
Abstract
Purpose of Review
Spontaneous pneumothoraces can be the presenting phenotype in a variety of different syndromic genetic conditions. Respiratory physicians therefore have the opportunity to diagnose and manage these patients early to prevent serious complications associated with these syndromes.
Recent Findings
The genetic syndromes that present with pneumothoraces can be split broadly between those resulting from defective extracellular matrix formation and those caused by defective tumour-suppressor pathways. When connective tissues are weakened, lifelong surveillance for arterial dilatation can be life-saving as surgical intervention is effective. Long-term aggressive treatment of blood pressure can also commence, although some controversy surrounds which drugs are most effective and precisely how these drugs modify disease progression. Rational treatments of syndromes in which tumour suppressor function is lost are being developed and, in some instances, can already be offered.
Summary
Careful clinical assessment of spontaneous pneumothorax may identify an underlying causal condition and facilitate life-saving intervention. Respiratory physicians must therefore be aware of these diseases and their diagnostic criteria.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pneumothorax, the presence of air within the pleural space, has been a medical diagnosis since Jean Itard coined the term early in the nineteenth century. When pneumothorax occurs without obvious lung pathology or trauma, it is termed primary spontaneous pneumothorax and happens to approximately 1 in 10,000 of the population [1]. A typical patient will be tall and slender, the so-called asthenic phenotype. Males, who outnumber females with pneumothorax 3:1, tend to present in their late teens to late twenties, while the ages of affected women are more evenly spread from late teens onward [2].
Despite the relative rarity of primary spontaneous pneumothorax, 10% of patients report having an affected family member indicating a strong genetic component to this condition [3]. Such families are said to have ‘familial pneumothorax’, and unlike sporadic cases, there appears to be a more equal distribution between males and females [4]. We previously suggested that familial pneumothoraces be categorised into those syndromes resulting from a weakened extracellular matrix and those that affect tumour suppressor genes, the former typically causing emphysematous changes, the latter cause pulmonary cysts [4].
Familial pneumothorax frequently represents only one part of a broader syndrome. Given the essential role of connective tissues in providing structural integrity to organs, it is unsurprising that mutations of the extracellular matrix have diverse effects, in particular, in the cardiovascular system. By contrast, mutations that impair tumour suppressor pathways permit excessive cellular proliferation leading to the development of both benign and malignant neoplasms.
Targeted preemptive management of individuals with specific genetic defects can reduce morbidity and mortality, so-called precision medicine. By helping to identify individuals with genetic defects, pneumothorax can enable precision medicine to be practiced and lead to patient benefit. It is therefore important that respiratory physicians are able to identify individuals with a syndromic cause for their pneumothoraces and ensure onward referral to appropriate specialist healthcare providers. In this review, we describe the diagnosis and management of the most frequently encountered genetic syndromes linked with the development of pneumothorax.
Connective Tissue Disorders
Marfan Syndrome
Genetics
Marfan syndrome is perhaps one of the best known causes of familial pneumothorax and has an incidence of 1:5000 to 1:10,000 [5]. It was first described by Antoine Marfan at the end of the nineteenth century when he treated a young girl with unusually long thin limbs [6]. Five decades later, a case of pneumothorax in the context of Marfan syndrome was reported, subsequently followed by many others [7]. Marfan syndrome is an autosomal dominant disorder and in 1991 three groups simultaneously reported causative mutations in the FBN1 gene, which encodes the glycoprotein fibrillin-1 [8,9,10]. In addition to a role in extracellular matrix structure, fibrillin-1 helps to regulate TGFβ signalling and both of these functions account for the complex thoracic and extrathoracic manifestations of the disorder.
Clinical features
FBN1 is a large and highly polymorphic gene, and so sequencing is a complex and often confusing process [11]. For this reason, a diagnosis of Marfan syndrome depends on identification of its clinical features as laid out by the revised Ghent nosology (Table 1). The abnormal bone growth first noted by Marfan over a century ago leads to tall stature, chest wall abnormalities (pectus carinatum or excavatum), scoliosis and arachnodactyly. The excessive length of long bones relative to others, e.g. vertebrae, can make the arm span disproportionally long compared with the height. In the respiratory clinic, it is straightforward to measure arm span relative to height as a screening tool. An arm span that is more than 1.05-fold that of the height raises the possibility of aberrant bone growth (Table 1, systemic features). Arachnodactyly is similarly easy to check using two clinical signs: the Walker-Murdoch sign is positive if the thumb and small finger of one hand can completely encircle the opposite wrist; Steinberg’s sign is positive when the patient can adduct the thumb across the palm sufficiently far that the distal phalanx of the thumb protrudes when the fingers are closed over it. Ocular manifestations include lens subluxation and retinal detachment, which can be appreciated by slit-lamp examination. However, the most clinically significant manifestations of Marfan syndrome are its cardiovascular features including cardiomyopathy, mitral or aortic valve degeneration and aortic root dilatation [11].
