
Abstract
Four new sesquiterpenoids, artemyrianins A–D (1–4), and three new norlignans, artemyrianins E–G (5–7), together with five known compounds (8–12), were isolated from the aerial parts of Artemisia myriantha (Asteraceae). The new compounds were established by spectroscopic data analyses (HRMS, IR, 1D and 2D NMR), and their absolute configurations were confirmed by the single-crystal X-ray diffraction or ECD calculations. The isolates showed cytotoxicity against HepG2 cells with IC50 values ranging from 33.3 to 145.2 μM.
Graphic Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Hepatocellular carcinoma (HCC) resulting in 780000 deaths every year is the fourth cause of cancer mortality [1]. The treatment for advanced HCC has long been unsatisfied until the authorization of sorafenib in 2007. Afterwards, six drugs involving nivolumab, regorafenib, lenvatinib, pembrolizumab, ramucirumab and cabozantinib have been approved by the Food and Drug Administration (FDA). Whereas, the drug resistance and side effects are inevitable [2]. Natural products, especially sesquiterpenoids, such as artemisinin, thapsigargin, parthenolide [3] and arglabin [4] are leading compounds for the development of potent therapeutic agents for neoplastic diseases, which inspired us to search for anti-HCC active compounds with high efficiency and low toxicity from natural sources.
Artemisia myriantha Wall. ex Bess., a folk medicine herb, has been used to treat inflammation and menorrhagia diseases [5]. According to previous investigation, sesquiterpenoids [5,6,7,8,9], dimeric sesquiterpenoids [5], flavones [7] and volatile oils [10,11,12] with antibacterial, anticancer and immunomodulating activity have been reported from this plant. In order to investigate the bioactive constituents of A. myriantha, our phytochemical study led to the isolation of four new sesquiterpenoids, artemyrianins A–D (1–4), three new norlignans, artemyrianins E–G (5–7), as well as five known compounds (Fig. 1). Herein, we reported their isolation, structural identification and cytotoxicity on HepG2 cells.

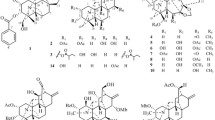
Chemical structures of compounds 1–12
2 Results and Discussion
Artemyrianin A (1) was obtained as colorless prismatic crystals and had a molecular formula of C15H20O2 with six indices of hydrogen deficiency from the (+)-HRESIMS (m/z 233.1518 [M+H]+, calcd for 233.1536). The IR spectrum indicated the presence of carbonyl (1771 and 1671 cm−1) and double bond (1606 cm−1) functionalities. The 1H NMR data (Table 1) exhibited signals for three singlet methyls at δH 1.28 (H3-14), 1.16 (H3-13) and 1.01 (H3-12), a double bond at δH 7.00 (1H, d, J = 2.2 Hz, H-4) and a formyl group at δH 9.54 (1H, s, H-15). The 13C NMR (DEPT) spectrum displayed 15 carbon resonances, including three methyls at δC 30.5 (C-13), 27.5 (C-14) and 26.5 (C-12), three methylenes at δC 43.2 (C-7), 33.8 (C-9) and 21.6 (C-8), four methines [including an olefinic at δC 148.7 (C-4)], three quaternary carbons [including an olefinic at δC 145.5 (C-3)], a formyl group at δC 189.9 (C-15) and a keto carbonyl at δC 203.9 (C-1). Inspection of its 1H and 13C NMR (DEPT) data suggested the similar structure with vulgarone A [13] except that a methyl (δH 1.78, δC 23.5) in vulgarone A was replaced by a formyl group (δH 9.54, δC 189.9) in 1. The HMBC correlations (Fig. 2) from H-15 to C-2 (δC 61.2), C-3 and C-4 confirmed the position of the formyl group at C-3. The single-crystal X-ray diffraction experiment using Cu Kα radiation (Fig. 3) unambiguously determined its structure as 3E-(2R,5S,10S,11R)-15-oxovulgarone A.

