
Abstract
A culture isolated from ascospores of Hypoxylon rickii, a xylariaceous ascomycete collected in Martinique, had yielded botryane, noreremophilane and abietane-type terpenoids in a preceding study, but additional metabolites were detected by extensive HPLC–MS analysis in other fractions. Herein we report the further isolation of four new sesquiterpenoids with a silphiperfol-6-ene skeleton from extracts of H. rickii. The planar structures were elucidated by NMR and HRMS data as 13-hydroxysilphiperfol-6-ene (1), 9-hydroxysilphiperfol-6-en-13-oic acid (2), 2-hydroxysilphiperfol-6-en-13-oic acid (3) and 15-hydroxysilphiperfol-6-en-13-oic acid (4). For compounds 2–4 we propose the trivial names rickinic acids A–C. Their stereochemistry was assigned by ROESY correlations as well as by the specific optical rotation. Additionally, the known compounds, botryenanol, dehydrobotrydienol, cyclo(Phe-Pro), cyclo(Pro-Leu), (+)-ramulosin and α-eleostearic acid were isolated. The antimicrobial and cytotoxic activities of the new compounds are also reported.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The exploration of the highly diverse fungal family Xylariaceae in terms of secondary metabolite production has revealed a tremendous amount of natural products from most of the biosynthetic pathways. Whereas polyketides and PKS-NRPS hybrid molecules occur in the stromata [1–6] and cultures of the family members [7, 8], terpenes were so far exclusively reported from cultures. Important examples of the latter structural class produced by the Xylariaceae are the antifungal sordarins from Rosellinia and Hypoxylon spp. [9], the neuropeptide gamma receptor antagonists, xylarenals A and B from Xylaria [10], the phytotoxic hymatoxins from “Hypoxylon” (current name Entoleuca) mammatum [11], the antihypertensive vinigrol from Virgaria [12], the antibacterial hypocoprins from Hypocopra [13], or botryanes from Daldinia concentrica [14].
Although the genus Hypoxylon is one of the largest within the Ascomycota and its representatives are frequently encountered as endophytes, little is known about their secondary metabolite production capabilities in cultures. Recently, we evaluated the diversity of natural products produced by the ex-epitype strain of H. rickii. Subsequently, various terpenoids of the botryane, noreudesmane and abietane scaffold were isolated from a single large scale fermentation of the strain [15]. We now report the isolation, structure elucidation and biological activity of four new silphiperfolene-type terpenoids and six known natural products from the same fungus.
2 Results and Discussion
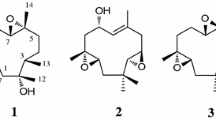
A 70 L fermentation of a H. rickii strain was processed by separating the mycelia from the culture broth and extraction of the corresponding biomass with acetone. The crude extract was pre-fractionized by MPLC and we focused on the isolation of the hydrophobic components by using HPLC. Besides several not further characterized fatty acids, we isolated the new metabolite 1 (Fig. 1) by subsequent HPLC. Compound 1 was obtained as a colorless oil; its molecular formula C15H24O was deduced from the molecular ion cluster [M+H]+ at m/z 219.1737 in the HRESIMS spectrum, which is implying 4 degrees of unsaturation. Proton and 1H,13C-HSQC NMR experiments revealed the presence of three methyls, six methylenes (one of which was oxygenated) and two methines. Furthermore, the carbon NMR spectrum suggested two sp 2 hybridized and two sp 3 hybridized quaternary carbons. The structural backbone of 1 was determined by 1H,1H COSY and 1H,13C HMBC correlations. Starting form methyl H3-15 the extensive spin system H2-11/H2-10/H-9(H3-15)/H-1/H2-2/H2-3 was determined by 1H,1H COSY and TOCSY correlations (Fig. 2). By 1H,13C HMBC correlations from methylenes H2-5 and H2-13 along with methyl H3-14 the C-5/C-6(C-13)/C-7/C-14 partial structure was determined. Finally, 1H,13C HMBC correlations from H-1, H2-2, H2-3, H2-5, H2-9, H2-10, H2-11, H3-12, H3-14 to quaternary carbon C-8 demonstrated the 13-hydroxysilphiperfol-6-ene structure. 1H,1H ROESY correlations between H3-15 and Hβ-11 on the β-face and H3-12 and Hα-11 on the α-face specify the typical 1S*,4S*,8S*,9R* configuration of silphiperfolene-type sesquiterpenoids. Because compound 1 bears no additional optical centers, the negative specific optical rotation of 1 defines the absolute configuration as 1S,4S,8S,9R, since (−)-silphiperfol-6-ene has been synthesized from (R)-(+)-pulegone [16].