Pneumothoraces have been estimated to occur in up to 10% of patients with Marfan syndrome, although their aetiology is controversial. Although 5–9.6% of Marfan patients have evidence of blebs on CT [5], this is not significantly different to the general population [12]. It has been theorised that skeletal deformities may alter the lung parenchymal tension, predisposing individuals to pneumothorax [13].
Management
Prompt diagnosis and management are critical to decrease the risk of life-threatening complications associated with aortic aneurysms. Annual echocardiography allows monitoring of the aortic root, although more frequent imaging might be necessary when the aortic root is widening [14]. Therapy with beta-blockade was initially shown to slow the rate of aortic dilatation and reduce complications associated with aortic aneurysms [15]; however, metaanalyses have concluded both in favour and against their use in Marfan syndrome [16, 17]. A recent Cochrane review determined that while there is evidence that beta-blockers reduce the rate of aortic diameter change, the quality of evidence is relatively poor [18]. On balance, until definitive trials can be performed, it is recommended that individuals with Marfan syndrome continue to be treated with beta-blockade.
Angiotensin receptor blockade has been proposed for the treatment of patients with Marfan syndrome since; in addition to beneficial effects on aortic haemodynamics, these drugs may also inhibit the overactive TGFβ pathway in Marfan syndrome. A small-scale study showed benefit of losartan in slowing aortic-root enlargement in young individuals resistant to beta-blockers [19]; however, subsequent studies failed to show a difference between losartan and beta-blockers therapy in progression of aortic disease [20, 21].
Surgery is indicated if pharmacological treatment fails and the aortic root dilates to 50 mm (or 45 mm if there is a family history of dissection or severe aortic regurgitation) [22]. Total root replacement or valve sparing root replacement can be performed, the latter having the advantage of sparing the individual from lifelong anticoagulation. Recent meta-analyses and systematic reviews showed comparable outcomes between the two procedures [23, 24], although surgeon experience and case-specific features mean valve–sparing surgery will not be appropriate for all patients [23, 25].
Annual ophthalmologic examination has been recommended for individuals with Marfan syndrome [26]. If clinically significant lens subluxation is noted, corrective lenses or surgical extraction followed by artificial lens replacement can be offered [26].
Vascular Ehlers-Danlos Syndrome
Genetics
Although “Ehlers-Danlos syndrome” has been recognised since the 1930s, it is in fact a group of weakly related conditions that share a propensity toward hypermobile joints. At present, only vascular Ehlers-Danlos syndrome (vEDS or EDS type 4) is known to be a risk factor for the development of pneumothorax. vEDS is rare (1 case per 100,000 population) but is potentially a more serious connective tissue disorder than Marfan syndrome. It is caused by autosomal dominant mutations in the COL3A1 gene, which encodes collagen type III [27]. This protein is found in many connective tissues including the pulmonary vasculature and lung parenchyma [28]. In contrast to Marfan syndrome, molecular testing is often helpful in establishing a diagnosis of vEDS, but if genetic testing proves negative yet clinical suspicion remains high, biochemical abnormalities of collagen type III can be sought using cultured fibroblasts [29].
Clinical features
Diagnostic criteria developed in 2017 advise that vEDS should be suspected in patients with unexplained arterial aneurysms, dissection or rupture; intestinal rupture; uterine rupture during pregnancy; or a family history of vEDS [30]. However, pneumothorax is also a relatively frequent presenting feature of vEDS and offers a crucial opportunity to diagnose the condition before life-threatening complications occur [31, 32]. Other pulmonary features of vEDS reflect vascular fragility with spontaneous haemothorax and haemoptysis. Radiology may reveal pulmonary cavitation, which often leads clinicians to suspect pulmonary vasculitis. Consequently, a majority of vEDS diagnoses are made only after lung biopsy shows no vasculitis but evidence of recurrent haemorrhage, such as haemosiderin-laded macrophages and fibrous nodules with osseous metaplasia (Fig. 1a) [34].

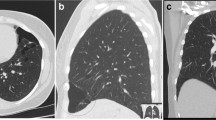
Syndromic causes of pneumothorax. a Vascular Ehlers Danlos syndrome. Lung parenchyma stained with haematoxylin and eosin. Magnification × 16 objective. Note hemosiderin-laden intraalveolar macrophages, suggesting prior pulmonary haemorrhage, and foci of intraalveolar organisation. b Loeys-Dietz syndrome. Bifid uvula. Reproduced with permission from [33•]. c Lymphangioleiomyomatosis. Axial CT image of a patient with lymphangioleiomyomatosis. Note the many thin-walled cysts of varying size. Bilateral recurrent pneumothoraces following previously bilateral pleurodesis. d Birt-Hogg-Dubé syndrome. Axial CT image of the lung bases of a patient with Birt-Hogg-Dubé syndrome. Note the irregular, multiseptated cysts that of frequently subpleural. Small contralateral anterior pneumothorax
A diagnosis of vEDS should be suspected clinically in patients who are hypermobile or have relatives with hypermobility. For this reason, we recommend that Beighton scores are recorded for all patients with spontaneous pneumothorax (Table 2). Other more rare skeletal features include clubfoot, congenital hip dislocation and facial dysmorphism with narrow nose, deep-set eyes, micrognathia and thin lips. A simple dermatological examination can reveal thin skin that bruises easily. A history (or family history) of spontaneous bowel or uterine rupture may also be elicited.
