
Abstract
Purpose of Review
Hypersensitivity pneumonitis (HP) is an immune-mediated disease triggered by a known or unknown antigen. While reversible in the early stages of disease, progression toward irreversible pulmonary fibrosis may occur. This narrative review summarizes recent publications highlighting a methodical approach toward the diagnosis, classification, and management of fibrotic and nonfibrotic HP.
Recent Findings
Establishing the diagnosis of HP is often challenging given its variable clinical course, extensive inciting agents, and overlapping features with other interstitial lung diseases. Recently, HP has been re-classified into nonfibrotic and fibrotic subtypes based on radiographic and histopathological features. Chronic fibrotic HP is associated with significant functional impairment and increased mortality. In addition to antigen avoidance, immunosuppression is the cornerstone of management in nonfibrotic HP. Antifibrotic agents have emerged as a therapeutic option in halting the progression of chronic fibrotic HP.
Summary
The combination of clinical, radiographical, and histopathological data will assist in increasing the diagnostic certainty of HP. The new dichotomization of HP is thought to provide better prognostication for patients. This review provides clinicians with a current and evidence-based approach toward the management of patients with HP.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hypersensitivity pneumonitis (HP) is subtype of interstitial lung disease (ILD). The rate of HP varies from 2 to 47%, depending on occupational or environmental exposure, geographical location, and inherent genetic host risk factors [1, 2]. The highest prevalence is noted in patients in the fifth or sixth decade of life [3•]. In the United States (US), HP accounts for less than 2% of ILD [4]. The annual incidence of HP is approximately 30 per 100,000 persons [5]. ILD registries in Europe indicate that HP accounts for 4 to 15% of all ILD cases [1]. The National Center for Health Statistics of the US shows an increase in HP mortality from 0.09 to 0.29 per million between 1980 and 2002 for residents aged 15 years or older [6].
Clinical Presentation
Patients with HP can present with a variety of symptoms, depending on the time course of the disease. Common symptoms and signs include dyspnea, cough, chest tightness, wheezing, and mid-inspiratory squeaks [7•]. Infrequently, constitutional symptoms such as low-grade fever, weight loss, and malaise may occur. These symptoms may be episodic and recurrent. Due to its non-specific nature, the diagnosis of HP is often delayed or missed. Physical examination may be completely normal or show findings of high-pitched end-inspiratory wheeze, “squeaks,” crackles, or rales [8]. Patients with acute HP present with symptoms of several weeks to months (less than 6 months) whereas symptoms for chronic hypersensitivity pneumonitis (CHP) are present for a longer duration [7•].
Pathophysiology of HP
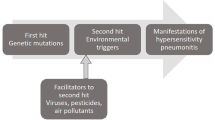
Pathophysiology and progression of HP are dependent on duration and type of exposure, and the host inflammatory response. Genetic predilection, combined with environmental factors, may yield a robust and significant immune response in the lungs. Increased expression of Th1 cytokines, TNF-alpha, IL-12, interferon-gamma, and toll-like receptor 9-mediated dendritic cell response appears to be implicated in the immune response to the inflammatory antigen (Fig. 1) in HP [7•]. The etiological agent that triggers the host response may be challenging to identify. Since its discovery in the 1700s, HP has been associated with many culprits [9]. Yet, the definite diagnosis of HP remains challenging as the inciting agent is not identified in up to 60% of patients [10].

Proposed mechanisms in the pathogenesis of hypersensitivity pneumonitis. This image illustrates the proposed mechanisms in the pathogenesis of hypersensitivity pneumonitis. It includes the interplay of genetic and environmental factors, inflammatory response in the alveoli, and fibroblastic activity leading to irreversible changes and fibrosis
Common infectious, organic, and inorganic trigger agents–ranging from most common to least common–are detailed in Table 1. It is critical that a thorough history, and an exploration of all possible exposures is conducted, as this may halt or reduce the likelihood of disease progression. Recurrent exposure leads to persistent inflammation and, consequently, disease progression into lung fibrosis.
