved in the pathogenesis of hepatic fibrogenesis. TNF-α induces the production of transforming growth factor-beta (TGF-β) by the hepatocytes and KCs, which triggers the activation, differentiation, and proliferation of hepatic stellate cells (HSCs) [26]. In addition, TNF-α was shown to upregulate the expression of periostin in hepatocytes [27] and tissue inhibitor of metalloproteinase (TIMP)-1 in HSCs [28], which were suggested to facilitate collagen deposition and extracellular matrix stabilization, respectively. Other authors proposed that TNF-α enhances the survival of HSCs, but it may not be directly involved in the activation and differentiation of HSCs into myofibroblasts [29]; this may imply that TNF-α requires the contribution of other factors to exert a fully fibrogenic effect to the liver. Thus, though TNF-α appears to favor a fibrogenic response in the liver, the precise underlying mechanisms by which TNF-α affects HSCs and interplays with other fibrogenic contributors are not fully understood.
Contribution of TNF-α to Insulin Resistance and NAFLD-Related Complications
TNF-α may link inflammation and IR, two hallmarks of NAFLD pathogenesis. Mice lacking TNF-α or TNFR were, at least partly, protected from IR in several “loss-of-function” experimental studies [30]. In line, hepatocyte-specific deficiency of TNFR1 protected mice from IR, but not NASH, as TNFR1 was deleted only in the hepatocytes, but not in KCs [31]. Furthermore, TNF-α administration was shown to induce IR, whereas inhibition of ΤNF-α was associated with improved insulin sensitivity [7]. Indeed, TNF-α may block insulin signaling in the hepatocytes at the post-receptor level; it triggers the expression of the suppressor of cytokine signaling (SOCS) proteins, which prevents tyrosine phosphorylation of insulin receptor substrates (IRS)-1 and IRS-2 and promotes their early degradation [32, 33]. Likewise, TNF-α-mediated activation of JNK contributes to the inhibition of insulin signaling (i.e., IR) through serine phosphorylation of IRS-1 and IRS-2 [24]. Moreover, TNF-α-induced SREBP-1c, in addition to promoting lipogenesis, also suppresses the IRS-1/2 synthesis, thus contributing to IR [24]. Notably, TNF-α also antagonizes adiponectin in NAFLD and suppresses its insulin-sensitizing effect on the hepatocytes [5, 8].
Therefore, TNF-α may integrate both inflammatory and metabolic signals, potentially connecting inflammation with IR, which synergically deteriorate NAFLD. Of note, these interconnecting mechanisms may also predispose to the development of NAFLD-related comorbidities, including CVD and HCC. Both IR and increased circulating TNF-α have been associated with NAFLD and CVD. Based on mechanistic studies, TNF-α may be one of the factors linking NAFLD, IR, and CVD [34, 35], the latter being the leading cause of mortality in patients with NAFLD [36]. In addition, we have recently focused on the interplay between inflammation, IR, and hepatic carcinogenesis, highlighting that IR, inflammation, and carcinogenesis in NAFLD may move hand-in-hand both intra- and extra-hepatically [37]. It has been supported that IR may directly be associated with HCC, independently from the classical inflammation-fibrosis cascade, by initiating intracellular mitogenic and anti-apoptotic pathways [38]. In this regard, TNF-α may be one of the mediators of this triadic interplay. TNF-α promotes hepatocellular tumorigenesis through the activation of hepatic progenitor cells [39]. Moreover, TNF-α-induces JNK, regarded as an oncogenic transcription factor, which is commonly activated in HCC [40].
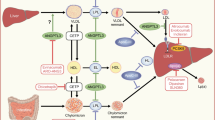
The above considering, TNF-α seems to be an important cytokine that potentiates hepatic IR and exhibits potential steatogenic, inflammatory, fibrogenic, and apoptotic effects in the liver. As a result, increased circulating and hepatic TNF-α may be associated with the onset and progression of NAFLD and its connection with hepatic complications and extrahepatic comorbidities. Figure 1 illustrates the potential molecular mechanisms through which TNF-α is possibly implicated in the pathogenesis of NAFLD.

The role of TNF-α in the pathogenesis of NAFLD. In the liver, TNF-α is primarily produced by the KCs, which respond to two types of stimuli: the DAMPs, which are released by lipid-infiltrated and damaged hepatocytes, and the gut-derived PAMPs, which translocate from the intestine to the liver, due to an impaired intestinal epithelial barrier. Both DAMPs and PAMPs bind TLRs on the surface of the KCs and activate the NF-κB, which is the key signaling pathway for the transcription of TNF-α. Furthermore, fat-stressed hepatocytes also contribute to TNF-α production via the interaction between DAMPs/PAMPs and TLRs, albeit to a lesser extent. Once released, TNF-α binds TNFR and stimulates the assembly of complex I, which initiates two important downstream signaling pathways, i.e., the JNK and NF-κB pathways, through which TNF-α induces the transcription of target genes involved in inflammation, cell proliferation, and survival. Besides these cellular responses, TNF-α also induces cell death by enabling the assembly of the death-inducing protein complex II, leading to either apoptosis or necroptosis. Besides, inflammation, survival, and apoptosis, TNF-α also contributes to IR and NAFL development; TNF-α perpetuates IR in hepatocytes as it blocks insulin signaling at the post-receptor level; TNF-α triggers the expression of SOCS, which prevent tyrosine phosphorylation of IRS-1 and IRS-2 and promote their early degradation. Likewise, TNF-α-mediated activation of JNK contributes to the phosphorylation of serine of IRS-1 and IRS-2, which inhibits their signaling. In addition, TNF-α is proposed as a potential positive regulator of the mTORC1 pathway in the hepatocytes, inducing the expression of the SREBP-1c, the main transcription factor of de novo lipogenesis. TNF-α-induced SREBP-1c, in addition to promoting de novo lipogenesis, also suppresses the IRS-1/2 synthesis, thus contributing to IR. Notably, TNF-α also antagonizes adiponectin and suppresses its insulin-sensitizing effect on the hepatocytes. Finally, TNF-α is involved in hepatic fibrogenesis; TNF-α induces the production of TGF-β by the hepatocytes and KCs, upregulates the expression of periostin in the hepatocytes and TIMP-1 in HSCs, which facilitate collagen deposition and EM stabilization
TNF-α in NAFLD: Evidence from Clinical Studies
Although most experimental and mechanistic studies suggest a causal role of TNF-α in the onset and progression of NAFLD from the early to advanced disease, clinical observational studies on the association between circulating TNF-α and NAFLD have yielded contradictory results. Most studies suggested that NAFLD patients had higher levels of circulating TNF-α than controls [41,42,43]. However, some studies did not show significant difference in circulating TNF-α concentrations between individuals with and without NAFLD [44, 45]. Of note, among studies with histologic confirmation of NAFLD, some found higher plasma TNF-α levels in NASH compared with NAFL patients [46, 47], while other studies showed no difference in circulating TNF-α between NAFL and NASH patients [48, 49].