Management
Owing in large part to its rarity, the management for vEDS is yet to be codified in definitive guidelines. Centres with experience of treating vEDS report that management is optimised through development of multidisciplinary teams comprising physicians, vascular surgeons and general surgeons [29]. In our own hospital, this team also includes respiratory, cardiology and genetics consultants, neurosurgeons and radiologists.
Surveillance for cardiovascular complications by echocardiography every 3 years has been recommended [36]. A randomised controlled trial and a subsequent observational study using celiprolol, a combined beta-1 antagonist and beta-2 partial agonist, showed a reduction in adverse arterial events, so this drug is often used [37, 38]. In the absence of clear guidelines, decisions on elective vascular procedures have to be made on a case-by-case basis. Owing to fragility of the blood vessels, open surgery can be challenging in vEDS, and so elective procedures are performed less frequently than in other diseases affecting connective tissues. The rise of endovascular approaches, however, may alter this balance in the future [39].
For patients that have suffered from gastrointestinal complications, a recent systematic review suggested that end ileostomies or ileorectal anastomoses are preferable over colostomies due to the risk of reperforation and anastomotic leaks with the latter [40].
Loeys-Dietz Syndrome
Genetics
Loeys-Dietz syndrome is a relative newcomer to the list of pneumothorax-associated disorders. It was first described in 2005 as a syndrome comprising craniofacial midline fusion defects, vascular tortuosity and neurodevelopmental defects [41••] but is now recognised to occur in more subtle forms. It is an autosomal dominant disorder caused by mutations in genes of the TGFβ signalling pathway: SMAD3, TGFB2, TGFB3, TGFBR1 and TGFBR2 [42].
Clinical features
We reported pneumothorax as the presenting feature in an individual with normal neurodevelopment ultimately confirmed to have Loeys-Dietz syndrome (LDS) with a TGFBR2 mutation [33•]. It is important to make an early diagnosis to permit preventive treatment of the cardiovascular risk. Arterial dissection may arise in a broader range of vessels in patients with LDS; this should therefore be considered even if the former arises below the diaphragm [43]. Other clinical features suggestive of LDS include midline fusion defects, with severely affected individuals having hypertelorism or cleft lip, while milder cases may have only a bifid uvula. The latter can easily been seen in the respiratory clinic by inspecting the oropharynx (Fig. 1b). Other features include skeletal abnormalities (pectus excavatum or carinatum, scoliosis, joint laxity or contracture) and skin changes (easy bruising, translucency and dystrophic scars) [41••]. A lack of ocular changes distinguishes LDS from Marfan syndrome.
It has been suggested that LDS requires evidence of aneurysm or dissection and a pathogenic mutation in any of the genes outlined above [43]. This guidance expands on previous reports suggesting that only mutations in TGFBR1 and TGFBR2 be considered [44]. Current guidance stresses that craniofacial features and cutaneous features may be very mild; however, the presence of severe craniofacial deformity predicts a poor prognosis [44].
Management
As with other connective tissue disorders, cardiovascular surveillance is important in LDS. Echocardiography should be performed at least annually and more regularly in severely affected individuals. Since a wider range of vessels can be affected, we recommend periodic magnetic resonance angiography from the head to lower limbs in selected patients. Prophylactic antihypertensives are often used albeit with a smaller evidence base than exists for Marfan syndrome. Angiotensin receptor blockers have the theoretical added benefit of antagonising the TGFβ pathway offering a scientifically plausible advantage over beta-blockers; as yet, head to head trials are lacking. Unlike Marfan syndrome, dissections have frequently been reported with aneurysmal diameters of only 40 mm with prophylactic surgery advocated for aneurysms of this size [44, 45].
α1-Antitrypsin Deficiency
Alpha1-antitrypsin deficiency is a recessive condition caused by mutations of the SERPINA1 gene [46]. This encodes α1-antitrypsin, which is an abundant plasma protein that inhibits neutrophil elastase. Mutations in SERPINA1 reduce the level of circulating α1-antitrypsin allowing uninhibited neutrophil elastase to degrade elastin fibres in the lungs. This can cause pulmonary emphysema even in nonsmokers and in some cases leads to pneumothorax [47]. The retention of polymerised α1-antitrypsin in the liver may cause cirrhosis or hepatocellular carcinoma; hence, surveillance imaging of the liver is recommended. In some countries, treatment with α1-antitrypsin augmentation therapy is available, but in all countries, lifestyle changes (smoking cessation, avoidance of excessive alcohol) can be suggested. If necessary, lung and/or liver transplantation can be considered.
Homocystinuria
Homocystinuria is a rare metabolic disorder that affects only 1 in 200,000 people and is inherited in an autosomal recessive manner. Several genes can be mutated to cause the condition, but the most common is CBS encoding cystathionine β-synthase. This is responsible for the homocysteinylation of various proteins, including fibrillin-1, which likely explains the skeletal and ocular features homocystinuria shares with Marfan syndrome [4]. Patients also carry a markedly increased risk of thrombosis, which is responsible for much of the morbidity of the condition. Neurological complications including intellectual disability and seizures, and cutaneous abnormalities such as livedo reticularis or malar flush are frequently observed [4]. There are rare reported cases in the literature of pneumothorax due to homocystinuria [48].