Key 1H–1H COSY and HMBC correlations of compounds 1–7

The X-ray ORTEP drawing of compound 1
Artemyrianin B (2) was purified as a white amorphous powder with a molecular formula of C15H20O5 by the [M−H]− ion at m/z 279.1242 (calcd for 279.1238) in the (−)-HRESIMS, exhibiting six degrees of unsaturation. The absorption bands at 3435, 1771, 1716 and 1633 cm−1 in the IR spectrum explained the existence of hydroxy, carbonyl and double bond groups. Two olefinic protons at δH 6.59 (d, J = 9.5 Hz, H-6) and 6.19 (d, J = 9.5 Hz, H-5), along with three methyls at δH 2.15 (s, H3-15), 1.89 (s, H3-13) and 1.01 (d, J = 7.0 Hz, H3-14) were recognized in the 1H NMR data. The 13C NMR (DEPT) data showed 15 resonances ascribed to three methyls at δC 30.0 (C-15), 15.5 (C-14) and 8.2 (C-13), three methylenes at δC 38.7 (C-9), 38.1 (C-3) and 28.9 (C-2), three methines [including two olefinic at δC 142.6 (C-5) and 119.5 (C-6)], an oxygenated tertiary carbon at δC 87.2 (C-1), a dioxygenated secondary carbon at δC 106.4 (C-8), two olefinic quaternary carbons at δC 155.7 (C-7) and 117.2 (C-11), and two carbonyls at δC 207.8 (C-4) and 172.0 (C-12), indicating a sesquiterpenoid scaffold. The spin systems of H2-2/H2-3, H-5/H-6 and H2-9/H-10/H3-14 inferred from the 1H–1H COSY spectrum, together with the HMBC correlations from H3-15 to C-3 and C-4, from H3-13 to C-7, C-11 and C-12, from H-6 to C-7 and C-8, from H2-9 (δH 2.60, 1.63) to C-8, and from H2-2 (δH 2.24, 1.94) to C-1, C-5 and C-10 (δC 41.7) constructed its planar structure with a 7-membered ring. An oxygen bridge linking C-1 and C-8 was affirmed by the de-shielded shifts of C-1 (δC 87.2) and C-8 (δC 106.4), and thus fulfilled the unsaturation degrees. The small coupling constant between H-5 and H-6 (J = 9.5 Hz) indicated the cis-configuration of Δ5. The cis-configuration of Δ7(11) was deduced with the fact that the cross-peak between H-6 and H3-13 was observed in the ROESY spectrum. In addition, the ROESY correlations (Fig. 4) of H2-2 (δH 2.24, 1.94) with H3-14 (δH 1.01) revealed their homolateral orientation. Its absolute configuration was established by the electronic circular dichroism (ECD) experiment which fitted well with the calculated ECD curve (Fig. 5). Therefore, compound 2 was characterized as 5Z,7(11)Z-(1R,8S,10S)-1,8-epoxy-8-hydroxy-4,5-secoguaian-5,7(11)-dien-12-oic acid.

Key ROESY correlations of compounds 1–4 and 7

The experimental and calculated ECD spectra of compounds 2–7
Artemyrianin C (3) was assigned a molecular formula of C18H26O5 by the analysis of the (+)-HRESIMS (m/z 345.1675 [M+Na]+, calcd for 345.1672), with six double bond equivalents. The characteristic absorption bands at 3434 (hydroxy), 1735 and 1720 (carbonyl) and 1629 cm−1 (double bond) were recognized in the IR spectrum. Compared with 2α-acetoxy-4α-hydroxy-1β-guai-11(13),10(14)-dien-12,8α-olide [14], the acetoxy group at C-2 was absent and the α-methylene-γ-lactone ring was opened in 3, which connected to a methoxy [δH 3.76 (s, H3-1″) and δC 52.2 (C-1″)] and an extra acetyl group [δH 1.98 (s, H3-2′) and δC 170.6 (C-1′), 21.3 (C-2′)]. The HMBC correlations from H-8 (δH 5.09) to C-1′, and from H3-1″ to C-12 (δC 167.3) verified the acetyl group at C-8 and the presence of methyl ester in the structure. Moreover, the shielded shift of C-2 from δC 75.6 (CH) in 2α-acetoxy-4α-hydroxy-1β-guai-11(13),10(14)-dien-12,8α-olide to δC 27.0 (CH2) in 3 interpreted the absence of the acetoxy group in 3. In the ROESY experiment, the correlations of H-5 with H-1, H-7 and H3-15, and of H-7 with H-8 explained their same orientation. Its absolute configuration was proposed as 1R,4S,5R,7R,8R by means of ECD calculation. Consequently, compound 3 was defined as (1R,4S,5R,7R,8R)-4-hydroxy-8-acetoxyguaian-10(14),11(13)-dien-12-oic acid methyl ester.