Structures of new metabolites 1–4

Selected 2D NMR correlations of 1

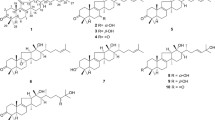
Structures of known metabolites isolated from H. rickii
To provide additional material H. rickii was cultivated in 10 L scale in YM medium. 13-Hydroxysilphiperfol-6-ene (1) could not be detected, but three oxidized derivatives (2–4) were isolated by preparative MPLC and HPLC. Metabolite 2 was obtained as a colorless oil; its molecular formula C15H22O3 was determined by the [M+H]+ molecular ion cluster at m/z 251.1649 in the HRESIMS spectrum. The main difference in the proton spectrum of 2 compared to 1 was the shortfall of signals for methine H-9 and methylene H2-13. This result was confirmed by the carbon spectrum, in which additional signals for a carboxyl and oxygenated quaternary sp 3 hybridized carbon atoms were observed. 1H,13C HMBC correlations from H2-5 to C-13 identified the carboxylic acid carbon atom as C-13. Singulet methyl H3-15 showed 1H,13C HMBC correlations to oxygen bearing C-9 besides C-1 and C-10. Therefore, 2 was identified as 9-hydroxysilphiperfol-6-en-13-oic acid. The configuration of stereocenter C-9 was assigned as S as a consequence of the 1H,1H ROESY correlation between methyls H3-15 and H3-14. This correlation demonstrates that the oxygenation occurred with retention of the configuration of methyl group CH3-15. 2 was named rickinic acid A.
Rickinic acid B (3) has the same molecular formula C15H22O3 as 2, which was obtained by HRESIMS data. Proton, carbon and 1H,13C HSQC NMR spectra were highly similar to that of 2. However, the key differences were the replacement of the methylene signal for C-2 by an oxygenated methine, and the replacement of oxygenated quaternary carbon C-9 by a methine. Therefore, 3 was identified as 2-hydroxysilphiperfol-6-en-13-oic acid. The 1H,1H ROESY correlations from H-2 to H-1/Hβ-3 and Hβ-5 on the β-face respectively from H3-12 to Hα-3 and Hα-5 on the α-face of the molecule indicated a 2R configuration.
With rickinic acid C (4) another metabolite was isolated with a C15H22O3 molecular formula. The proton and 1H,13C HSQC NMR spectra showed the oxidation of methyl CH3-15 to a hydroxymethyl group, thus identifying 4 as 15-hydroxysilphiperfol-6-en-13-oic acid.
Besides the new metabolites 1–4, we isolated several known metabolites (Fig. 3) botryenanol (5), dehydrobotrydienol (6), cyclo(Phe-Pro) (7), cyclo(Pro-Leu) (8), (+)-ramulosin (9) and α-eleostearic acid (10) from different cultivations of H. rickii [17–22].
The bioactivity of 1–4 was evaluated against S. cerevisiae, C. albicans, B. subtilis, E. coli and the mouse fibroblast cell line L929. We detected weak antifungal activity of 2 against the yeast S. cerevisiae (MIC = 66.7 µg/mL), weak antibacterial activity of 4 against B. subtilis (MIC = 33.3 µg/mL) and weak cytotoxic activity of 3 (IC50 = 20 µg/mL).
The sesquiterpenoids 1–4 belong to a family of compounds with a silphiperfolene core structure. These compounds are known to be produced by members of the plant family Asteraceae [23, 24]. However, no fungal metabolite with this particular core structure has been described to the best of our knowledge. The role of these metabolites for H. rickii in nature remains elusive. The compounds might be produced to modulate its host plant, similarly to the fungal production of gibberellin-type phytohormons [25]. Though, the silphiperfolene metabolites might also be side products in the biosynthesis of botryanes. Presilphiperfolanol has been proposed as precursor of triquinane [26] as well as botryane [27] type sesquiterpenes. Even though presilphiperfolanol as potential common precursor has not been detected so far, this might explain the co-occurrence of silphiperfolene and botryane-type metabolites in cultures of H. rickii.