Cutis Laxa
Cutis laxa is characterised by hypoelastic, sagging skin particularly prominent around the neck, armpits and groin. Various mutant genes are reported including ATP6V0A2, ATP7A, EFEMP2, ELN and FBLN5; the modes of inheritance are variable, recessive forms typically being more severe [49]. The morbidity of the condition depends on the organs affected, which can include the intestines, joints, blood vessels and lungs. In the lungs, it can cause emphysema and rarely pneumothorax [50].
Syndromes Related to Tumour Suppressor Mutations
Tuberous Sclerosis and Pulmonary Lymphangioleiomyomatosis
Genetics
Tuberous sclerosis complex (TSC) is an autosomal dominant disorder, which like Marfan syndrome dates back to the nineteenth century [51]. It is caused by loss-of-function mutations in either of two tumour suppressor genes, TSC1 and TSC2, which encode the proteins hamartin and tuberin respectively [52]. These proteins form a complex that inhibits mTORC1, a kinase involved in regulating cell proliferation, differentiation and adhesion [53]. Perhaps unsurprisingly, the dysfunction of these proteins leads to benign and malignant neoplasms affecting various tissues including the skin, central nervous system, bone, kidney, heart and lung [54].
Clinical features
TSC can be diagnosed either molecularly or clinically [55]. For the molecular route, a germline pathogenic mutation in TSC1 or TSC2 is required. Clinically, patients need two major features or one major feature with two or more minor features (Table 3) [55].
The most significant pulmonary manifestation of TSC is lymphangioleiomyomatosis (LAM). When individuals with TSC are subjected to thoracic CT imaging, up to 40% have pulmonary cysts consistent with a diagnosis of LAM, which in turn is the third commonest cause of morbidity in TSC [56]. Proliferation of abnormal smooth muscle cells, so-called ‘LAM cells’, causes the formation of cysts throughout the lungs (Fig. 1c). Around 60% of these patients experience pneumothorax, which is often the presenting feature of LAM [57].
Most patients with sporadic LAM (not associated with TSC) are women of childbearing age, which reflects the sensitivity of LAM cells to female sex hormones. LAM cells in sporadic disease are commonly found to have somatic mutations in TSC2 or more rarely TSC1 [58]. In patients with sporadic LAM, diagnosis is reached more easily if additional features are present such as angiomyolipomas, chylous effusions or lymphangioleiomyomas [59]. More recently, serum levels of vascular endothelial growth factor-D (VEGF-D) above 800 pg/ml have been shown to have diagnostic and prognostic value [59]. Although the literature has suggested that patients with sporadic LAM have more severe pulmonary manifestations compared with TSC-associated LAM, this may reflect lead-time bias since TSC patients will more often be diagnosed earlier because of preemptive screening [60].
Management
Patients found to have LAM should be screened for evidence of TSC and vice versa [58]. An early diagnosis of LAM is particularly important because the pulmonary disease can progress rapidly during pregnancy, and so individuals should be provided with appropriate counselling and support [61]. We have cared for several women in whom pregnancy-associated pneumothorax was the presenting feature of LAM.
LAM can run a benign course, and the 10-year transplant-free survival is reported to be 86% [62]. However, it is important to monitor women with the condition by pulmonary function testing. These tests can be challenging for patients with severe neurological complications of TSC, and in these patients, serial measurements of VEGF-D or regular radiological examinations may be offered instead [63, 64].
In those women who suffer progressive loss of lung function, treatment with an inhibitor mTORC1, such as sirolimus, can be given [65]. Rarely, lung transplantation is required, although LAM can recur in the transplanted organ [66].
Birt-Hogg Dubé Syndrome
Genetics
Folliculin is encoded by the FLCN gene, mutations of which cause the autosomal dominant disease Birt-Hogg-Dubé syndrome (BHDS) [67]. Like tuberin and hamartin in TSC, folliculin regulates mTORC1 although the precise mechanism is still incompletely understood. The large FLCN gene contains 14 exons and approximately 150 different pathogenic FLCN mutations have been identified [67]. These span the entire coding region, although several mutational ‘hot spots’ exist. Of these, a polycytosine tract in exon 11 is the most important and accounts for nearly 50% of all FLCN mutations, either as inserted (c.1285dupC) or deleted (c.1285delC) residues [68].
Clinical features
In contrast to tuberous sclerosis, Birt-Hogg-Dubé syndrome predominantly affects only a handful of tissues: the kidney, skin and lungs. In the kidney, it can manifest as renal cancers presenting typically in the fifth decade [67]. Only around a quarter of patients develop renal cancers because further random somatic mutations are required for oncogenesis, the so-called two-hit hypothesis [69]. Most of the renal tumours in Birt-Hogg-Dubé syndrome are atypical hybrid oncocytoma-chromophobe or chromophobe types. This is in contradistinction to other familial renal cancer syndromes, such as von-Hippel-Lindau syndrome in which more typical clear cell cancers are frequently seen [70]. An important consequence of this is that the atypical tumours of Birt-Hogg-Dubé syndrome are less easily detected by renal ultrasound scanning and can be missed unless renal MRI scanning is performed.