Artemyrianin D (4) was proposed a molecular formula of C16H26O4 based on the (+)-HRESIMS (m/z 305.1725 [M+Na]+, calcd for 305.1723). The IR spectrum suggested absorption bands for hydroxy, carbonyl and double bond functionalities at 3501, 3459, 1701 and 1626 cm−1. Its 1H and 13C NMR (DEPT) data exhibited similarity with those of 8-desacyl-4β-hydroxy-15-dihydrocarmanin [15] except for the absence of three hydroxyl groups in 4, which was proved by the shielded shifts of C-1 [from δC 76.1 (CH) to δC 41.3 (CH2)], C-6 [from δC 66.3 (CH) to δC 20.1 (CH2)] and C-15 [from δC 63.7 (CH2) to δC 23.0 (CH3)], respectively. The above deduction was further supported by the HMBC correlations of H2-1 (δH 1.45, 1.08) with C-3 (δC 43.6), C-5 (δC 56.0) and C-14 (δC 21.1), of H2-6 (δH 1.69) with C-8 (δC 68.0), C-10 (δC 34.8) and C-11 (δC 142.0), and of H3-15 (δH 1.20) with C-3, C-4 (δC 72.4) and C-5. The ROESY correlations of H-7 with H-5, H-8 and H-9b, and of H3-14 with H-9a and H3-15 suggested that H-5, H-7 and H-8 were on the same side, while the H3-14 and H3-15 were on the other side. The absolute configuration of 4 was constructed from the high match of experimental and calculated ECD data. Thus, compound 4 was identified as (4R,5R,7R,8R,10R)-4,8-dihydroxyeudesma-11(13)-en-12-oic acid methyl ester.
Artemyrianin E (5) was isolated as a yellowish oil and had a chemical composition of C19H18O5 with eleven degrees of unsaturation from the HREIMS (m/z 326.1148 [M]+, calcd for 326.1154). The IR spectrum displayed the existence of hydroxy (3384 cm−1) and phenyl ring (1606, 1503 and 1444 cm−1) groups. The similar 1H and 13C NMR (DEPT) data (Table 2) of 5 to those of (+)-artemisarin [16] revealed their closely related structures. The large coupling constant of H-7′ with H-8′ (\(J_{{7^{\prime},8^{\prime}}}\) = 15.7 Hz) indicated the E-configuration of \(\Delta^{{7^{\prime}}}\) in 5, instead of Z-configuration in (+)-artemisarin (\(J_{{7^{\prime},8^{\prime}}}\) = 11.2 Hz). The absolute stereochemistry of 5 was elucidated by the high agreement between the experimental and calculated ECD spectra. Hence, compound 5 was assigned as 7′E-(8S)-artemisarin.
Artemyrianin F (6) gave a molecular formula of C19H18O5 from the HREIMS at m/z 326.1152 ([M]+, calcd for 326.1154). Its IR spectrum presented the characteristic absorption bands of hydroxy (3404 cm−1) and phenyl ring (1607, 1503 and 1442 cm−1) functionalities. The high resemblance between the 1H and 13C NMR (DEPT) data of 6 and (+)-artemisarin [16] manifested their same planar structure. The geometry of \(\Delta^{{7^{\prime}}}\) was Z-form as deduced from the small \(J_{{7^{\prime},8^{\prime}}}\) value of 11.6 Hz. The ECD spectrum of 6 was superimposable to the calculated one, compound 6 was thus elucidated as 7′Z-(8R)-artemisarin.