Because of these heterogeneous and inconsistent findings, two meta-analyses on this topic have attempted to quantify the results of existing observational studies. In a meta-analysis of 56 studies, we demonstrated that circulating TNF-α was associated with the severity of NAFLD: TNF-α was higher in patients with NAFL than in non-NAFLD controls and higher in patients with NASH than NAFL or controls [50]. Ιn meta-regression analysis, male ratio was positively associated with TNF-α in the comparison between patients with NASH and NAFL and could explain 36% of the heterogeneity in this comparison [50]; therefore, the male to female ratio may influence the association between TNF-α levels and NAFLD, which should be taken into account in the design of relevant future studies. In line, another meta-analysis also demonstrated marginally higher circulating TNF-α levels in patients with NAFLD than non-NAFLD controls (11 studies) [51]. In addition, higher circulating TNF-α levels in patients with than without NAFLD-associated fibrosis were also reported in this study [51]; however, this finding should be carefully interpreted, because only two studies were included in this subgroup comparison. Apart from cross-sectional and case–control studies, which inherently cannot prove causal relationships, a prospective cohort study reported that higher serum TNF-α levels at baseline in apparently healthy participants were associated with increased adjusted risk of developing NAFLD after 4 years of follow-up [52]. A selection of the relevant observational studies, mainly those with largest sample size (> 100 participants) are summarized in Table 1, whereas a more comprehensive summary has been reported in the above-mentioned meta-analyses [50, 51].
Treatment Considerations
Due to the potential association between TNF-α and NAFLD, there are experimental and clinical studies targeting to evaluate the effect of various therapeutic interventions on TNF-α and NAFLD. Furthermore, anti-TNF-α therapies in various diseases have opened a possible therapeutic window for the management of advanced NAFLD.
Pharmacologic Strategies Downregulating TNF-α in NAFLD
This section provides an overview of current and emerging pharmacologic strategies for NAFLD that reduce circulating or hepatic TNF-α based on prevailing experimental or clinical evidence. These data are also summarized in Tables 2 and 3.
Obeticholic acid (OCA) is a potent farnesoid X receptor (FXR) agonist, an intriguing class of medication under investigation for the treatment of NAFLD. In addition to controlling a number of metabolic processes, including bile acid synthesis, glucose homeostasis, and lipid metabolism, OCA also exhibits anti-inflammatory and anti-fibrotic properties in the liver [53]. OCA has been reported to reduce hepatic TNF-α expression, either as monotherapy [54, 55] or more potently when combined with other agents (e.g., simvastatin, losartan, sitagliptin, elafibranor) [56,57,58,59] in experimental animal models of NASH. In clinical trials, OCA improved histological features of NASH and, more importantly, fibrosis [60, 61], thus being an important candidate for the treatment of NASH, as long as safety concerns, including unfavorable effects on lipid profile and pruritus are addressed.
Peroxisome proliferator-activated receptors (PPARs) are a superfamily of lipid-sensoring nuclear receptors, which are considered promising therapeutic targets for NAFLD, as they regulate glucose and lipid metabolism, inflammation and possibly fibrosis [62]. Pioglitazone, a PPAR-γ agonist that belongs to thiazolidinediones, is a long-standing antidiabetic medication, usually preferred as a second- or third-line option [63]. Although pioglitazone was shown to reduce TNF-α experimentally [64], in human NASH, it improved hepatic steatosis, inflammation, and possibly marginally hepatic fibrosis [65], by increasing the levels of circulating adiponectin but without affecting circulating TNF-α [66]. Pioglitazone is currently recommended for off-label treatment in selected patients with biopsy-proven NASH and fibrosis stage (F) ≥ 2 [67]. Similarly, rosiglitazone, another PPAR-γ activator belonging to thiazolidinediones, also reduced circulating TNF-α in NASH rat models [68, 69]; however, no data regarding its effect on TNF-α in human NASH are available. In addition, rosiglitazone did not improve NASH in the phase II trial (FLIRT) [70]. It should be also noted that the use of rosiglitazone, initially approved as anti-diabetic medication, was restricted because of an increase in myocardial infarction risk [62]. Fenofibrate, a commonly used drug against hypertriglyceridemia with an agonistic effect on PPAR-α, has been investigated against NAFLD, due to its pleiotropic properties, including lipid-lowering and anti-inflammatory action [71]. In line, fenofibrate was shown to decrease hepatic TNF-α expression in mouse models of NASH [72,73,74]. Similar to rosiglitazone, fenofibrate effect on TNF-α in human NASH has not been investigated to-date. Saroglitazar, which is a dual PPAR-α/γ agonist, i.e., exerting combined effects on PPAR-α and PPAR-γ, reduced hepatic TNF-α expression and improved histology in experimental NASH models [73,74,75]. Saroglitazar is under evaluation in NASH patients with fibrosis, following encouraging results in a recent randomized controlled trial (RCT), in which saroglitazar successfully decreased liver function tests (LFTs) and liver fat content in NAFLD patients [76]. Lanifibranor, a pan-PPAR agonist, ameliorated all histological features of NASH in mice, including fibrosis, and reduced activation of macrophages and TNF-α expression mainly via PPAR-δ agonism [77]. Lanifibranor is another promising therapeutic candidate as it achieved both primary and secondary endpoints in human NAFLD (resolution of NASH and fibrosis), therefore is currently under investigation in a phase 3 RCT (NCT04849728). Of note, beyond pre-clinical studies, clinical evidence on the effect of dual-PPAR agonist (saroglitazar) and pan-PPAR agonist (lanifibranor) on TNF-α are scarce.
Following favorable effects on non-invasive biomarkers of hepatic steatosis and fibrosis by sodium-glucose co-transporter-2 (SGLT-2) inhibitors in patients with NAFLD [78], a phase 3 RCT with dapagliflozin is ongoing in patients with biopsy-proven NASH (NCT03723252). SGLT-2 inhibitors act primarily by inducing glucosuria; however, data from T2DM preclinical and clinical studies have also revealed potential anti-inflammatory action and reduction of some circulating cytokines, including TNF-α [79]. Clinical data on the effect of SGLT-2 on TNF-α in the setting of NAFLD are lacking. Experimentally, remogliflozin was found to reduce hepatic TNF-α mRNA in mice with diet-induced NASH [80]; nevertheless, additional evidence is required to establish a potentially anti-TNF-α effect of SGLT2 in NAFLD. Glucagon-like peptide-1 receptor agonists (GLP-1RAs) represent another class of anti-diabetic medication showing many favorable metabolic effects that make them an appealing therapeutic option for NASH. Semaglutide and liraglutide achieved higher rates of NASH resolution compared with placebo in phase 2 clinical trials, but without improving fibrosis [2]. Besides metabolic properties, liraglutide was shown to exert anti-inflammatory action through downregulating TNF-α in a mouse model of NASH [81], a finding that was further supported in an RCT of patients with concomitant T2DM and NAFLD, where liraglutide, as well as metformin, reduced circulating TNF-α [82]. However, more studies are needed to establish any anti-TNF-α potential of liraglutide and other GLP-1RAs. On the other hand, sitagliptin, a dipeptidyl peptidase-4 (DPP-4) inhibitor, an approved anti-diabetic medication, failed to decrease hepatic TNF-α in an experimental NASH rat model [57]; it is highlighted that DPP-4 inhibitors showed minimal or null effects on NAFLD [2].