Individuals with Birt-Hogg-Dubé syndrome can also have skin changes including fibrofolliculomas, trichodiscomas, perifollicular fibromas and acrochordons. The most common are fibrofolliculomas, which are a useful marker of the condition when detected in the clinic. Respiratory physicians should be familiar with these lesions and when in doubt seek dermatological assistance. Fibrofolliculomas are benign tumours of the follicular epithelium that present as smooth, 2–4 mm, white or flesh-coloured papules found on the face, ears and upper torso.
The lung manifestations are the most commonly observed features of Birt-Hogg-Dubé syndrome, pulmonary cysts being almost universal [67]. Irregular, sometimes septated cysts occur characteristically toward the bases or at least below the level of the carina and tend to be medially sited. This distinguishes Birt-Hogg-Dubé syndrome from LAM which has round cysts that are more evenly spread throughout the lung fields (Fig. 1d) [67]. Although the pathogenesis of these cysts is uncertain, the ‘stretch theory’ arose because folliculin plays a role in regulating cell-cell adhesion through the formation of the folliculin-plakophilin-4 complex, perhaps independent of its role in mTORC1 regulation [71]. It is hypothesised that a lack of folliculin increases cell-cell adhesion and that, over time, the stress induced by respiratory movement causes the alveolar space to rupture and form cysts [71, 72]. Interestingly, this predicts that cyst numbers or size will increased over time, but evidence for this is currently lacking. Pneumothoraces can occur because the cysts are frequently subpleural. Indeed, Birt-Hogg-Dubé syndrome is by far the most common genetic cause of familial pneumothorax, accounting for around 50% of cases in which a diagnosis is made, ahead of Marfan syndrome and vEDS. The age at which Birt-Hogg-Dubé syndrome patients present with pneumothoraces is around a decade earlier than the age they develop renal carcinomas.
Management
If a diagnosis of Birt-Hogg-Dubé syndrome is made at the time of the pneumothorax, patients can be offered renal surveillance imaging (ideally with annual MRI) and thereby have early renal cancers diagnosed. This is especially appealing because these renal tumours tend to be curable if excised before reaching 3 cm. If the tumour is 3 cm or less then a partial nephrectomy can be performed, but for larger tumours, a full nephrectomy may be indicated [73]. At present, it is recommended that patients are screened annually from the age of 20 years [73], but recently, a 14-year old with Birt-Hogg-Dubé syndrome was reported as having developed a renal cancer, and so this guidance might change [74].
Fibrofolliculomas are benign, and so treatment is only undertaken for cosmetic reasons. Topical anti-mTORC1 has thus far proved unsuccessful in treating the lesions [75] and fibrofolliculomas often recur following laser therapy [76]. Electrocoagulation followed by curettage is therefore the treatment of choice [77].
Conclusion
Pneumothorax is an important presenting feature in several genetic disorders that carry high risks of morbidity and mortality later in life. By far, the most common syndromic cause of pneumothorax is Birt-Hogg-Dubé syndrome. Careful clinical assessment of spontaneous pneumothorax can identify this and many other conditions to enable life-saving interventions to be commenced. Because many of these conditions have autosomal inheritance, the diagnosis of one proband frequently leads to the diagnosis of many affected but subclinical family members, thus widening the potential clinical benefit. Respiratory physicians must therefore be alert to these diseases and their diagnostic implications.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Noppen M. Spontaneous pneumothorax: epidemiology, pathophysiology and cause. Eur Respir Rev. 2010;19(117):217–9.
Gupta D, Hansell A, Nichols T, Duong T, Ayres JG, Strachan D. Epidemiology of pneumothorax in England. Thorax. 2000;55(8):666–71.
Abolnik IZ, Lossos IS, Zlotogora J, Brauer R. On the inheritance of primary spontaneous pneumothorax. Am J Med Genet. 1991;40(2):155–8.
Scott RM, Henske EP, Raby B, Boone PM, Rusk RA, Marciniak SJ. Familial pneumothorax: towards precision medicine. Thorax. 2018;73(3):270–6.
Karpman C, Aughenbaugh GL, Ryu JH. Pneumothorax and bullae in Marfan syndrome. Respiration. 2011;82(3):219–24.
Marfan A. Un cas de déformation congénitale des quatre membres, plus prononcée aux extrémités, caractérisée par l'allongement des os avec un certain degré d'amincissement. Bull et Mém Soc Méd Hôp de Paris. 1896:220–6.
Adams RA, Porter WB. Marfan’s syndrome; report of a case. South Med J. 1949;42(10):844–8.
Lee B, Godfrey M, Vitale E, Hori H, Mattei MG, Sarfarazi M, et al. Linkage of Marfan syndrome and a phenotypically related disorder to two different fibrillin genes. Nature. 1991;352(6333):330–4.