In 2020, a list of potential inciting agents was derived from a Delphi systematic review of literature conducted by an international panel of ILD experts [11•]. All patients suspected of having CHP should be investigated for exposure to these agents. Examples include exposure to water damage, mold, air conditioners, hot tubs, organic matter, musical instruments, dentistry products, avian exposures, farming and food products, and chemical agents such as isocyanates, metal working fluids, and fumes [11•]. Patients should complete a comprehensive environmental and occupational questionnaire tailored to the local geographical prevalence. In a prospective study evaluating a cohort of 400 patients, exposure to identified inciting agent was found to be the strongest of six significant predictors of HP [12].
Subtypes of HP
HP is historically subcategorized into acute, subacute, or chronic subtypes. Acute HP is often reversible secondary to an acute inflammatory process with complete resolution of symptoms following antigen avoidance and/or therapy [13]. However, this categorization provides little prognostic value [13]. To better predict prognostication, the American Thoracic Society, Japanese Respiratory Society, and Asociación Latinoamericana del Tórax (ATS/JRS/ALAT) recently published guidelines categorizing HP into fibrotic and nonfibrotic subtypes, based on the presence or absence of radiographic or histopathological fibrosis [7•]. Table 2 illustrates key distinguishing features of the most recent dichotomization of HP.
Radiologic Features of HP
The radiographic features of HP are best determined by high-resolution computed tomogram (HRCT) of the chest [14]. Features of nonfibrotic HP include findings suggestive of parenchymal infiltration, such as ground glass opacity (GGO), and findings suggestive of air trapping, such as centrilobular diffuse micronodules on inspiration, and mosaicisms [15]. The distribution of abnormalities is often diffuse, with or without basal sparing. On the other hand, airspace consolidations, subtle GGO, and lung cysts are radiographic findings that are compatible with nonfibrotic HP but are not specific to HP [7•]. Figure 2A, B depicts common features of nonfibrotic HP. “Three-density pattern” or the “head cheese signs” describes the combination of GGO, mosaic attenuation, and normal appearing lung that is highly specific for HP [16].

A, B Computed-tomogram images of a patient with nonfibrotic hypersensitivity pneumonitis. A and B depict the “three-density pattern” commonly seen in nonfibrotic hypersensitivity pneumonitis. There is evidence of ground glass opacities, mosaic attenuation, and normal appearing lung
Meanwhile, the HRCT pattern of fibrotic HP is characterized by small airway diseases and fibrosis [17]. Examples include reticulation with architectural lung distortion, and traction bronchiectasis with or without honeycombing. The distribution of abnormalities can be patchy, peribronchovascular, subpleural, or in any zonal distribution. Honeycombing can be present and may have a subpleural distribution but is less frequently associated with a basal predominance as compared to idiopathic pulmonary fibrosis (IPF) [3•, 13]. Figure 3A, B depicts common features of fibrotic HP. Areas of mosaic attenuation that is suggestive of small airway disease can be helpful in distinguishing it from that of IPF [18, 19]. Meanwhile, HRCT findings of fibrosis in fibrotic HP are considered nonspecific, and differentials of other granulomatous fibrosing ILD such as sarcoidosis and connective tissue disease-ILD should be considered.

A, B Computed-tomogram images of a patient with fibrotic hypersensitivity pneumonitis. A depicts architectural distortions seen in fibrosis. There is evidence of honeycombing, traction bronchiectasis, septal thickening, and course reticulations. Fibrotic changes are predominantly located in upper lobes as seen in (B)
Histopathological Features of HP
The histopathological alterations of HP can be heterogeneous. Nonfibrotic HP histopathology features bronchiolocentric lymphohistocytic interstitial pneumonia, chronic bronchiolitis, and small poorly formed non-necrotizing granulomas [13]. The pulmonary infiltrate of interstitial pneumonia is typically polymorphic and includes predominantly small lymphocytes with smaller numbers of plasma cells and occasionally eosinophils [7•]. Cellular non-specific interstitial pneumonia (NSIP) pattern can also be present [20]. Histopathological features that suggest an alternative diagnosis include extensive lymphoid hyperplasia, more than focal peribronchiolar lymphoid aggregates with germinal centers, extensive well formed sarcoidal granulomas, necrotizing granuloma, and plasma cells predominant infiltrate [7•].