The identification of compounds 5 and 6 continues the list of known botryane-type terpenoids from H. rickii [15]. This class of secondary metabolites has already been reported from other xylariaceous fungi like Daldinia concentrica and a Geniculisporium sp. [14, 28] and seems therefore common within the family. The same is true for the isocoumarin derivative (+)-ramulosin (9) which frequently occurs in the genus Hypoxylon [29]. The latter has been shown to exhibit phytotoxic and antifungal effects [30]. The production of diketopiperazines in the Xylariaceae is so far only known from Rosellinia necatrix [31] and therefore 7 and 8 are the first report of this particular compound class from another member of the family.
3 Experimental
3.1 General Experimental Procedures
Optical rotations were determined with a Perkin-Elmer 241 spectrometer and UV spectra were recorded with a Shimadzu UV–Vis spectrophotometer UV-2450. NMR spectra were recorded with Bruker Avance III 700 spectrometer with a 5 mm TCI cryoprobe (1H 700 MHz, 13C 175 MHz) and Avance III 500 (1H 500 MHz, 13C 125 MHz) spectrometers. HRESIMS mass spectra were obtained as previously described [32]. Isolation of pure compounds was achieved if not indicated otherwise with a preparative HPLC (Gilson, Middleton, USA) equipped with a GX-271 Liquid Handler, a 172 DAD, a 305 and 306 pump (with 50SC Piston Pump Head). As stationary phase a VP Nucleodur C18 ec column (125 × 40 mm, 7 µm; Macherey–Nagel) was used. The mobile phase was composed of deionised water (Milli-Q, Millipore, Schwalbach, Germany) with 0.1 % acetic acid (solvent A1; Roth) and acetonitrile (ACN) with 0.1 % acetic acid (solvent B1). Flow rate was set to 15 ml/min.
3.2 Fungal Material
Stromata (fruiting bodies) of H. rickii MJF10324 were collected in 2010 from the Caribbean island Martinique by J. Fournier. The strain was designated as epitype of the species [33]. The culture was derived by multispore isolation on YMG medium (1.0 % malt extract, 0.4 % glucose, 0.4 % yeast extract, pH 6.3) using the method outlined by Stadler et al. [34] and has been deposited in public culture collections (MUCL 53309, CBS 129345).
3.3 Cultivation in 70 L Scale and Isolation of 1 and 10
Large-scale fermentation of the strain was carried out as previously described [14] in HLX media (3.0 % sucrose, 1.0 % casamino acids, 0.1 % K2HPO4, 0.1 % yeast extract, 0.05 % MgSO4 × 7H2O, 0.05 % KCl, 0.001 % FeSO4 × 7H2O). The fermentation was aborted after 7 days as sugars (sucrose, fructose) were depleted. Compound 1 and 10 were obtained from the acetone crude extract of the biomass (12 g) by preparative RP MPLC and subsequent HPLC. The conditions for the RP MPLC were as follows: a ODS/AQ C18 column (480 × 30 mm, Kronlab) as stationary phase, mobile phase composed of solvent A2 [90 % deionised water (Milli-Q), 10 % methanol] and solvent B2 (methanol), linear gradient of solvent B2 from 10 to 100 % in 60 min, followed by isocratic conditions at 100 % solvent B2 for 20 min, flow rate of 30 ml/min, UV peak detection at 210 nm. Compound 1 (2 mg) was purified from the MPLC fraction with a retention time (RT) of 58.5–64.0 min (390 mg) using a linear gradient from 55 to 100 % solvent B1 in 25 min followed by isocratic conditions at 100 % for 15 min at a RT = 26.0 min. Compound 10 (RT: 14.0 min; 2 mg) was isolated from another MPLC fraction (RT: 64.0–67.0 min; 1.8 g) by RP HPLC (linear gradient from 90 to 100 % B1 in 20 min, followed by 15 min isocratic conditions).