Maslen CL, Corson GM, Maddox BK, Glanville RW, Sakai LY. Partial sequence of a candidate gene for the Marfan syndrome. Nature. 1991;352(6333):334–7.
Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352(6333):337–9.
Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47(7):476–85.
Amjadi K, Alvarez GG, Vanderhelst E, Velkeniers B, Lam M, Noppen M. The prevalence of blebs or bullae among young healthy adults: a thoracoscopic investigation. Chest. 2007;132(4):1140–5.
Saita K, Murakawa T, Kawano H, Sano A, Nagayama K, Nakajima J. Chest wall deformity found in patients with primary spontaneous pneumothorax. Asian Cardiovasc Thorac Ann. 2013;21(5):582–7.
Cañadas V, Vilacosta I, Bruna I, Fuster V. Marfan syndrome. Part 2: treatment and management of patients. Nat Rev Cardiol. 2010;7(5):266–76.
Shores J, Berger KR, Murphy EA, Pyeritz RE. Progression of aortic dilatation and the benefit of long-term beta-adrenergic blockade in Marfan’s syndrome. N Engl J Med. 1994;330(19):1335–41.
Gao L, Mao Q, Wen D, Zhang L, Zhou X, Hui R. The effect of beta-blocker therapy on progressive aortic dilatation in children and adolescents with Marfan’s syndrome: a meta-analysis. Acta Paediatr. 2011;100(9):e101–5.
Gersony DR, McClaughlin MA, Jin Z, Gersony WM. The effect of beta-blocker therapy on clinical outcome in patients with Marfan’s syndrome: a meta-analysis. Int J Cardiol. 2007;114(3):303–8.
Koo HK, Lawrence KA, Musini VM. Beta-blockers for preventing aortic dissection in Marfan syndrome. Cochrane Database Syst Rev. 2017;11:CD011103.
Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC 3rd. Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med. 2008;358(26):2787–95.
Lacro RV, Dietz HC, Sleeper LA, Yetman AT, Bradley TJ, Colan SD, et al. Atenolol versus losartan in children and young adults with Marfan’s syndrome. N Engl J Med. 2014;371(22):2061–71.
Teixido-Tura G, Forteza A, Rodríguez-Palomares J, González Mirelis J, Gutiérrez L, Sánchez V, et al. Losartan versus atenolol for prevention of aortic dilation in patients with Marfan syndrome. J Am Coll Cardiol. 2018;72(14):1613–8.
Vahanian A, Alfieri O, Andreotti F, Antunes MJ, Barón-Esquivias G, Baumgartner H, et al. Guidelines on the management of valvular heart disease (version 2012): the Joint Task Force on the Management of Valvular Heart Disease of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS). Eur J Cardiothorac Surg. 2012;42(4):S1–44.
Flynn CD, Tian DH, Wilson-Smith A, David T, Matalanis G, Misfeld M, et al. Systematic review and meta-analysis of surgical outcomes in Marfan patients undergoing aortic root surgery by composite-valve graft or valve sparing root replacement. Ann Cardiothorac Surg. 2017;6(6):570–81.
Benedetto U, Melina G, Takkenberg JJ, Roscitano A, Angeloni E, Sinatra R. Surgical management of aortic root disease in Marfan syndrome: a systematic review and meta-analysis. Heart. 2011;97(12):955–8.
Hughes GC, Zhao Y, Rankin JS, Scarborough JE, O’Brien S, Bavaria JE, et al. Effects of institutional volumes on operative outcomes for aortic root replacement in North America. J Thorac Cardiovasc Surg. 2013;145(1):166–70.
Esfandiari H, Ansari S, Mohammad-Rabei H, Mets MB. Management strategies of ocular abnormalities in patients with Marfan syndrome: current perspective. J Ophthalmic Vis Res. 2019;14(1):71–7.
Tsipouras P, Byers PH, Schwartz RC, Chu ML, Weil D, Pepe G, et al. Ehlers-Danlos syndrome type IV: cosegregation of the phenotype to a COL3A1 allele of type III procollagen. Hum Genet. 1986;74(1):41–6.
Watanabe A, Kawabata Y, Okada O, Tanabe N, Kimura H, Hatamochi A, et al. Ehlers-Danlos syndrome type IV with few extrathoracic findings: a newly recognized point mutation in the COL3A1 gene. Eur Respir J. 2002;19(1):195–8.
Byers PH, Belmont J, Black J, De Backer J, Frank M, Jeunemaitre X, et al. Diagnosis, natural history, and management in vascular Ehlers-Danlos syndrome. Am J Med Genet C Semin Med Genet. 2017;175(1):40–7.
Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):8–26.
Shalhub S, Neptune E, Sanchez DE, Dua A, Arellano N, McDonnell NB, et al. Spontaneous pneumothorax and hemothorax frequently precede the arterial and intestinal complications of vascular Ehlers-Danlos syndrome. Am J Med Genet A. 2019;179(5):797–802.
Lim R, Marciniak SJ, Marcadier J, Rassl D, Mitchell PD. Time is of the essence: a young man with recurrent pneumothorax and cavitating lung lesions. Ann Am Thorac Soc. 2018;15(8):988–91.