Fibrotic HP histopathology is associated with less inflammatory changes but exhibit more fibrotic changes with the presence of chronic interstitial pneumonia and bronchiolitis. These include airway fibrosis and poorly formed non-necrotizing granulomas. Subpleural and centriacinar fibrosis are typically seen, and there may be an absence of or sparsity of airway-centered granulomas [13]. Although the histopathological pattern might overlap with those of usual interstitial pneumonia (UIP), the presence of abnormality within and/or around the small airways is highly suggestive of HP [13]. Lung tissue for histopathological analysis may be obtained in several ways including fiberoptic transbronchial biopsy, transbronchial lung cryobiopsy, and surgical lung biopsy [7•]. The need for and the preferred method of obtaining tissue samples is often determined by individual patients’ profile, physicians’ clinical judgement, and the shared decision-making process between patient and physician. Balancing the risk vs. benefits of invasive procedures, and awareness of local expertise and availability of facilities, is important. If obtaining histopathological samples of the lung is important in determining treatment, and thus improving patients’ outcomes, then invasive procedures should be pursued.
Serum and Bronchoscopic Evaluation
Bronchoalveolar lavage (BAL) fluid cellular analysis often demonstrates an inflammatory pattern, predominantly lymphocytosis (> 30% in nonsmokers/former smokers and > 20% in current smokers) [21]. BAL lymphocytosis is strongly supportive of HP diagnosis, although not specific [22]. Microbiological investigations must be pursued prior to further workup. Other diagnostic modalities to consider include serum immunoglobulin G (sIgG). sIgG testing may assist with the identification of potentially relevant exposures; however, its superiority over a comprehensive history and/or the use of exposure questionnaires is debatable [23]. It is also an unreliable test when attempting to distinguish HP from other types of ILD [23].
Pulmonary Function Testing (PFT)
The PFT profile of patients with HP share similarities with other subtypes of ILD. Patients may exhibit a restrictive PFT pattern with a low diffusion capacity of the lung for carbon monoxide (DLCO). In certain types of CHP, including farmer’s lung, an obstructive defect secondary to emphysema may observed [24]. Specific inhalation challenges (SIC) have been explored as a confirmatory test. The sensitivity and specificity of the test were 72.7% and 84%, respectively [25]. Thus, positive SIC testing virtually confirms the diagnosis of HP, while a negative testing does not rule it out, especially when the antigenic sources are not birds or fungi [25].
The Role of the ILD Multidisciplinary Meeting (MDM)
The ILD multidisciplinary meeting (MDM) is broadly accepted as the gold standard for ILD diagnosis worldwide [26]. Team members include pulmonologists, radiologists, pathologists, and/or rheumatologists. Due to the heterogeneity of ILD, accurately diagnosing ILD may be challenging [27]. The role of the MDD is to collaboratively discuss and integrate available clinical data to generate a consensus diagnosis for the patient [27]. MDM has consistently been shown to change ILD diagnosis in approximately 50% of patients presented and consequently is more reflective of patient outcomes [28, 29].
It is noteworthy that the ILD MDD is not universally available, especially outside large academic centers. In practice, utilization of an algorithmic approach (Fig. 4) for the diagnosis, treatment, and management of patients with HP is recommended to avoid delays which may adversely affect patient outcomes [30]. For instance, should clinical, radiological, and exposure history align with the diagnosis of HP, further testing is unnecessary. However, should there be a discordance, additional testing, like BAL fluid cellular analysis, and histopathological testing should be considered.