Artemyrianin G (7) had a molecular formula of C19H18O6 based on the HREIMS at m/z 342.1104 ([M]+, calcd for 342.1103). The existence of hydroxy (3406 cm−1) and phenyl ring (1608, 1503 and 1443 cm−1) groups was recognized in the IR spectrum. Its 1H and 13C NMR (DEPT) spectroscopic data were close to those of mandshuricol A [17], but the main difference was that C-8′ in 7 was a methine attached to a hydroxy with chemical shift of δC 79.6 and C-8′ in mandshuricol A was a quaternary carbon attached to a hydroxy and a hydroxymethyl group with chemical shift of δC 83.4, which suggested that 7 was de-hydroxymethyl derivative at C-8′ of mandshuricol A. The above deduction was supported by the 1H–1H COSY correlations of H2-9/H-8/H-8′/H-7′. The ROESY correlations of H-8/H-8′, H-7′/H-8′ and H-8/H-7′ suggested the same orientation of H-8, H-7′ and H-8′. Its absolute configuration was proposed owning to the well match between the experimental and calculated ECD curves. Accordingly, compound 7 was established as (8R,7′R,8′R)-8′-dehydroxymethylmandshuricol A.
By comparing their 1H and 13C NMR (DEPT) spectroscopic data with those reported in literatures, five known compounds were identified as (−)-hinokinin (8) [18], (8R,8′R)-4,4′,5,5′-bis(methylenedioxy)-7′-oxolignano-9,9′-lactone (9) [19], isofraxidin (10) [20], dracunculin (11) [21] and 5-acetyl-6-hydroxy-2-(1-hydroxy-1-methylethyl)benzofuran (12) [22], respectively.
In addition to four sesquiterpenoids (1–4), five lignans (5–9), two coumarins (10 and 11) and a benzofuran (12) were isolated from A. myriantha for the first time. The isolates were assayed for their cytotoxicity against HepG2 cells with sorafenib as the positive control. As a result (Table 3), all compounds showed cytotoxic activity with IC50 values ranging from 33.3 to 145.2 μM.
3 Experimental
3.1 General Experimental Procedures
HRESIMS data were obtained from an LCMS-IT-TOF mass spectrometer (Shimadzu, Kyoto, Japan) or a UPLC/Q-TOF mass spectrometer (Agilent Technologies, Santa Clara, USA). HREIMS data were acquired on an Agilent G6230 mass spectrometer (Agilent Technologies, Santa Clara, USA). 1D and 2D NMR spectra were recorded on Avance III HD 400 (Bruker, Bremerhaven, Germany) or III 600 (Bruker, Fällanden, Switzerland) spectrometers with TMS as the internal standard. IR spectra were performed on a NICOLET iS10 spectrometer (Thermo Fisher Scientific, Madison, USA) with KBr pellets or ATR ITX-DIAMOND mode. UV spectra were measured on a Shimadzu UV-2700 spectrophotometer (Shimadzu, Suzhou, China). X-ray crystallographic data with Cu Kα radiation were collected on a Bruker D8 QUEST apparatus (Bruker, Karlsruher, Germany). ECD experiments were conducted with an Applied Photophysics Chirascan instrument (Applied Photophysics, Surrey, UK). Optical rotations were obtained on an Autopol VI automatic polarimeter (Rudolph Research Analytical, Hackettstown, NJ, USA). Melting point was acquired on an SGW® X-4B microscopic melting point instrument (Shanghai Precision and Scientific Instrument Co., Ltd., Shanghai, China). Silica gel (200–300 mesh) and Sephadex LH-20 gel (20–50 μm) for column chromatography were purchased from Linyi Haixiang Co., Ltd. (Haixiang, Linyi, China) and Pharmacia Fine Chemical Co., Ltd., (Pharmacia, Uppsala, Sweden), respectively. TLC detection was carried out using silica gel HSGF254 plates (Jiangyou, Chemical Co., Ltd., Yantai, China), and compounds were visualized by heating after spraying with 10% H2SO4 in EtOH (v/v). MPLC separation was achieved on a Dr-Flash II instrument (Lisure Science Co., Ltd., Suzhou, China) with an MCI gel CHP 20P column (75–150 μm, Mitsubishi Chemical Corporation, Tokyo, Japan). Semi-preparative HPLC purification was performed on a Shimadzu HPLC system equipped with LC-20AR pumps and a model SPD-M20A UV detector (Shimadzu, Kyoto, Japan) using an Agilent Eclipse XDB-C18 column (5 μm, 9.4 × 250 mm, Agilent Technologies, Santa Clara, USA).