• Chambers JE, Dalton LE, Subramanian DN, Gooptu B, Balan A, Park SM, et al. Spontaneous pneumothorax can be associated with TGFBR2 mutation. Eur Respir J. 2015;46(6):1832–5 This paper describes the first case fo a patient with Loeys Dietz syndrome presenting as spontaneous pneumothorax. A novel in vitro test to demonstrate the functional significance of a TGFPR2 receptor mutation is described.
Badawi RA, Brent LH, Feinstein DE. Mimics of vasculitis: vascular Ehlers-Danlos syndrome masquerading as polyarteritis nodosa. J Rheumatol. 2009;36(8):1845–7.
Beighton P, Horan F. Orthopaedic aspects of the Ehlers-Danlos syndrome. J Bone Joint Surg Br. 1969;51(3):444–53.
Eagleton MJ. Arterial complications of vascular Ehlers-Danlos syndrome. J Vasc Surg. 2016;64(6):1869–80.
Ong KT, Perdu J, De Backer J, Bozec E, Collignon P, Emmerich J, et al. Effect of celiprolol on prevention of cardiovascular events in vascular Ehlers-Danlos syndrome: a prospective randomised, open, blinded-endpoints trial. Lancet. 2010;376(9751):1476–84.
Frank M, Adham S, Seigle S, Legrand A, Mirault T, Henneton P, et al. Vascular Ehlers-Danlos syndrome: long-term observational study. J Am Coll Cardiol. 2019;73(15):1948–57.
Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al. GeneReviews. 1993.
Speake D, Dvorkin L, Vaizey CJ, Carlson GL. Management of colonic complications of type IV Ehlers-Danlos syndrome: a systematic review and evidence-based management strategy. Color Dis. 2019.
•• Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37(3):275–81 This paper is the first description of the Loeys Dietz syndrome and a clinical entity.
Bertoli-Avella AM, Gillis E, Morisaki H, Verhagen JMA, de Graaf BM, van de Beek G, et al. Mutations in a TGF-β ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J Am Coll Cardiol. 2015;65(13):1324–36.
MacCarrick G, Black JH, Bowdin S, El-Hamamsy I, Frischmeyer-Guerrerio PA, Guerrerio AL, et al. Loeys-Dietz syndrome: a primer for diagnosis and management. Genet Med. 2014;16(8):576–87.
Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006;355(8):788–98.
van de Laar IM, Oldenburg RA, Pals G, Roos-Hesselink JW, de Graaf BM, Verhagen JM, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet. 2011;43(2):121–6.
Greene CM, Marciniak SJ, Teckman J, Ferrarotti I, Brantly ML, Lomas DA, et al. α1-Antitrypsin deficiency. Nat Rev Dis Primers. 2016;2:16051.
Lin YC, Chiu WK, Chang H, Cheng YL, Chen JC. Spontaneous pneumothorax in flight as first manifestation of alpha-1 antitrypsin deficiency. Aviat Space Environ Med. 2008;79(7):704–6.
Bass HN, LaGrave D, Mardach R, Cederbaum SD, Fuster CD, Chetty M. Spontaneous pneumothorax in association with pyridoxine-responsive homocystinuria. J Inherit Metab Dis. 1997;20(6):831–2.
Berk DR, Bentley DD, Bayliss SJ, Lind A, Urban Z. Cutis laxa: a review. J Am Acad Dermatol. 2012;66(5):842.e1–17.
Nascimento GM, Nunes CS, Menegotto PF, Raskin S, Almeida N. Cutis laxa: case report. An Bras Dermatol. 2010;85(5):684–6.
Brigo F, Lattanzi S, Trinka E, Nardone R, Bragazzi NL, Ruggieri M, et al. First descriptions of tuberous sclerosis by Désiré-Magloire Bourneville (1840-1909). Neuropathology. 2018;38(6):577–82.
Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet. 2008;372(9639):657–68.
Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;169(2):361–71.
Giannikou K, Malinowska IA, Pugh TJ, Yan R, Tseng YY, Oh C, et al. Whole exome sequencing identifies TSC1/TSC2 Biallelic loss as the primary and sufficient driver event for renal angiomyolipoma development. PLoS Genet. 2016;12(8):e1006242.
Northrup H, Krueger DA, Group ITSCC. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49(4):243–54.
Franz DN, Bissler JJ, McCormack FX. Tuberous sclerosis complex: neurological, renal and pulmonary manifestations. Neuropediatrics. 2010;41(5):199–208.
Johnson SR. The ERS guidelines for LAM: trying a rationale approach to a rare disease. Respir Med. 2010;104(Suppl 1):S33–41.
Carsillo T, Astrinidis A, Henske EP. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci U S A. 2000;97(11):6085–90.
Gupta N, Finlay GA, Kotloff RM, Strange C, Wilson KC, Young LR, et al. Lymphangioleiomyomatosis diagnosis and management: high-resolution chest computed tomography, transbronchial lung biopsy, and pleural disease management. An Official American Thoracic Society/Japanese respiratory society clinical practice guideline. Am J Respir Crit Care Med. 2017;196(10):1337–48.