Algorithm for the diagnosis of fibrotic and nonfibrotic hypersensitivity pneumonitis adapted from Pérez et al. [30]. This conceptual model aids the practicing pulmonologist in the diagnosis and management of patients with hypersensitivity pneumonitis. BAL, bronchoalveolar lavage; HRCT, high-resolution computed tomogram; HP, hypersensitivity pneumonitis
Treatment
The early diagnosis of HP, and the institution of strict measures toward antigen elimination are key components of the management of nonfibrotic HP to halt the inflammatory cascade of the host immune response. Lifestyle modifications to mitigate antigen exposure is warranted to aid in minimizing symptoms and improve quality of life (QoL) [31]. Depending on the causative antigen, some studies suggest that Bird Fancier’s HP may be associated with a worse prognosis than Farmer’s Lung [32]. Prolonged exposure to antigen, older age, digital clubbing, a histopathologic pattern of fibrotic NSIP, and UIP pattern on imaging are associated with worse outcomes [33].
Corticosteroids (CS) have been used to treat HP; however, it may not necessarily affect long-term outcomes of the disease [34]. In a cohort study, a recommended dose of oral prednisone of 40 to 60 mg (0.5 mg/kg/d) was administered for 2 weeks in acute versus 4 to 6 weeks in subacute to chronic forms of HP, followed by a gradual taper to a daily maintenance dose of 10 mg prior to discontinuation based on clinical response [35]. Four weeks of CS was found be as efficacious as an extended course of CS [35]. A large single-center cohort study demonstrated that nonfibrotic HP patients exhibited an overall increase in forced vital capacity (FVC) and DLCO with CS use, but no therapeutic effect was noted in patients with fibrotic HP [36•]. Additionally, patients with fibrotic HP had a dismal prognosis (medial survival of 9.2 years) while nonfibrotic HP patients demonstrated an excellent survival [36•]. Notably, long-term outcomes of CS treatment have not been validated by randomized control studies [2].
Immunosuppressive and/or immunomodulatory agents have been utilized as an alternative to CS. Unfortunately, evidence for their efficacy is scant. Mycophenolate mofetil (MMF) and azathioprine (AZA) use has been associated with an increase in DLCO and a decrease in treatment-associated adverse effects compared with CS therapy in HP [37]. However, no consistent improvement in the incidence of death, lung transplantation, and respiratory hospitalization was demonstrated. In a single-center retrospective study, the addition of leflunomide to prednisone, AZA, or MMF showed an increase in FVC after 12 months of treatment in patients with nonfibrotic HP [38]. Rituximab, an anti-CD20 monoclonal antibody, has been shown to improve 6-min walk distance and stabilize HRCT progression in patients with CHP [39]. In a more recent retrospective study, rituximab use in patients with CHP was also associated with stabilization of FVC and improvement of DLCO [40].
Macrolides have been used for their immunomodulatory and CS-sparing effect in cases of organizing pneumonia and IPF [41]. However, high-quality research is needed to evaluate its role in HP [42]. Supportive therapy including oxygen supplementation for persistent hypoxia below 90%, use of bronchodilators, and opiates may be considered for refractory dyspnea.
Treatment response failure may lead to progression of fibrosis with the eventual consideration for lung transplantation. Compared to patients with IPF, patients with HP who underwent lung transplant have reduced risk for death in 1, 3, and 5 years [43]. However, there remains significant risk for continued allergic inflammatory response against the transplanted lung [43]. The recurrence of HP has been reported after lung transplantation, usually presenting in the form of bronchiolitis obliterans syndrome [43]. This signifies the importance of exposure avoidance as well as the challenges in preventing recurrence of HP in cases where the inciting agent cannot be identified [43].
Recently, antifibrotics have been studied in patients who experience disease progression despite antigen avoidance and immunosuppressive therapy. Nintedanib is an intracellular inhibitor of tyrosine kinase that has shown promise in reducing rate of decline of FVC in patients with IPF and systemic sclerosis-associated ILD [44]. The INBUILD trial was a multicenter, randomized double blinded, placebo-controlled trial. Twenty-six percent of participants in this study had CHP. It demonstrated that in patients with progressive fibrosing ILD (> 10% of the lung volume affected on HRCT, FVC of > 45%, DLCO of 30 to 80%), the annual rate of decline in FVC in patients who received at least one dose of Nintedanib was significantly lower than those who received placebo [45•]. Despite this, there was no significant changes in QoL measures [45•]. Nintedanib was associated with a higher frequency of GI adverse events such as diarrhea, nausea, and vomiting [45•].