3.2 Plant Material
The aerial parts of A. myriantha Wall. ex Bess. were obtained from Lijiang, Yunnan Province, China, in September 2018, and identified by Dr. Zhuo Zhou (Key Laboratory for Plant Diversity and Biogeography of East Asia, Kunming Institute of Botany, Chinese Academy of Sciences). A voucher specimen (No. 201809AM) was stored in the Laboratory of Antivirus and Natural Medicinal Chemistry, Kunming Institute of Botany, Chinese Academy of Sciences.
3.3 Extraction and Isolation
The air-dried aerial parts of A. myriantha (24.7 kg) were smashed and extracted with 90% EtOH for two times at room temperature (each 100 L, four days). The combined extracts were concentrated and suspended in H2O, which was extracted with EtOAc. The EtOAc portion (1.2 kg) was fractionated into six fractions A–F (300, 120, 150, 230, 165, 220 g) via silica gel column chromatography (CC) using acetone–petroleum ether (PE) gradient (5:95, 10:90, 20:80, 30:70, 40:60 and 50:50, v/v). Fr. C (150 g) was separated through MPLC on an MCI gel CHP 20P column and eluted with a gradient of H2O–MeOH (50:50, 30:70, 20:80 and 0:100) to give four subfractions (Frs. C1–C4). Fr. C1 (29 g) was subjected to silica gel CC (EtOAc–PE, 10:90 to 30:70) to afford three subfractions (Frs. C1-1‒C1-3). Compound 4 (1.9 g) was obtained by recrystallization in MeOH from Fr. C1-1 (12.5 g). Fr. C1-2 (7 g) was chromatographed over Sephadex LH-20 CC (MeOH–CHCl3, 50:50) and further purified by semi-preparative HPLC (H2O–MeCN, 38:62) to provide compound 2 (6 mg, tR = 27.5 min). Fr. C2 (35 g) was separated into three subfractions (Frs. C2-1‒C2-3) via silica gel CC with a gradient of EtOAc–PE (15:85 to 30:70). From Fr. C2-1 (1.5 g), compounds 1 (30 mg, tR = 13.0 min) and 12 (8 mg, tR = 39.5 min) was obtained by semi-preparative HPLC with H2O–MeCN (42:58) as the mobile phase. Fr. C2-3 (12 g) was submitted on silica gel CC with a step gradient of acetone–PE (20:80 to 30:70) and further purified by semi-preparative HPLC (H2O–MeOH, 51:49) to yield compounds 3 (62 mg, tR = 23.5 min) and 10 (9 mg, tR = 35.0 min). Fr. C4 (16.5 g) was separated on silica gel CC (EtOAc–PE, 15:85 to 30:70) to give three subfractions (Frs. C4-1–C4-3). Compounds 5 (85 mg, tR = 25.0 min), 6 (15 mg, tR = 21.5 min), 8 (7 mg, tR = 14.0 min), and 11 (88 mg, tR = 32.0 min) were provided from Fr. C4-2 (1.3 g) by semi-preparative HPLC (H2O–MeCN, 42:58). Fr. C4-3 (2.7 g) was subjected to Sephadex LH-20 CC (MeOH–CHCl3, 50:50) and further purified by semi-preparative HPLC (H2O–MeCN, 47:53) to afford compounds 7 (4 mg, tR = 19.0 min) and 9 (4 mg, tR = 15.0 min).