A number of RCTs have evaluated the efficacy and safety of probiotics in the treatment of NAFLD. The rationale lies on the proposed ability of probiotics to modulate the gut microbiome, thus beneficially affecting the gut-liver axis. At present, various strains, preparations, dosage schemes, and durations of treatment have been investigated on different NAFLD-related endpoints, which increases the heterogeneity between studies, thus rendering hard any secure conclusions. Probiotics improved LFTs, hepatic steatosis, plasma glucose and insulin levels, and lipid profile, but they did not affect body mass index (BMI) or circulating TNF-α, according to a meta-analysis of 21 RCTs involving 1037 patients with NAFLD [83]. In contrast, synbiotic supplementation (i.e., nutritional supplements that combine probiotics and prebiotics), apart from improving LFTs, lipid profile, and glucose metabolism, had favorable effect on circulating TNF-α, in another meta-analysis of 7 RCTs including 419 NAFLD patients [84].
Rifaximin is a broad-spectrum minimally absorbable antibiotic, which targets dysbiosis of intestinal microbiota and related endotoxemia. A 6-week course of 800 mg rifaximin daily in 15 histologically-proven NASH patients was not associated with robust changes in LFTs, hepatic steatosis, insulin sensitivity, and pro-inflammatory serum cytokines, including TNF-α [85]. A previous 4-week trial using a higher dose of rifaximin (1200 mg) reduced lipopolysaccharides (LPS) and improved BMI and LFTs in 27 biopsy-proven NASH patients, but serum TNF-α concentrations remained unaffected [86]. The authors suggested that the higher dose of rifaximin is most likely to reduce endotoxemia, but the relatively short treatment period may probably be insufficient to suppress cytokines, including TNF-α. In accordance with this hypothesis, a longer (6-month) RCT in histologically-confirmed NASH patients showed that daily administration of 1100 mg rifaximin reduced circulating TNF-α along with LFTs, IR, and presumable hepatic steatosis. [87].
Vitamin E is a potent antioxidant, which may also exhibit anti-TNF-α action, as shown in rats with diet-induced NASH [88]. Indeed, the anti-inflammatory properties of vitamin E were also evidenced in an RCT of patients with ultrasound-defined NAFLD, where vitamin E, mainly in the form of δ-tocotrienol reduced inflammatory mediators, including TNF-α [89]. Of note, vitamin E at a daily dose of 800 IU resolves NASH, but not hepatic fibrosis, and may be prescribed off-label for selected NASH patients with F ≥ 2 for maximum 2 years [71].
Pentoxifylline is a xanthine derivative, which is a non-selective phosphodiesterase inhibitor, shown to histologically improve NASH in two meta-analyses [90, 91]. These meta-analyses, however, do not agree on the effect of pentoxifylline on circulating TNF-α; one of them, including 3 placebo-controlled RCTs and 2 ursodeoxycholic acid (UDCA)-controlled prospective cohort studies, reported that pentoxifylline decreases circulating TNF-α [90], while the latter, limited to RCTs (n = 5), did not show a significant difference in TNF-α levels between pentoxifylline and placebo [91]. Thus, more and probably more focused on TNF-α studies are needed to elucidate the potential effect of pentoxifylline on TNF-α.
UDCA, a secondary bile acid, was shown efficacy to reduce pro-inflammatory cytokines in animal studies [92]. Clinical trials have shown some effects of UDCA on LFTs and possibly hepatic steatosis, but minimal, if any effect on hepatic inflammation and fibrosis [71]. However, the combination of UDCA with vitamin E improved histology in patients with NASH, by increasing adiponectin levels and reducing hepatocellular apoptosis, but without affecting circulating TNF-α and other mediators of inflammation [93]. Currently, existing evidence does not support the use of UDCA in patients with NASH.
Fibroblast growth factor-21 (FGF-21) analogs or mimetics act at the same receptors as the endogenous hepatokine FGF-21, which is regarded as a promising molecule against hepatic steatosis, inflammation and apoptosis [94]. B1344, an FGF-21 analog, reduced the expression of TNF-α and other cytokines in the liver of mice with diet-induced NASH [95], corroborating earlier evidence showing FGF-21 analogs to be effective in both animal models and humans with NASH, although their direct effect on TNF-α has not been investigated in human NASH to-date [94]. As a result, numerous clinical trials with FGF-21 analogs are currently underway [94].
Atorvastatin, a strong and widely used statin for the treatment of dyslipidemia [96], was shown to improve lipid profile, LFTs, and NAFLD activity score (NAS) in an uncontrolled, interventional trial with 42 biopsy-confirmed NASH patients at a daily dose of 10 mg for 12 months; this effect was partly attributed to its lowering effect on circulating TNF-α [97]. Similarly, rosuvastatin and simvastatin both decreased hepatic TNF-α mRNA in experimental mice models of NASH, implying their potential anti-inflammatory properties on the liver [58, 98]. Overall, statin therapy appears to be safe in NAFLD patients and should be used at least to treat dyslipidemia and prevent cardiovascular events in patients with NAFLD, although their effects on hepatic histology are not well documented [96].
Due to their triglyceride lowering effect, N-3 polyunsaturated fatty acids (n-3 PUFAs) were considered to be beneficial for NAFLD [99]. Supplementation with eicosapentaenoic acid (EPA), one of the major components of n-3 PUFAs, or a mixture of EPA with docosahexaenoic (DHA) and alpha lipoic acid (ALA) plus caloric restriction, significantly reduced circulating TNF-α receptor or TNF-α, respectively, in NAFLD patients [100, 101]. However, RCTs with histological endpoints did not provide favorable results in NAFLD [102] and, therefore, n-3 PUFAs are not currently recommended for the treatment of NASH [71]. Different compositions and formulas used in research and in clinical practice further complicate secure conclusions on their effects on NAFLD.
Angiotensin receptor blockers (ARBs) have been proposed as alternative therapeutic option for NAFLD, mainly owing to their potentially anti-inflammatory and anti-fibrotic effects in the liver [103]. Both valsartan [104] and olmesartan [105] were reported to reduce hepatic TNF-α in experimental studies, and telmisartan was more effective than valsartan in reducing hepatic expression of TNF-α in rats with diet-induced NASH in a comparative study [106]. In contrast, losartan had no effect on the hepatic TNF-α mRNA in another experimental NASH rat model [56]. These variations may be attributed to structural differences between ARBs, as well as to telmisartan properties beyond angiotensin receptor, e.g., the activation of PPAR-γ [103]. The experimentally observed anti-TNF action of some of ARBs has not been demonstrated to-date in human NASH. However, an open-label prospective study with short-term administration of telmisartan to hypertensive patients with metabolic syndrome (MetS) showed an increase in circulating adiponectin and improvement in IR but no effect on serum TNF-α levels [107]; however, the results of this study could not directly be extrapolated to NAFLD patients.
Anti-TNF-α Therapies: a Promising Therapeutic Approach for NAFLD Treatment
Given the potential steatotic, inflammatory, and fibrogenic effects of TNF-α in the liver, targeting TNF-α may be promising for the treatment of advanced NAFLD. TNF inhibitors are a class of biologic agents, which include infliximab, adalimumab, etanercept, golimumab, and certolizumab pegol, and have been widely used to treat chronic immune-related diseases, such as rheumatoid arthritis (RA), psoriatic arthritis (PsA), and inflammatory bowel diseases (IBD) [108]. Importantly, the extensive use of these agents in clinical practice over the last few decades has resulted in considerable experience on their efficacy and safety.