Pirfenidone is an antifibrotic agent frequently used for the treatment of IPF [46]. It is known to reduce the decline in vital capacity (VC) and improve progression-free survival [47]. Pirfenidone demonstrated decreased disease progression in the ASCEND study, as reflected by lung function, exercise tolerance, and progression-free survival [48]. It has also been considered effective therapy for other fibrotic lung diseases such as amyopathic dermatomyositis and scleroderma [49, 50]. In the RELIEF study, patients with fibrotic HP demonstrated a slower disease progression as measured by loss of FVC upon addition of Pirfenidone [51]. In 2018, 23 patients with avian-related CHP were enrolled in a study to monitor change in VC with administration of pirfenidone [52]. Pirfenidone reduced the decline of VC in patients with chronic fibrotic HP without significant adverse events, as observed in patients with IPF [47, 52]. Table 3 illustrates ongoing clinical trials involving patients with HP.
Conclusion
HP is an immune-mediated disease caused by exposure to a large variety of organic and inorganic materials in genetically susceptible patients. The disease has a heterogenous clinical presentation with a variable radiological and histopathological pattern on chest imaging and lung biopsy. The severity, persistence, and duration of antigen exposure influence the severity and progression of HP. It is imperative to obtain an extensive and exhaustive history to identify the inciting antigen. Utilizing published HP-specific questionnaires may augment the search to identify trigger agents. The combination of clinical, radiographical, and histopathological data will assist in determining the diagnosis in a timely manner [53]. More randomized control trials are needed to better understand and characterize HP. This would aid in standardizing management, identifying prognostic factors, and determining targets for future therapeutic interventions to improve survival and QoL.
Abbreviations
- ATS/JRS/ALAT:
-
American Thoracic Society, Japanese Respiratory Society, and Asociación Latinoamericana del Tórax
- AZA:
-
Azathioprine
- BAL:
-
Bronchoalveolar lavage
- CHP:
-
Chronic hypersensitivity pneumonitis
- CS:
-
Corticosteroids
- DLCO:
-
Diffusion capacity for carbon monoxide
- FVC:
-
Forced vital capacity
- GGO:
-
Ground glass opacity
- HRCT:
-
High-resolution computed tomogram
- HP:
-
Hypersensitivity pneumonitis
- ILD:
-
Interstitial lung disease
- IPF:
-
Idiopathic pulmonary fibrosis
- MMF:
-
Mycophenolate mofetil
- MDM:
-
Multidisciplinary meeting
- NSIP:
-
Non-specific interstitial pneumonia
- PFT:
-
Pulmonary function testing
- QoL:
-
Quality of life
- sIgG:
-
Serum immunoglobulin G
- SIC:
-
Specific inhalation challenges
- UIP:
-
Usual interstitial pneumonia
- US:
-
United States
- VC:
-
Vital capacity
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Thomeer MJ, Costabe U, Rizzato G, Poletti V, Demedts M. Comparison of registries of interstitial lung diseases in three European countries. Eur Respir J Suppl. 2001;32:114s-s118.
Selman M, Pardo A, King TE. Hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2012;186(4):314–24. Available from: https://www.atsjournals.org/doi/10.1164/rccm.201203-0513CI.
• Fernández Pérez ER, Kong AM, Raimundo K, Koelsch TL, et al. Epidemiology of hypersensitivity pneumonitis among an insured population in the United States: a claims-based cohort analysis. Ann Am Thorac Soc [Internet]. 2018;15(4):460–9. Available from: www.atsjournals.org. This review highlights the changing trends of HP in the U.S.
Riario Sforza GG, Marinou A. Hypersensitivity pneumonitis: a complex lung disease. Clinical and Molecular Allergy. 2017;15(1).
Lacasse Y, Cormier Y. Hypersensitivity pneumonitis. Orphanet J Rare Dis. 2006;1(1):25.