Gupta N, Henske EP. Pulmonary manifestations in tuberous sclerosis complex. Am J Med Genet C Semin Med Genet. 2018;178(3):326–37.
Logginidou H, Ao X, Russo I, Henske EP. Frequent estrogen and progesterone receptor immunoreactivity in renal angiomyolipomas from women with pulmonary lymphangioleiomyomatosis. Chest. 2000;117(1):25–30.
Oprescu N, McCormack FX, Byrnes S, Kinder BW. Clinical predictors of mortality and cause of death in lymphangioleiomyomatosis: a population-based registry. Lung. 2013;191(1):35–42.
Yao J, Taveira-DaSilva AM, Jones AM, Julien-Williams P, Stylianou M, Moss J. Sustained effects of sirolimus on lung function and cystic lung lesions in lymphangioleiomyomatosis. Am J Respir Crit Care Med. 2014;190(11):1273–82.
Young L, Lee HS, Inoue Y, Moss J, Singer LG, Strange C, et al. Serum VEGF-D a concentration as a biomarker of lymphangioleiomyomatosis severity and treatment response: a prospective analysis of the Multicenter International Lymphangioleiomyomatosis Efficacy of Sirolimus (MILES) trial. Lancet Respir Med. 2013;1(6):445–52.
McCormack FX, Inoue Y, Moss J, Singer LG, Strange C, Nakata K, et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med. 2011;364(17):1595–606.
Karbowniczek M, Astrinidis A, Balsara BR, Testa JR, Lium JH, Colby TV, et al. Recurrent lymphangiomyomatosis after transplantation: genetic analyses reveal a metastatic mechanism. Am J Respir Crit Care Med. 2003;167(7):976–82.
Schmidt LS, Linehan WM. Molecular genetics and clinical features of Birt-Hogg-Dubé syndrome. Nat Rev Urol. 2015;12(10):558–69.
Toro JR, Wei MH, Glenn GM, Weinreich M, Toure O, Vocke C, et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dube syndrome: a new series of 50 families and a review of published reports. J Med Genet. 2008;45(6):321–31.
Khoo SK, Giraud S, Kahnoski K, Chen J, Motorna O, Nickolov R, et al. Clinical and genetic studies of Birt-Hogg-Dubé syndrome. J Med Genet. 2002;39(12):906–12.
Gupta S, Kang HC, Ganeshan D, Morani A, Gautam R, Choyke PL, et al. The ABCs of BHD: an in-depth review of Birt-Hogg-Dubé syndrome. AJR Am J Roentgenol. 2017;209(6):1291–6.
Medvetz DA, Khabibullin D, Hariharan V, Ongusaha PP, Goncharova EA, Schlechter T, et al. Folliculin, the product of the Birt-Hogg-Dube tumor suppressor gene, interacts with the adherens junction protein p0071 to regulate cell-cell adhesion. PLoS One. 2012;7(11):e47842.
Kennedy JC, Khabibullin D, Henske EP. Mechanisms of pulmonary cyst pathogenesis in Birt-Hogg-Dube syndrome: the stretch hypothesis. Semin Cell Dev Biol. 2016;52:47–52.
Menko FH, van Steensel MA, Giraud S, Friis-Hansen L, Richard S, Ungari S, et al. Birt-Hogg-Dubé syndrome: diagnosis and management. Lancet Oncol. 2009;10(12):1199–206.
Schneider M, Dinkelborg K, Xiao X, Chan-Smutko G, Hruska K, Huang D, et al. Early onset renal cell carcinoma in an adolescent girl with germline FLCN exon 5 deletion. Familial Cancer. 2018;17(1):135–9.
Gijezen LM, Vernooij M, Martens H, Oduber CE, Henquet CJ, Starink TM, et al. Topical rapamycin as a treatment for fibrofolliculomas in Birt-Hogg-Dubé syndrome: a double-blind placebo-controlled randomized split-face trial. PLoS One. 2014;9(6):e99071.
Gambichler T, Wolter M, Altmeyer P, Hoffman K. Treatment of Birt-Hogg-Dubé syndrome with erbium:YAG laser. J Am Acad Dermatol. 2000;43(5 Pt 1):856–8.
Farrant PB, Emerson R. Letter: hyfrecation and curettage as a treatment for fibrofolliculomas in Birt-Hogg-Dube syndrome. Dermatol Surg. 2007;33(10):1287–8.
Funding
SJM is supported by the MRC (MR/R009120/1, G1002610), EPSRC (EP/R03558X/1), British Lung Foundation, Cambridge NIHR BRC, the Alpha1 Foundation (pC ID 614939 and pC ID 395467).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Kenki Matsumoto and Stefan J. Marciniak declare no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Pleural Diseases and Mesothelioma
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Matsumoto, K., Marciniak, S.J. The Importance of Genetic Factors in the Management of Spontaneous Pneumothorax. Curr Pulmonol Rep 9, 47–55 (2020). https://doi.org/10.1007/s13665-020-00248-w
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13665-020-00248-w