13-Hydroxysilphiperfol-6-ene (1): amorphous powder, [α] 25D −29 (c 0.2 MeOH); 1H (700 MHz) and 13C (175 MHz) NMR data (methanol-d 4), see Tables 1 and 2; HRESIMS: m/z 203.1791 (calcd for C15H23, [M+H−H20]+, 203.1794), 219.1737 (calcd for C15H23O, [M+H]+, 219.1743).
α-Eleostearic acid (10): colorless oil; 13C NMR (chloroform-d, 125 MHz) δ 14.1, 22.4, 24.8, 28.0, 29.1, 29.2, 29.7, 29.9, 31.5, 32.7, 33.9, 126.2, 129.0, 130.5, 132.1, 133.2, 135.5, 178.8; HRESIMS: m/z 279.2319 (calcd for C18H31O2, [M+H]+, 279.2319); spectroscopic and spectrometric data are in good agreement with the literature [22].
3.4 Cultivation in 10 L Scale and Isolation of 2–4 and 6–8
A seed culture of the strain with a total volume of 200 mL was prepared in YMG medium (1.0 % malt extract, 0.4 % glucose, 0.4 % yeast extract, pH 6.3), incubated at 22 °C and 140 rpm for 5 days, homogenized with an ultratorax and incubated again for 2 days (22 °C, 140 rpm). A BR 15.4 bioreactor (B. Braun Melsungen AG, Germany) filled with 10 L YMG medium and supplemented with 0.5 % talcum powder (Sigma-Aldrich, St. Louis, USA) was inoculated with 100 mL of the seed culture. The temperature was set at 26 °C. The stirrer speed was set to 150 rpm, aeration rate was set to 0.07 vvm and remained constant during fermentation. The culture was harvested after 7 days as sugars (sucrose, fructose) were depleted. Thereafter, the mycelium was separated from the culture fluid by vacuum filtration to yield a total amount of 525 g wet biomass, which was later extracted with 1.8 L acetone in an ultrasonic bath for 1 h. The acetone extract was filtered and evaporated to yield an aqueous phase, which was further processed by extraction with 3 × 100 mL ethyl acetate in a separating funnel. Subsequently the organic phases were combined and evaporated to yield 221 mg of oily mycelial crude extract (ME) in total.
The culture filtrate was extracted by 2 % XAD-16 (200 g) over 2 h at room temperature. The XAD was separated by filtration and extracted with methanol (500 mL). The extract was evaporated to yield an aqueous phase, which was further processed by extraction with 3 × 50 mL ethyl acetate. The combined organic extracts were filtrated over Strata X column to yield 643 mg crude extract.
The crude extract was subjected to silica gel chromatography using gradient elution with portions of dichloromethane/methanol mixtures (250 mL) of 100/0, 99/1, 98/2, 97/3, 95/5, 90/10, 85/15, 80/20, 70/30, 50/50, 25/75, 0/100 to give 12 fractions. Fraction 2 (303 mg) was fractionized with preparative HPLC (PLC 2020, Gilson, Middleton, USA). A VP Nucleodur C18 ec column (150 × 40 mm, 7 µm; Macherey–Nagel) was used as stationary phase. The mobile phase was composed of deionised water (Milli-Q) as solvent A and methanol (MeOH) as solvent B. A flow rate of 30 ml min−1 was used for the following gradient: 10–80 % solvent B in 30 min, afterwards in 5 min to 100 % B, isocratic conditions at 100 % for 10 min. UV detection was carried out at 210 and 254 nm and fractions were collected and combined according to the observed peaks. 2 (0.9 mg) was obtained at a retention time (RT) = 30.7–31.0 min.
Fraction 5 (89 mg) was subjected to preparative HPLC as described above (but gradient: 30–100 % solvent B in 30 min, isocratic conditions at 100 % for 10 min) to yield 3 (0.6 mg) at a RT = 18.7–21.7 min and 4 (0.3 mg) at RT = 14.3–15.5 min.
Fraction 1 (242 mg) subjected to preparative HPLC as described above (but gradient: 0–70 % solvent B in 40 min, afterwards in 5 min to 100 % B, isocratic conditions at 100 % for 5 min) to yield 7 (0.8 mg) and 8 at a RT = 19.6–20.2 and RT = 17.7–18.6 min, respectively. Fractions from 35.1 to 36.4 min were combined and fractionated again (gradient: 40–80 % acetonitril in 40 min, afterwards in 5 min to 100 % ACN, isocratic conditions at 100 % for 5 min, VP Nucleodur C18 ec column (250 × 21 mm, 7 µm; Macherey–Nagel) to yield 6 (2.4 mg) at a RT = 20.5–22 min.