Bang KM, Weissman DN, Pinheiro GA, Antao VCS, Wood JM, Syamlal G. Twenty-three years of hypersensitivity pneumonitis mortality surveillance in the United States. Am J Ind Med. 2006;49(12):997–1004.
• Raghu G, Remy-Jardin M, Ryerson CJ, Myers JL, Kreuter M, Vasakova M, et al. American Thoracic Society Documents. Diagnosis of hypersensitivity pneumonitis in adults. An Official ATS/JRS/ALAT Clinical Practice Guideline. 2020; Available from: http://www.atsjournals.org/doi/suppl/10.1164/rccm.202005-2032ST. This review discusses updated recommendation for the diagnosis of HP.
Reich JM. Chirping rales in bird-fancier’s lung. Chest. 1993;104(1):326–7. Available from: https://www.sciencedirect.com/science/article/pii/S0012369216475458.
Zergham AS, Heller D. Farmers lung: StatPearls. Florida: Treasure Island. 2022.
Ryerson CJ, Vittinghoff E, Ley B, Lee JS, Mooney JJ, Jones KD, et al. Predicting survival across chronic interstitial lung disease. Chest. 2014;145(4):723–8.
• Barnes H, Morisset J, Molyneaux P, et al. A systematically derived exposure assessment instrument for chronic hypersensitivity pneumonitis. Chest. 2020;158(3):1292. This review features the most important exposures to investigate during an evaluation of an HP patient.
Lacasse Y, Selman M, Costabel U, et al. Clinical diagnosis of hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2003;168(8):952–8. Available from: www.atsjournals.org.
Vasakova M, Morell F, Walsh S, Leslie K, Raghu G. Hypersensitivity pneumonitis: perspectives in diagnosis and management. 2017. Available from: www.atsjournals.org.
Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. American Thoracic Society Documents. Diagnosis of idiopathic pulmonary fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2018;198(5):44–68. Available from: http://www.atsjournals.org/doi/suppl/.
Magee AL, Montner SM, Husain A, Adegunsoye A, Vij R, Chung JH. Imaging of hypersensitivity pneumonitis. Radiol Clin North Am. 2016;54(6):1033–46. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0033838916300665.
Barnett J, Molyneaux PL, Rawal B, Abdullah R, Hare SS, Vancheeswaran R, et al. Variable utility of mosaic attenuation to distinguish fibrotic hypersensitivity pneumonitis from idiopathic pulmonary fibrosis. Eur Respir J. 2019;54(1):1900531. Available from: http://erj.ersjournals.com/lookup/doi/10.1183/13993003.00531-2019.
Salisbury ML, Myers JL, Belloli EA, Kazerooni EA, Martinez FJ, Flaherty KR. Diagnosis and treatment of fibrotic hypersensitivity pneumonia where we stand and where we need to go. 2017. Available from: www.atsjournals.org.
Silva CIS, Müller NL, Lynch DA, Curran-Everett D, Brown KK, Lee KS, et al. Chronic hypersensitivity pneumonitis: differentiation from idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia by using thin-section CT. Radiology [Internet]. 2008 Jan 1;246(1):288–97. Available from: https://doi.org/10.1148/radiol.2453061881.
Buschman DL, Gamsu G, Waldron JA Jr, et al. Chronic hypersensitivity pneumonitis: use of CT in diagnosis. AJR Am J Roentgenol. 1992;159(5):957–60.
Tafti SF, Cheraghvandi A, Mokri B, Talischi F. Cases non-specific interstitial pneumonia and hypersensitivity pneumonia: a new pathologic diagnosis or overlap syndrome. Respir Med Case Rep. 2012;5:45–8. Available from: https://pubmed.ncbi.nlm.nih.gov/26029588.
Meyer KC, Raghu G. Bronchoalveolar lavage for the evaluation of interstitial lung disease: is it clinically useful? Eur Respir J. 2011;38(4):761. Available from: http://erj.ersjournals.com/content/38/4/761.abstract.