Experimental evidence has shown favorable effects of anti-TNF approaches on NAFLD outcomes; administration of infliximab in rats with diet-induced NASH resulted in histological regression of hepatic inflammation and fibrosis [109]. Hepatic steatosis, although improved, seems to be less affected by anti-TNF agents than other histological features of NAFLD [110].
In the clinical setting, initial reports on the metabolic and hepatic effects of anti-TNF biologics derive mostly from observational studies in patients with RA and PsA. PsA patients receiving a 24-month course of etanercept and adalimumab showed an improvement in metabolic syndrome components, like waist circumference, triglycerides, high-density lipoprotein-cholesterol (HDL-C), and glucose, when compared with those treated with methotrexate [111]. In line, other studies reported that anti-TNF reduced IR [112] and cardiovascular risk [113], both closely associated with NAFLD. Of note, administration of adalimumab to a young woman with rheumatoid arthritis and coexisting biopsy-proven NASH resulted in a remarkable biochemical response in terms of long-lasting improvements in LFTs [114]. Importantly, liver stiffness, which is associated with hepatic fibrosis, was lower in PsA patients treated with anti-TNF agents compared with those not on anti-TNF treatment, suggesting a possible antifibrotic effect [115]. On the contrary, some studies have raised the possibility that anti-TNF may increase body weight, which is a major risk factor for the development and progression of NAFLD [116]. There are also studies showing adverse hepatic effect after treatment with anti-TNF biologics. Administration of anti-TNF agents for 12 months in patients with PsA and US-defined NAFLD resulted in higher rates of worsening of hepatic steatosis compared with controls (with NAFLD but without PsA); the worsening of hepatic steatosis was greater in PsA patients with more active disease [117].
Concerning IBD populations, most studies showed higher prevalence of NAFLD in IBD patients than the general population [118]. Most studies do not reveal an association between biologic therapy and NAFLD incidence or severity [119]; however, existing studies are observational and were not designed towards this aim, i.e., the hepatic effect of biologics on NAFLD in IBD patients. It is highlighted that any possible benefit of anti-TNF inhibitors on NAFLD may be negated by a “rebound” weight gain observed following anti-TNF treatment, particularly in CD patients [108]. Anti-TNF medications may, in fact, ameliorate intestinal inflammation and achieve disease remission, which may lead to increased BMI and visceral adiposity due to restored nutrient absorption and increased appetite [108].
Clinical trials assessing anti-TNF medications specifically in NASH patients are not yet available. Before initiating such studies, observational studies with patients on anti-TNF agents for other conditions, who also have concomitant NASH, would be an excellent starting point [108]. Also, post hoc evaluations of existing clinical trials in patients with other diseases and concomitant NASH at baseline receiving anti-TNF agents may provide indirect insights into the effectiveness of anti-TNF agents on NASH. Positive outcomes in such observational studies may possibly support clinical trials with anti-TNF agents specifically in NASH, ideally with histological endpoints or non-invasive biomarkers of hepatic steatosis and fibrosis as acceptable alternatives [108]. Undoubtedly, we anticipate future studies designed to evaluate the role of anti-TNF-α agents in the treatment of NASH.
Conclusions
Collectively, based on current mechanistic and experimental studies, TNF-α seems to be a contributor, critically implicated in the onset and progression of NAFLD. Furthermore, clinical data are showing higher circulating concentrations of TNF-α in NAFLD patients. Importantly, TNF-α may be associated with disease severity; more advanced phenotypes (i.e., NASH or NAFLD-related cirrhosis) seem to be associated with higher circulating TNF-α, which may also link NAFLD with extrahepatic manifestations, such as CVD and malignancies.
Based on these considerations, this topic may have valuable perspectives and clinical implications, but also considerable challenges. TNF-α has been suggested as a potentially useful biomarker for the non-invasive stratification of patients with NAFLD [120], an hypothesis, however, warranting diagnostic accuracy studies to be verified and possibly to provide specific cut-offs of TNF-α to rule out or rule in the diagnosis of NASH or NAFLD-associated fibrosis. In our opinion, the incorporation of TNF-α in combined diagnostic algorithms seems to be a more realistic potential rather the use of TNF-α alone. Therapeutically, a number of NAFLD pharmacotherapies appear to lower TNF-α, thus their effects may be mediated, at least in part, through suppressing TNF-α, which, however, remains to be elucidated. Interestingly, anti-TNF biologics have shown favorable metabolic and hepatic effects both in experimental models of NASH, as well as in observational studies involving patients with immune-related diseases, which may provide the rationale for repurposing ant-TNF agents in the treatment of NASH, thus opening a new therapeutic window for the management of advanced NAFLD. However, we should also bear in mind that TNF-α is a complex molecule with diverse functions, thus blocking its activity may have unintended consequences. In this regard, it has been reported that anti-TNF agents may increase the risk of serious infections, as well cancer risk [121]. In addition there are concerns about weight gain during treatment with anti-TNF medications, which should also be taken into account when treating patients with NAFLD, the majority of whom are obese. Thus, before proceeding with further research in the field, a balanced assessment of risk–benefit ratio is highly required. It seems that an individualized approach is needed in NAFLD patients, so different management may be needed for different patients [122]; in this aspect, anti-TNF medications may possibly be proven valuable for selected only NAFLD patient in the future.
Data Availability
No datasets were generated or analyzed during the current review article.
Abbreviations
- AdipoR:
-
Adiponectin receptor
- DAMPs:
-
Danger-associated molecular patterns
- EM:
-
Extracellular matrix
- HSCs:
-
Hepatic stellate cells
- IR:
-
Insulin resistance
- IRS-1/2:
-
Insulin receptor substrate-1/2
- JNK:
-
C-Jun N-terminal kinase
- KCs:
-
Kupffer cells
- mTORC1:
-
Mammalian target of rapamycin complex
- NAFL:
-
Nonalcoholic fatty liver
- NF-κΒ:
-
Nuclear factor-kappa B
- PAMPs:
-
Pathogen-associated molecular patterns
- SOCS:
-
Suppressor of cytokine signaling proteins
- SREBP-1c:
-
Sterol regulatory element-binding protein-1c
- TGF-β:
-
Transforming growth factor-beta
- TIMP-1:
-
Tissue inhibitor of metalloproteinase-1
- TNF-α:
-
Tumor necrosis factor-α
- TNFR:
-
TNF receptor
- TLR:
-
Toll-like receptor
References
Papers of particular interest, published recently, have been highlighted as: •Of importance ••Of major importance
Henry L, Paik J, Younossi ZM. Review article: the epidemiologic burden of non-alcoholic fatty liver disease across the world. Aliment Pharmacol Ther. 2022;56:942–56.
Polyzos SA, Kang ES, Boutari C, Rhee E-J, Mantzoros CS. Current and emerging pharmacological options for the treatment of nonalcoholic steatohepatitis. Metabolism. 2020;111S:154203.
Makri E, Goulas A, Polyzos SA. Epidemiology, pathogenesis, diagnosis and emerging treatment of nonalcoholic fatty liver disease. Arch Med Res. 2021;52:25–37.
••Lu S, Wang Y, Liu J. Tumor necrosis factor-α signaling in nonalcoholic steatohepatitis and targeted therapies. J Genet Genomics. 2022;49:269–278. TNF-α is regarded as one of the main contributors to NASH development, based on several gain-of-function and loss-of-function experimental approaches.