3.4 Spectroscopy Data of Compounds
The 1H NMR spectra of artemyrianins A–G (1–7) were recorded in CDCl3 at 600 MHz, and their 13C NMR (DEPT) spectra were recorded in CDCl3 at 150 MHz.
3.4.1 Artemyrianin A (1)
Colorless, prismatic crystals (MeOH); mp 95.8–97.5 °C; [α]22D + 45 (c 0.13, MeOH); UV (MeOH) λmax (log ε) 206 (3.69), 224 (3.73), 266 (3.49) nm; IR νmax 1771, 1671, 1606, 1454, 1132 cm−1; 1H NMR and 13C NMR (DEPT) data see Table 1; (+)-HRESIMS m/z 233.1518 [M+H]+ (calcd for C15H21O2, 233.1536).
3.4.2 Artemyrianin B (2)
White amorphous powder; [α]22D − 205 (c 0.11, MeOH); UV (MeOH) λmax (log ε) 263 (3.06) nm; ECD (MeOH) λmax (Δε) 213 (− 0.14), 238 (+ 3.00), 272 (− 20.81) nm; IR (KBr) νmax 3435, 1771, 1716, 1633, 1291, 1171, 1085, 1026 cm−1; 1H NMR and 13C NMR (DEPT) data see Table 1; ( −)-HRESIMS m/z 279.1242 [M−H]− (calcd for C15H19O5, 279.1238).
3.4.3 Artemyrianin C (3)
White amorphous powder; [α]24D − 110 (c 0.11, MeOH); UV (MeOH) λmax (log ε) 204 (4.08) nm; ECD (MeOH) λmax (Δε) 219 (+ 2.17) nm; IR (KBr) νmax 3434, 1735, 1720, 1629, 1440, 1376, 1247, 1148 cm−1; 1H NMR and 13C NMR (DEPT) data see Table 1; (+)-HRESIMS m/z 345.1675 [M+Na]+ (calcd for C18H26O5Na, 345.1672).
3.4.4 Artemyrianin D (4)
White amorphous powder; [α]24D − 111 (c 0.16, MeOH); UV (MeOH) λmax (log ε) 206 (3.82) nm; ECD (MeOH) λmax (Δε) 196 (− 5.96), 254 (+ 0.56) nm; IR (KBr) νmax 3501, 3459, 1701, 1626, 1290, 1245 cm−1; 1H NMR and 13C NMR (DEPT) data see Table 1; (+)-HRESIMS m/z 305.1725 [M+Na]+ (calcd for C16H26O4Na, 305.1723).
3.4.5 Artemyrianin E (5)
Yellowish oil; [α]27D − 185 (c 0.22, MeOH); UV (MeOH) λmax (log ε) 215 (4.18), 267 (4.04), 290 (3.87) nm; ECD (MeCN) λmax (Δε) 201 (+ 18.46), 224 (− 4.18), 241 (− 2.24), 262 (− 5.89), 282 (− 3.15), 300 (− 5.71) nm; IR νmax 3384, 1606, 1503, 1489, 1444, 1250, 1190, 1039, 929, 804 cm−1; 1H NMR and 13C NMR (DEPT) data see Table 2; HREIMS m/z 326.1148 [M]+ (calcd for C19H18O5, 326.1154).
3.4.6 Artemyrianin F (6)
Yellowish oil; [α]19D + 152 (c 0.15, MeOH); UV (MeCN) λmax (log ε) 262 (3.03), 290 (2.95) nm; ECD (MeCN) λmax (Δε) 200 (− 40.56), 215 (+ 15.69), 240 (+ 2.92), 297 (+ 5.89) nm; IR (KBr) νmax 3404, 1607, 1503, 1489, 1442, 1245, 1189, 1040, 928, 869, 812 cm−1; 1H NMR and 13C NMR (DEPT) data see Table 2; HREIMS m/z 326.1152 [M]+ (calcd for C19H18O5, 326.1154).