Adderley N, Humphreys CJ, Barnes H, Ley B, Premji ZA, Johannson KA. Bronchoalveolar lavage fluid lymphocytosis in chronic hypersensitivity pneumonitis: a systematic review and meta-analysis. European Respir J. 2020;56(2):2000206. Available from: http://erj.ersjournals.com/content/56/2/2000206.abstract.
Jenkins AR, Chua A, Chami H, Diaz-Mendoza J, Duggal A, Knight S, et al. Systematic Reviews. Questionnaires or serum immunoglobulin G testing in the diagnosis of hypersensitivity pneumonitis among patients with interstitial lung disease. Ann Am Thorac Soc. 2021;18(1):130–47. Available from: www.atsjournals.org.
Soumagne T, Chardon ML, Dournes G, Laurent L, Degano B, Ois Laurent F, et al. Emphysema in active farmer’s lung disease. 2017. Available from: https://doi.org/10.1371/journal.pone.0178263.
Muñoz X, Sánchez-Ortiz M, Torres F, Villar A, Morell F, Cruz MJ. Diagnostic yield of specific inhalation challenge in hypersensitivity pneumonitis. Eur Respir J. 2014;44:1658–65. Available from: http://ow.ly/zxszi.
Lee CT. Multidisciplinary meetings in interstitial lung disease: polishing the gold standard. Ann Am Thorac Soc. 2022;19(1):7–9. Available from: https://www.atsjournals.org/doi/10.1513/AnnalsATS.202108-979ED.
Ageely G, Souza C, de Boer K, Zahra S, Gomes M, Voduc N. The impact of multidisciplinary discussion (MDD) in the diagnosis and management of fibrotic interstitial lung diseases. 2020. Available from: https://doi.org/10.1155/2020/9026171.
de Sadeleer LJ, Meert C, Yserbyt J, Slabbynck H, Verschakelen JA, Verbeken EK, et al. Diagnostic ability of a dynamic multidisciplinary discussion in interstitial lung diseases. Chest. 2018;153(6):1416–23. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0012369218304732.
Jo HE, Glaspole IN, Levin KC, Mccormack SR, Mahar AM, Cooper WA, et al. Clinical impact of the interstitial lung disease multidisciplinary service. 2016.
Pérez ERF, Travis WD, Lynch DA, Brown KK, Johannson KA, Selman M, et al. Diagnosis and evaluation of hypersensitivity pneumonitis. Chest. 2021;160(2):e97-156.
Lacasse Y, Fraser RS, Fournier M, Cormier Y. Diagnostic accuracy of transbronchial biopsy in acute farmer’s lung disease. Chest. 1997;112(6):1459–65.
Vourlekis JS, Schwarz MI, Cherniack RM, Curran-Everett D, Cool CD, Tuder RM, et al. The effect of pulmonary fibrosis on survival in patients with hypersensitivity pneumonitis. Am J Med. 2004;116(10):662–8.
Lacasse Y, Girard M, Cormier Y. Recent advances in hypersensitivity pneumonitis. Chest. 2012;142(1):208–17.
Kokkarinen JI, Tukiainen HO, Terho EO. Effect of corticosteroid treatment on the recovery of pulmonary function in farmer’s lung. Am Rev Respir Dis. 1992;145(1):3–5.
Mönkäre S, Haahtela T. Farmer’s lung-a 5-year follow-up of eighty-six patients. Clin Allergy. 1987;17(2):143–51.
• de Sadeleer L, Hermans F, de Dycker E, Yserbyt J, Verschakelen J, Verbeken E, et al. Effects of corticosteroid treatment and antigen avoidance in a large hypersensitivity pneumonitis cohort: a single-centre cohort study. J Clin Med. 2018;8(1):14. Available from: http://www.mdpi.com/2077-0383/8/1/14. This review demonstrates the effects of corticosteroid therapy and antigen elimination in patients with HP.
Morisset J, Johannson KA, Vittinghoff E, Aravena C, Elicker BM, Jones KD, et al. Use of mycophenolate mofetil or azathioprine for the management of chronic hypersensitivity pneumonitis. Chest. 2017;151(3):619–25.