Polyzos SA, Kountouras J, Zavos C. The multi-hit process and the antagonistic roles of tumor necrosis factor-alpha and adiponectin in non alcoholic fatty liver disease. Hippokratia. 2009;13:127.
Polyzos SA, Kountouras J, Mantzoros CS. Adipose tissue, obesity and non-alcoholic fatty liver disease. Minerva Endocrinol. 2017;42:92–108.
Polyzos SA, Kountouras J, Zavos C. Nonalcoholic fatty liver disease: the pathogenetic roles of insulin resistance and adipocytokines. Curr Mol Med. 2009;9:299–314.
Polyzos SA, Kountouras J, Mantzoros CS. Adipokines in nonalcoholic fatty liver disease. Metabolism. 2016;65:1062–79.
Machado MV, Coutinho J, Carepa F, Costa A, Proença H, Cortez-Pinto H. How adiponectin, leptin, and ghrelin orchestrate together and correlate with the severity of nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol. 2012;24:1166–72.
Wen F, Shi Z, Liu X, Tan Y, Wei L, Zhu X, et al. Acute elevated resistin exacerbates mitochondrial damage and aggravates liver steatosis through AMPK/PGC-1α signaling pathway in male NAFLD mice. Horm Metab Res. 2021;53:132–44.
Alisi A, Carsetti R, Nobili V. Pathogen- or damage-associated molecular patterns during nonalcoholic fatty liver disease development. Hepatology. 2011;54:1500–2.
Liu T, Zhang L, Joo D, Sun S-C. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2:17023.
••Tiegs G, Horst AK. TNF in the liver: targeting a central player in inflammation. Semin Immunopathol. 2022;44:445–59.TNF-α exhibits multiple biological functions in the liver, including hepatocyte apoptosis and necroptosis, liver inflammation and regeneration, and autoimmunity, but also progression to hepatocellular carcinoma.
Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65.
Rada P, González-Rodríguez Á, García-Monzón C, Valverde ÁM. Understanding lipotoxicity in NAFLD pathogenesis: is CD36 a key driver? Cell Death Dis. 2020;11:802.
Tomita K, Tamiya G, Ando S, Ohsumi K, Chiyo T, Mizutani A, et al. Tumour necrosis factor alpha signalling through activation of Kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut. 2006;55:415–24.
Kanuri G, Spruss A, Wagnerberger S, Bischoff SC, Bergheim I. Role of tumor necrosis factor α (TNFα) in the onset of fructose-induced nonalcoholic fatty liver disease in mice. J Nutr Biochem. 2011;22:527–34.
Kakino S, Ohki T, Nakayama H, Yuan X, Otabe S, Hashinaga T, et al. Pivotal role of TNF-α in the development and progression of nonalcoholic fatty liver disease in a murine model. Horm Metab Res. 2018;50:80–7.
Li Z, Yang S, Lin H, Huang J, Watkins PA, Moser AB, et al. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology. 2003;37:343–50.
•Wandrer F, Liebig S, Marhenke S, Vogel A, John K, Manns MP, et al. TNF-Receptor-1 inhibition reduces liver steatosis, hepatocellular injury and fibrosis in NAFLD mice. Cell Death Dis. 2020;11:212.Administration of anti-TNF receptor 1 antibody improved insulin resistance and histological features of NASH in NAFLD mice.
Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol. 2018;15:349–64.
Gautheron J, Gores GJ, Rodrigues CMP. Lytic cell death in metabolic liver disease. J Hepatol. 2020;73:394–408.
Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol. 2021;18:1106–21.
Bessone F, Razori MV, Roma MG. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell Mol Life Sci. 2019;76:99–128.
Polyzos SA, Kountouras J, Zavos C, Stergiopoulos C. Adipocytokines in insulin resistance and non-alcoholic fatty liver disease: the two sides of the same coin. Med Hypotheses. 2010;74:1089–90.
Yang YM, Seki E. TNFα in liver fibrosis. Curr Pathobiol Rep. 2015;3:253–61.
Amara S, Lopez K, Banan B, Brown S-K, Whalen M, Myles E, et al. Synergistic effect of pro-inflammatory TNFα and IL-17 in periostin mediated collagen deposition: potential role in liver fibrosis. Mol Immunol. 2015;64:26–35.
Osawa Y, Hoshi M, Yasuda I, Saibara T, Moriwaki H, Kozawa O. Tumor necrosis factor-α promotes cholestasis-induced liver fibrosis in the mouse through tissue inhibitor of metalloproteinase-1 production in hepatic stellate cells. PLoS ONE. 2013;8: e65251.
Pradere J-P, Kluwe J, De Minicis S, Jiao J-J, Gwak G-Y, Dapito DH, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58:1461–73.
Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature. 1997;389:610–4.
Bluemel S, Wang Y, Lee S, Schnabl B. Tumor necrosis factor alpha receptor 1 deficiency in hepatocytes does not protect from non-alcoholic steatohepatitis, but attenuates insulin resistance in mice. World J Gastroenterol. 2020;26:4933–44.
Galic S, Sachithanandan N, Kay TW, Steinberg GR. Suppressor of cytokine signalling (SOCS) proteins as guardians of inflammatory responses critical for regulating insulin sensitivity. Biochem J. 2014;461:177–88.
Polyzos SA, Kountouras J, Zavos C, Deretzi G. The potential adverse role of leptin resistance in nonalcoholic fatty liver disease. J Clin Gastroenterol. 2011;45:50–4.
Lim S, Oh TJ, Koh KK. Mechanistic link between nonalcoholic fatty liver disease and cardiometabolic disorders. Int J Cardiol. 2015;201:408–14.
Ajmal MR, Yaccha M, Malik MA, Rabbani MU, Ahmad I, Isalm N, et al. Prevalence of nonalcoholic fatty liver disease (NAFLD) in patients of cardiovascular diseases and its association with hs-CRP and TNF-α. Indian Heart J. 2014;66:574–9.
Polyzos SA, Kechagias S, Tsochatzis EA. Review article: non-alcoholic fatty liver disease and cardiovascular diseases: associations and treatment considerations. Aliment Pharmacol Ther. 2021;54:1013–25.
Nonalcoholic fatty liver disease and hepatocellular carcinoma. insights in epidemiology, pathogenesis, imaging, prevention and therapy. Semin Cancer Biol. 2023;93:20–35.
Chettouh H, Lequoy M, Fartoux L, Vigouroux C, Desbois-Mouthon C. Hyperinsulinaemia and insulin signalling in the pathogenesis and the clinical course of hepatocellular carcinoma. Liver Int. 2015;35:2203–17.
Jing Y, Sun K, Liu W, Sheng D, Zhao S, Gao L, et al. Tumor necrosis factor-α promotes hepatocellular carcinogenesis through the activation of hepatic progenitor cells. Cancer Lett. 2018;434:22–32.
Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma Nat Rev Dis Primers. 2021;7:6.
Chellali S, Boudiba A, Griene L. Koceir E-A [Incretins-adipocytokines interactions in type 2 diabetic subjects with or without non-alcoholic fatty liver disease: interest of GLP-1 (glucagon-like peptide-1) as a modulating biomarker]. Ann Biol Clin. 2019;77:261–71.