3.4.7 Artemyrianin G (7)
White amorphous powder; [α]26D − 18 (c 0.12, MeOH); UV (MeOH) λmax (log ε) 235 (3.63), 287 (3.59) nm; ECD (MeCN) λmax (Δε) 206 (+ 5.76), 248 (+ 0.20), 282 (+ 0.29) nm; IR νmax 3406, 1608, 1503, 1490, 1443, 1247, 1189, 1099, 1039, 927, 811 cm−1; 1H NMR and 13C NMR (DEPT) data see Table 2; HREIMS m/z 342.1104 [M]+ (calcd for C19H18O6, 342.1103).
3.5 X-ray Crystallographic Analysis of Artemyrianin A (1)
Single crystals of compound 1 were obtained from MeOH. X-ray crystallographic data were collected on a Bruker D8 Quest apparatus employing graphite-monochromated Cu Kα radiation at 100.(2) K. Cell refinement and data reduction were carried out by the aid of Bruker SAINT. The structure was solved by direct methods with SHELXS-97. All non-hydrogen atoms were refined anisotropically by means of the least-squares method, and all hydrogen atoms were positioned using difference Fourier overlapping calculation and geometric calculations. Flack parameter = 0.18(14). The crystallographic data for 1 have been deposited at the Cambridge Crystallographic Data Centre (CCDC) as Supplementary Publication No. CCDC 1985641. Copy of the data can be obtained free of charge from CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [fax: +44 (0) 1223 336033; e-mail: deposit@ccdc.cam.ac.uk].
3.5.1 Crystal Data for 1
C15H20O2, M = 232.31, a = 6.7424(3) Å, b = 6.9858(3) Å, c = 7.7309(3) Å, α = 105.3440(10)°, β = 101.9110(10)°, γ = 106.6430(10)°, V = 320.37(2) Å3, T = 100.(2) K, space group P1, Z = 1, μ(Cu Kα) = 0.615 mm−1, 4616 reflections measured, 2090 independent reflections (Rint = 0.0519). The final R1 values were 0.0484 [I > 2σ(I)]. The final wR(F2) values were 0.1244 [I > 2σ(I)]. The final R1 values were 0.0485 (all data). The final wR(F2) values were 0.1245 (all data). The goodness of fit on F2 was 1.040. Flack parameter = 0.18(14).
3.6 Computational Details
The ECD calculations for compounds 2–7 were achieved by Gaussian 09 program. The configurations of compounds 2–4 and 7 were initially established on the basis of their ROESY data, and further optimized by means of the DFT calculation at b3lyp/6-31G(d,p) level in the gas phase. The imaginary frequencies were excluded by the aid of frequency calculations at the same level. Taking the solvent effects into consideration, ECD calculations were carried out using the TDDFT methodology at b3lyp/6-311+g(d,p) level. The ECD curves were plotted by the Origin Pro 9 program (OriginLab Corporation, Northampton, USA).
3.7 Cytotoxicity Assay
MTT assays were conducted to evaluate the cytotoxic activity of the isolates on HepG2 cells [23]. HepG2 cells were seeded onto 96-well plates at a density of 1 × 104 cells per well. Following 24 h incubation at 37 °C with 5% CO2, cells were treated with 100 μL of culture medium containing tested samples at gradient concentrations for 48 h. Next, the medium was removed and replaced by 100 μL MTT solution (1 mg/mL), and the plates were further incubated in dark for four h. After removing the medium, 100 μL DMSO was added into each well to dissolve the MTT formazan salt. Then the plates were measured at 490 nm on a microplate reader (Bio-Rad, USA). The cytotoxicity of compounds was expressed as IC50 values which calculated by GraphPad prism 5 (GraphPad Software, San Diego, California, USA). The HepG2 cells (Jining Biotechnology Co., Ltd., Shanghai, China) were maintained at 37 °C with 5% CO2 and grown in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, Thermo Fisher Scientific Co., Ltd., Suzhou, China) supplemented with 10% fetal bovine serum (FBS) (Gibco, Life Technologies, NY, USA) (Table 3).