Noh S, Yadav R, Li M, Wang X, Sahoo D, Culver DA, et al. Use of leflunomide in patients with chronic hypersensitivity pneumonitis. BMC Pulm Med. 2020;20(1):199. Available from: https://bmcpulmmed.biomedcentral.com/articles/10.1186/s12890-020-01227-2.
Morell F, Ojanguren I, Villar A, Ramon MA, Muñoz X, Cruz MJ. Addition of rituximab to oral corticosteroids in the treatment of chronic hypersensitivity pneumonitis. Arch Bronconeumol. 2020;56(4):254–6.
Ferreira M, Borie R, Crestani B, Rigaud P, Wemeau L, Israel-Biet D, et al. Efficacy and safety of rituximab in patients with chronic hypersensitivity pneumonitis (cHP): A retrospective, multicentric, observational study. Respir Med. 2020;172: 106146.
Faverio P, Bini F, Vaghi A, Pesci A. Long-term macrolides in diffuse interstitial lung diseases. Eur Respir Rev. 2017;26(146):170082.
Jaffe A, Bush A. State of the Art. Anti-inflammatory effects of macrolides in lung disease. Pediatr Pulmonol. 2001;31.
Kern RM, Singer JP, Koth L, Mooney J, Golden J, Hays S, et al. Lung transplantation for hypersensitivity pneumonitis. Chest. 2015;147(6):1558–65.
Lamb YN. Nintedanib: a review in fibrotic interstitial lung diseases. Drugs. 2021;81(5):575–86. Available from: https://link.springer.com/10.1007/s40265-021-01487-0.
• Wells AU, Flaherty KR, Brown KK, Inoue Y, Devaraj A, Richeldi L, et al. Nintedanib in patients with progressive fibrosing interstitial lung diseases–subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: a randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Respir Med. 2020;8(5):453–60. The INBUILD trial demonstrates the efficacy of Nintedanib in halting ILD progression by lung function.
Takeda Y, Tsujino K, Kijima T, Kumanogoh A. Patient Preference and Adherence Dovepress Efficacy and safety of pirfenidone for idiopathic pulmonary fibrosis. Patient Prefer Adherence. 2014;8:361–70. Available from: https://doi.org/10.2147/PPA.S37233.
Taniguchi H, Ebina M, Kondoh Y, Ogura T, Azuma A, Suga M, et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2010;35(4):821–9.
King TE, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;22:2083–92.
Li T, Guo L, Chen Z, Gu L, Sun F, Tan X, et al. Pirfenidone in patients with rapidly progressive interstitial lung disease associated with clinically amyopathic dermatomyositis OPEN. 2016. Available from: www.nature.com/scientificreports.
Miura Y, Saito T, Fujita K, Tsunoda Y, Tanaka T, Takoi H, et al. Clinical experience with pirfenidone in five patients with scleroderma-related interstitial lung disease. Sarcoidosis Vasc Diffuse Lung Dis. 2014;31(3):235–8.
Behr J, Prasse A, Kreuter M, Johow J, Rabe KF, Bonella F, et al. Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): a double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir Med. 2021;9(5):476–86.
Shibata S, Furusawa H, Inase N. Pirfenidone in chronic hypersensitivity pneumonitis: a real-life experience. Sarcoidosis Vasc Diffuse Lung Dis. 2018;35(2):139–42.
Vasakova M, Selman M, Morell F, Sterclova M, Molina-Molina M, Raghu G. Hypersensitivity pneumonitis: current concepts of pathogenesis and potential targets for treatment. 2019. Available from: www.atsjournals.org.
Acknowledgements
We thank Carmen Fullmer, MS, for her assistance and support in the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors have no relevant financial or non-financial interests to disclose.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Ethical Statement
Ethical approval was not required because this study retrieved and synthesized data from already published studies.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Environmental and Occupational Health
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Anwar, J., Kong, W.T. & Balakrishnan, B. Updates in Hypersensitivity Pneumonitis: A Narrative Review. Curr Pulmonol Rep 11, 106–115 (2022). https://doi.org/10.1007/s13665-022-00294-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13665-022-00294-6