Federico A, Dallio M, Masarone M, Gravina AG, Di Sarno R, Tuccillo C, et al. Evaluation of the effect derived from silybin with vitamin D and vitamin E administration on clinical, metabolic, endothelial dysfunction, oxidative stress parameters, and serological worsening markers in nonalcoholic fatty liver disease patients. Oxid Med Cell Longev. 2019;2019:8742075.
Kapil S, Duseja A, Sharma BK, Singla B, Chakraborti A, Das A, et al. Small intestinal bacterial overgrowth and toll-like receptor signaling in patients with non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2016;31:213–21.
Loguercio C, De Simone T, D’Auria MV, de Sio I, Federico A, Tuccillo C, et al. Non-alcoholic fatty liver disease: a multicentre clinical study by the Italian Association for the Study of the Liver. Dig Liver Dis. 2004;36:398–405.
Kashyap SR, Diab DL, Baker AR, Yerian L, Bajaj H, Gray-McGuire C, et al. Triglyceride levels and not adipokine concentrations are closely related to severity of nonalcoholic fatty liver disease in an obesity surgery cohort. Obesity. 2009;17:1696–701.
Wong VW-S, Hui AY, Tsang SW-C, Chan JL-Y, Tse AM-L, Chan K-F, et al. Metabolic and adipokine profile of Chinese patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2006;4:1154–61.
Abdel-Razik A, Mousa N, Shabana W, Refaey M, ElMahdy Y, Elhelaly R, et al. A novel model using mean platelet volume and neutrophil to lymphocyte ratio as a marker of nonalcoholic steatohepatitis in NAFLD patients: multicentric study. Eur J Gastroenterol Hepatol. 2016;28:e1–9.
Koehler E, Swain J, Sanderson S, Krishnan A, Watt K, Charlton M. Growth hormone, dehydroepiandrosterone and adiponectin levels in non-alcoholic steatohepatitis: an endocrine signature for advanced fibrosis in obese patients. Liver Int. 2012;32:279–86.
Ajmera V, Perito ER, Bass NM, Terrault NA, Yates KP, Gill R, et al. Novel plasma biomarkers associated with liver disease severity in adults with nonalcoholic fatty liver disease. Hepatology. 2017;65:65–77.
••Potoupni V, Georgiadou M, Chatzigriva E, Polychronidou G, Markou E, Zapantis Gakis C, et al. Circulating tumor necrosis factor-α levels in non-alcoholic fatty liver disease: a systematic review and a meta-analysis. J Gastroenterol Hepatol. 2021;36:3002–14. Circulating TNF-α was associated with the severity of NAFLD; its concentrations increased gradually from controls to patients with NAFL, and then, to patients with NASH.
••Duan Y, Pan X, Luo J, Xiao X, Li J, Bestman PL, et al. Association of inflammatory cytokines with non-alcoholic fatty liver disease. Front Immunol. 2022;13:880298. Circulating TNF-α levels, as well as CRP, IL-1β, IL-6 and ICAM-1 were higher in patients with NAFLD compared with non-NAFLD controls.
••Seo YY, Cho YK, Bae J-C, Seo MH, Park SE, Rhee E-J, et al. Tumor necrosis factor-α as a predictor for the development of nonalcoholic fatty liver disease: a 4-year follow-up study. Endocrinol Metab (Seoul). 2013;28:41–5. A prospective cohort study, which reported that higher serum TNF-α levels at baseline in apparently healthy participants were associated with increased risk of developing NAFLD after 4 years of follow-up.
Polyzos SA, Kountouras J, Mantzoros CS. Obeticholic acid for the treatment of nonalcoholic steatohepatitis: expectations and concerns. Metabolism. 2020;104:154144.
Morrison MC, Verschuren L, Salic K, Verheij J, Menke A, Wielinga PY, et al. Obeticholic acid modulates serum metabolites and gene signatures characteristic of human NASH and attenuates inflammation and fibrosis progression in Ldlr-/-.Leiden Mice. Hepatol Commun. 2018;2:1513–32.
Goto T, Itoh M, Suganami T, Kanai S, Shirakawa I, Sakai T, et al. Obeticholic acid protects against hepatocyte death and liver fibrosis in a murine model of nonalcoholic steatohepatitis. Sci Rep. 2018;8:8157.
Namisaki T, Moriya K, Kitade M, Takeda K, Kaji K, Okura Y, et al. Effect of combined farnesoid X receptor agonist and angiotensin II type 1 receptor blocker on hepatic fibrosis. Hepatol Commun. 2017;1:928–45.
Shimozato N, Namisaki T, Kaji K, Kitade M, Okura Y, Sato S, et al. Combined effect of a farnesoid X receptor agonist and dipeptidyl peptidase-4 inhibitor on hepatic fibrosis. Hepatol Res. 2019;49:1147–61.
Li W-C, Zhao S-X, Ren W-G, Zhang Y-G, Wang R-Q, Kong L-B, et al. Co-administration of obeticholic acid and simvastatin protects against high-fat diet-induced non-alcoholic steatohepatitis in mice. Exp Ther Med. 2021;22:830.
Roth JD, Veidal SS, Fensholdt LKD, Rigbolt KTG, Papazyan R, Nielsen JC, et al. Combined obeticholic acid and elafibranor treatment promotes additive liver histological improvements in a diet-induced ob/ob mouse model of biopsy-confirmed NASH. Sci Rep. 2019;9:9046.
Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385:956–65.
Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2019;394:2184–96.
Polyzos SA, Mantzoros CS. Adiponectin as a target for the treatment of nonalcoholic steatohepatitis with thiazolidinediones: a systematic review. Metabolism. 2016;65:1297–306.
Upadhyay J, Polyzos SA, Perakakis N, Thakkar B, Paschou SA, Katsiki N, et al. Pharmacotherapy of type 2 diabetes: an update. Metabolism. 2018;78:13–42.
Zhao J-S, Zhu F-S, Liu S, Yang C-Q, Chen X-M. Pioglitazone ameliorates nonalcoholic steatohepatitis by down-regulating hepatic nuclear factor-kappa B and cyclooxygenases-2 expression in rats. Chin Med J. 2012;125:2316–21.
Mahady SE, Webster AC, Walker S, Sanyal A, George J. The role of thiazolidinediones in non-alcoholic steatohepatitis - a systematic review and meta analysis. J Hepatol. 2011;55:1383–90.
Lutchman G, Promrat K, Kleiner DE, Heller T, Ghany MG, Yanovski JA, et al. Changes in serum adipokine levels during pioglitazone treatment for nonalcoholic steatohepatitis: relationship to histological improvement. Clin Gastroenterol Hepatol. 2006;4:1048–52.
European Association for the Study of the Liver (EASL), European Association for the Study of Diabetes (EASD), European Association for the Study of Obesity (EASO). EASL-EASD-EASO clinical practice guidelines for the management of non-alcoholic fatty liver disease. Obes Facts. 2016;9:65–90.
Liu S, Wu H-J, Zhang Z-Q, Chen Q, Liu B, Wu J-P, et al. The ameliorating effect of rosiglitazone on experimental nonalcoholic steatohepatitis is associated with regulating adiponectin receptor expression in rats. Eur J Pharmacol. 2011;650:384–9.