References
F. Bray, J. Ferlay, I. Soerjomataram, R.L. Siegel, L.A. Torre, A. Jemal, CA Cancer J. Clin. 68, 394–424 (2018)
Q.Z. Dai, C.L. Zhang, Z.G. Yuan, Q.S. Sun, Y.Y. Jiang, Expert Opin. Drug Discov. 15, 243–258 (2020)
A. Ghantous, H. Gali-Muhtasib, H. Vuorela, N.A. Saliba, N. Darwiche, Drug Discov. Today 15, 668–678 (2010)
S.H. Lone, K.A. Bhat, M.A. Khuroo, Chem. Biol. Interact. 240, 180–198 (2015)
H.F. Wong, G.D. Brown, J. Nat. Prod. 65, 481–486 (2002)
H.F. Wong, G.D. Brown, Phytochemistry 59, 529–536 (2002)
Q.J. Li, W.P. Chen, J.H. Fan, Q. Zhao, X.H. Li, Q. Yang, C.Y. Liu, Chin. J. Mod. Appl. Pharm. 31, 706–710 (2014)
K. Zan, X.Q. Chen, M.B. Zhao, P.F. Tu, China J. Chin. Mater. Med. 41, 2833–2837 (2016)
K. Zan, X.Q. Chen, P.F. Tu, China J. Chin. Mater. Med. 43, 2295–2299 (2018)
G.C. Shah, C.S. Mathela, J. Essent. Oil Res. 18, 633–634 (2006)
G.D. Bagchi, F. Haider, N. Kumar, S.C. Singh, A.A. Naqvi, J. Essent. Oil Res. 21, 43–45 (2009)
X.Y. Dai, X. Li, X.R. Dong, B.L. Zhou, M. Xie, J. Anhui, Agric. Sci. 40, 11562–11564, 11597 (2012)
Y. Uchio, Tetrahedron 34, 2893–2899 (1978)
L.Y. Nie, J.J. Qin, Y. Huang, L. Yan, Y.B. Liu, Y.X. Pan, H.Z. Jin, W.D. Zhang, J. Nat. Prod. 73, 1117–1120 (2010)
L. Cardona, R.A. Aleman, B. Garcia, J.R. Pedro, Phytochemistry 31, 3630–3632 (1992)
C. Song, Y.Q. Li, Y.M. Yan, M. Hu, Q.Z. Zhang, Chem. Nat. Compd. 50, 414–416 (2014)
Q.M. Xu, Q. Li, Y.L. Liu, Y.L. Feng, S.L. Yang, X.R. Li, Chem. Nat. Compd. 46, 366–369 (2010)
G. Zhang, S. Shimokawa, M. Mochizuki, T. Kumamoto, W. Nakanishi, T. Watanabe, T. Ishikawa, K. Matsumoto, K. Tashima, S. Horie, Y. Higuchi, O.P. Dominguez, J. Nat. Prod. 71, 1167–1172 (2008)
B. Raffaelli, K. Wähälä, T. Hase, Org. Biomol. Chem. 4, 331–341 (2006)
P. Panichayupakaranant, H. Noguchi, W. De-Eknamkul, U. Sankawa, Phytochemistry 40, 1141–1143 (1995)
J.H. Kwak, W.Y. Jang, O.P. Zee, K.R. Lee, Planta Med. 63, 474–476 (1997)
M. Talbi, B. Saadali, D. Boriky, L. Bennani, M. Elkouali, T. Ainane, J. Asian Nat. Prod. Res. 18, 724–729 (2016)
Y. Li, X.Y. Cheng, C.L. Chen, H.J. Wu, H. Zhao, W. Liu, Z. Xiang, Q. Wang, Pathol. Res. Pract. 216, 152701 (2020)
Acknowledgements
This work was financially supported by the Yunnan Wanren Project (YNWR-KJLJ-2019-002), the Program of Yunling Scholarship, the Reserve Talents of Young and Middle-aged Academic and Technical Leaders in Yunnan Province, and the Youth Innovation Promotion Association, CAS (2013252).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tang, S., Ma, YB., Geng, CA. et al. Artemyrianins A–G from Artemisia myriantha and Their Cytotoxicity Against HepG2 Cells. Nat. Prod. Bioprospect. 10, 251–260 (2020). https://doi.org/10.1007/s13659-020-00255-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-020-00255-z