Tahan V, Eren F, Avsar E, Yavuz D, Yuksel M, Emekli E, et al. Rosiglitazone attenuates liver inflammation in a rat model of nonalcoholic steatohepatitis. Dig Dis Sci. 2007;52:3465–72.
Ratziu V, Charlotte F, Bernhardt C, Serfaty L, Podevin P, Moussalli J, et al. 56 long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis (Nash): results of the extension phase of the flirt-2 trial. J Hepatol. 2009;50:S24–5.
Mintziori G, Polyzos SA. Emerging and future therapies for nonalcoholic steatohepatitis in adults. Expert Opin Pharmacother. 2016;17:1937–46.
Hong XZ, Li LD, Wu LM. Effects of fenofibrate and xuezhikang on high-fat diet-induced non-alcoholic fatty liver disease. Clin Exp Pharmacol Physiol. 2007;34:27–35.
Jain MR, Giri SR, Bhoi B, Trivedi C, Rath A, Rathod R, et al. Dual PPARα/γ agonist saroglitazar improves liver histopathology and biochemistry in experimental NASH models. Liver Int. 2018;38:1084–94.
Akbari R, Behdarvand T, Afarin R, Yaghooti H, Jalali MT, Mohammadtaghvaei N. Saroglitazar improved hepatic steatosis and fibrosis by modulating inflammatory cytokines and adiponectin in an animal model of non-alcoholic steatohepatitis. BMC Pharmacol Toxicol. 2021;22:53.
Kumar DP, Caffrey R, Marioneaux J, Santhekadur PK, Bhat M, Alonso C, et al. The PPAR α/γ agonist saroglitazar improves insulin resistance and steatohepatitis in a diet induced animal model of nonalcoholic fatty liver disease. Sci Rep. 2020;10:9330.
Gawrieh S, Noureddin M, Loo N, Mohseni R, Awasty V, Cusi K, et al. Saroglitazar, a PPAR-α/γ agonist, for treatment of NAFLD: a randomized controlled double-blind phase 2 trial. Hepatology. 2021;74:1809–24.
Lefere S, Puengel T, Hundertmark J, Penners C, Frank AK, Guillot A, et al. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages☆. J Hepatol. 2020;73:757–70.
Makri ES, Goulas A, Polyzos SA. Sodium-glucose co-transporter 2 inhibitors in nonalcoholic fatty liver disease. Eur J Pharmacol. 2021;907: 174272.
Wang D, Liu J, Zhong L, Li S, Zhou L, Zhang Q, et al. The effect of sodium-glucose cotransporter 2 inhibitors on biomarkers of inflammation: a systematic review and meta-analysis of randomized controlled trials. Front Pharmacol. 2022;13:1045235.
Nakano S, Katsuno K, Isaji M, Nagasawa T, Buehrer B, Walker S, et al. Remogliflozin etabonate improves fatty liver disease in diet-induced obese male mice. J Clin Exp Hepatol. 2015;5:190–8.
Luo Y, Yang P, Li Z, Luo Y, Shen J, Li R, et al. Liraglutide improves non-alcoholic fatty liver disease in diabetic mice by modulating inflammatory signaling pathways. Drug Des Devel Ther. 2019;13:4065–74.
Ying X, Rongjiong Z, Kahaer M, Chunhui J, Wulasihan M. Therapeutic efficacy of liraglutide versus metformin in modulating the gut microbiota for treating type 2 diabetes mellitus complicated with nonalcoholic fatty liver disease. Front Microbiol. 2023;14:1088187.
Zhou X, Wang J, Zhou S, Liao J, Ye Z, Mao L. Efficacy of probiotics on nonalcoholic fatty liver disease: a meta-analysis. Medicine. 2023;102:e32734.
Hadi A, Mohammadi H, Miraghajani M, Ghaedi E. Efficacy of synbiotic supplementation in patients with nonalcoholic fatty liver disease: a systematic review and meta-analysis of clinical trials: synbiotic supplementation and NAFLD. Crit Rev Food Sci Nutr. 2019;59:2494–505.
Cobbold JFL, Atkinson S, Marchesi JR, Smith A, Wai SN, Stove J, et al. Rifaximin in non-alcoholic steatohepatitis: an open-label pilot study. Hepatol Res. 2018;48:69–77.
Gangarapu V, Ince AT, Baysal B, Kayar Y, Kılıç U, Gök Ö, et al. Efficacy of rifaximin on circulating endotoxins and cytokines in patients with nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol. 2015;27:840–5.
Abdel-Razik A, Mousa N, Shabana W, Refaey M, Elzehery R, Elhelaly R, et al. Rifaximin in nonalcoholic fatty liver disease: hit multiple targets with a single shot. Eur J Gastroenterol Hepatol. 2018;30:1237–46.
Lahmi A, Oryan S, Eidi A, Rohani AH. Comparative effects of thymol and vitamin E on nonalcoholic fatty liver disease in male Wistar rats. Braz J Biol. 2023;84:e268781.
Pervez MA, Khan DA, Mirza SA, Slehria AUR, Nisar U, Aamir M. Comparison of delta-tocotrienol and alpha-tocopherol effects on hepatic steatosis and inflammatory biomarkers in patients with non-alcoholic fatty liver disease: a randomized double-blind active-controlled trial. Complement Ther Med. 2022;70:102866.
Du J, Ma Y-Y, Yu C-H, Li Y-M. Effects of pentoxifylline on nonalcoholic fatty liver disease: a meta-analysis. World J Gastroenterol. 2014;20:569–77.
Zeng T, Zhang C-L, Zhao X-L, Xie K-Q. Pentoxifylline for the treatment of nonalcoholic fatty liver disease: a meta-analysis of randomized double-blind, placebo-controlled studies. Eur J Gastroenterol Hepatol. 2014;26:646–53.
Li H, Wang Q, Chen P, Zhou C, Zhang X, Chen L. Ursodeoxycholic acid treatment restores gut microbiota and alleviates liver inflammation in non-alcoholic steatohepatitic mouse model. Front Pharmacol. 2021;12:788558.
Balmer ML, Siegrist K, Zimmermann A, Dufour J-F. Effects of ursodeoxycholic acid in combination with vitamin E on adipokines and apoptosis in patients with nonalcoholic steatohepatitis. Liver Int. 2009;29:1184–8.
Raptis DD, Mantzoros CS, Polyzos SA. Fibroblast growth factor-21 as a potential therapeutic target of nonalcoholic fatty liver disease. Ther Clin Risk Manag. 2023;19:77–96.
Cui A, Li J, Ji S, Ma F, Wang G, Xue Y, et al. The effects of B1344, a novel fibroblast growth factor 21 analog, on nonalcoholic steatohepatitis in nonhuman primates. Diabetes. 2020;69:1611–23.
Athyros VG, Alexandrides TK, Bilianou H, Cholongitas E, Doumas M, Ganotakis ES, et al. The use of statins alone, or in combination with pioglitazone and other drugs, for the treatment of non-alcoholic fatty liver disease/non-alcoholic steatohepatitis and related cardiovascular risk. An Expert Panel Statement Metabolism. 2017;71:17–32.
Hyogo H, Yamagishi S-I, Maeda S, Kimura Y, Ishitobi T, Chayama K. Atorvastatin improves disease activity of nonalcoholic steatohepatitis partly through its tumour necrosis factor-α-lowering property. Dig Liver Dis. 2012;44:492–6.
Okada Y, Yamaguchi K, Nakajima T, Nishikawa T, Jo M, Mitsumoto Y, et al. Rosuvastatin ameliorates high-fat and high-cholesterol diet-induced nonalcoholic steatohepatitis in rats. Liver Int. 2013;33:301–11.
de Castro GS, Calder PC. Non-alcoholic fatty liver disease and its treatment with n-3 polyunsaturated fatty acids. Clin Nutr. 2018;37:37–55.
Tanaka N, Sano K, Horiuchi A, Tanaka E, Kiyosawa K, Aoyama T. Highly purified eicosapentaenoic acid treatment improves nonalcoholic steatohepatitis. J Clin Gastroenterol. 2008;42:413–8.
Spadaro L, Magliocco O, Spampinato D, Piro S, Oliveri C, Alagona C, et al. Effects of n-3 polyunsaturated fatty acids in subjects with nonalcoholic fatty liver disease. Dig Liver Dis. 2008;40:194–9.
Sanyal AJ, Abdelmalek MF, Suzuki A, Cummings OW, Chojkier M, EPE-A Study Group. No significant effects of ethyl-eicosapentanoic acid on histologic features of nonalcoholic steatohepatitis in a phase 2 trial. Gastroenterology. 2014;147:377–84.e1.
Paschos P, Tziomalos K. Nonalcoholic fatty liver disease and the renin-angiotensin system: implications for treatment. World J Hepatol. 2012;4:327–31.
Assy N, Grozovski M, Bersudsky I, Szvalb S, Hussein O. Effect of insulin-sensitizing agents in combination with ezetimibe, and valsartan in rats with non-alcoholic fatty liver disease. World J Gastroenterol. 2006;12:4369–76.
Hirose A, Ono M, Saibara T, Nozaki Y, Masuda K, Yoshioka A, et al. Angiotensin II type 1 receptor blocker inhibits fibrosis in rat nonalcoholic steatohepatitis. Hepatology. 2007;45:1375–81.
Fujita K, Yoneda M, Wada K, Mawatari H, Takahashi H, Kirikoshi H, et al. Telmisartan, an angiotensin II type 1 receptor blocker, controls progress of nonalcoholic steatohepatitis in rats. Dig Dis Sci. 2007;52:3455–64.
Kiyici S, Guclu M, Budak F, Sigirli D, Tuncel E. Even short-term telmisartan treatment ameliorated insulin resistance but had no influence on serum adiponectin and tumor necrosis factor-alpha levels in hypertensive patients with metabolic syndrome. Metab Syndr Relat Disord. 2019;17:167–72.
Papaefthymiou A, Potamianos S, Goulas A, Doulberis M, Kountouras J, Polyzos SA. Inflammatory bowel disease-associated fatty liver disease: the potential effect of biologic agents. J Crohns Colitis. 2022;16:852–62.
Barbuio R, Milanski M, Bertolo MB, Saad MJ, Velloso LA. Infliximab reverses steatosis and improves insulin signal transduction in liver of rats fed a high-fat diet. J Endocrinol. 2007;194:539–50.
•Koca SS, Bahcecioglu IH, Poyrazoglu OK, Ozercan IH, Sahin K, Ustundag B. The treatment with antibody of TNF-alpha reduces the inflammation, necrosis and fibrosis in the non-alcoholic steatohepatitis induced by methionine- and choline-deficient diet. Inflammation. 2008;31:91–8. Administration of anti-TNF-α antibody (infliximab) in rats with diet-induced NASH resulted in histological regression of hepatic inflammation and fibrosis, and, to a lesser extent, of hepatic steatosis.
Costa L, Caso F, Atteno M, Del Puente A, Darda MA, Caso P, et al. Impact of 24-month treatment with etanercept, adalimumab, or methotrexate on metabolic syndrome components in a cohort of 210 psoriatic arthritis patients. Clin Rheumatol. 2014;33:833–9.
Maruotti N, d’Onofrio F, Cantatore FP. Metabolic syndrome and chronic arthritis: effects of anti-TNF-α therapy. Clin Exp Med. 2015;15:433–8.
Barnabe C, Martin B-J, Ghali WA. Systematic review and meta-analysis: anti-tumor necrosis factor α therapy and cardiovascular events in rheumatoid arthritis. Arthritis Care Res. 2011;63:522–9.
Schramm C, Schneider A, Marx A, Lohse AW. Adalimumab could suppress the activity of non alcoholic steatohepatitis (NASH). Z Gastroenterol. 2008;46:1369–71.
Seitz M, Reichenbach S, Möller B, Zwahlen M, Villiger PM, Dufour J-F. Hepatoprotective effect of tumour necrosis factor alpha blockade in psoriatic arthritis: a cross-sectional study. Ann Rheum Dis. 2010;69:1148–50.
Alcorn N, Tierney A, Wu O, Gilmour H, Madhok R. Impact of anti-tumour necrosis factor therapy on the weight of patients with rheumatoid arthritis. Ann Rheum Dis. 2010;69:1571.
Di Minno MND, Iervolino S, Peluso R, Russolillo A, Lupoli R, Scarpa R, et al. Hepatic steatosis and disease activity in subjects with psoriatic arthritis receiving tumor necrosis factor-α blockers. J Rheumatol. 2012;39:1042–6.
Lin A, Roth H, Anyane-Yeboa A, Rubin DT, Paul S. Prevalence of nonalcoholic fatty liver disease in patients with inflammatory bowel disease: a systematic review and meta-analysis. Inflamm Bowel Dis. 2021;27:947–55.
Lapumnuaypol K, Kanjanahattakij N, Pisarcik D, Thongprayoon C, Wijarnpreecha K, Cheungpasitporn W. Effects of inflammatory bowel disease treatment on the risk of nonalcoholic fatty liver disease: a meta-analysis. Eur J Gastroenterol Hepatol. 2018;30:854–60.
Fontes-Cal TCM, Mattos RT, Medeiros NI, Pinto BF, Belchior-Bezerra M, Roque-Souza B, et al. Crosstalk between plasma cytokines, inflammation, and liver damage as a new strategy to monitoring NAFLD progression. Front Immunol. 2021;12: 708959.
Li J, Zhang Z, Wu X, Zhou J, Meng D, Zhu P. Risk of adverse events after anti-TNF treatment for inflammatory rheumatological disease. A meta-analysis Front Pharmacol. 2021;12:746396.
Makri ES, Makri E, Polyzos SA. Combination therapies for nonalcoholic fatty liver disease. J Pers Med. 2022;12:1166.
Funding
Open access funding provided by HEAL-Link Greece.
Author information
Authors and Affiliations
Contributions
IDV and SAP: concept and design; IDV: drafting the manuscript; IDV and SAP: critically revising the manuscript and approval of the final version to be submitted.
Corresponding author
Ethics declarations
Conflict of Interest
The authors have no competing interests to declare that are relevant to the content of this article.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vachliotis, I.D., Polyzos, S.A. The Role of Tumor Necrosis Factor-Alpha in the Pathogenesis and Treatment of Nonalcoholic Fatty Liver Disease. Curr Obes Rep 12, 191–206 (2023). https://doi.org/10.1007/s13679-023-00519-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13679-023-00519-y