Rickinic acid A (2): amorphous powder, [α] 25D +2.2 (c 0.02 CHCl3); UV (MeOH) λmax (log ε) 234 nm (4.7); 1H (700 MHz) and 13C (175 MHz) NMR data (chloroform-d), see Tables 1 and 2; ESIMS: m/z 250.95 [M+H]+, 248.86 [M−H]−; HRESIMS: m/z 251.1649 (calcd for C15H23O3,[M+H]+, 251.1642).
Rickinic acid B (3): amorphous powder, [α] 25D +12 (c 0.1 CHCl3); UV (MeOH) λmax (log ε) 234 nm (4.5); 1H (700 MHz) and 13C (175 MHz) NMR data (chloroform-d), see Tables 1 and 2; ESIMS: m/z 251.02 [M+H]+, 248.96 [M−H]−; HRESIMS: m/z 251.1648 (calcd for C15H23O3,[M+H]+, 251.1642).
Rickinic acid C (4): amorphous powder, [α] 25D +11 (c 0.1 CHCl3); UV (MeOH) λmax (log ε) 220 nm (sh); 1H (700 MHz) and 13C (175 MHz) NMR data (chloroform-d), see Tables 1 and 2; ESIMS: m/z 233.01 [M+H-H20]+, 248.94 [M−H]−; HRESIMS: m/z 251.1648 (calcd for C15H23O3,[M+H]+, 251.1642).
Dehydrobotrydienol (6): colorless oil; 13C NMR (chloroform-d, 125 MHz) δ 19.0, 26.3, 31.1, 32.2, 40.9, 50.4, 54.0, 58.8, 71.1, 123.1, 130.5, 134.4, 136.4, 144.1, 152.3; HRESIMS: m/z 257.1517 (calcd for C15H22O2Na, [M+H]+, 257.1512); spectroscopic and spectrometric data are in good agreement with the literature [18].
Cyclo(Phe-Pro) (7): colorless oil; 13C NMR (chloroform-d, 125 MHz) δ 22.6, 28.4, 36.8, 45.5, 56.2, 59.2, 127.6, 129.1, 129.4, 135.9, 165.1, 169.5; HRESIMS: m/z 245.1297 (calcd for C14H17N2O2, [M+H]+, 245.1285); spectroscopic and spectrometric data are in good agreement with the literature [19].
Cyclo(Pro-Leu) (8): colorless oil; 13C NMR (chloroform-d, 125 MHz) δ 21.2, 22.8, 23.3, 24.8, 28.2, 29.7, 38.7, 45.6, 53.4, 59.0, 166.1, 170.3; HRESIMS: m/z 211.1461 (calcd for C11H19N2O2, [M+H]+, 211.1441); spectroscopic and spectrometric data are in good agreement with the literature [20].
3.5 Small-Scale Cultivation and Isolation of 5 and 9
Two submerged cultures were grown in each case in 200 mL YMG medium supplemented with 3 g talcum powder on a rotary shaker at 140 rpm and 23 °C. Cultures were harvested after 12 days as free glucose was consumed. Afterwards the supernatant was separated from the biomass by vacuum filtration and extracted with 200 mL ethyl acetate. The organic phases were combined, dried over sodium sulfate and evaporated to yield 50 mg crude extract. The latter was fractionated by RP HPLC using the following conditions: a VP 250/21 Nucleodur100-5 C18 ec column (Macherey–Nagel) equipped with a Kromasil 100 C18 pre-column (50 × 20 mm, 7 μm; AkzoNobel) as stationary phase, solvents A1 and B1 as mobile phase, linear gradient from 20 to 70 % solvent B1 in 40 min, then from 70 to 100 % B1 in 5 min, followed by 10 min isocratic conditions, flow rate of 15 ml/min. Compound 5 (1.4 mg) was obtained at a RT = 33.0 min and 9 (1.5 mg) at a RT = 35.0 min.
Botryenanol (5): Colorless oil; 13C NMR (chloroform-d, 125 MHz) δ 20.9, 21.4, 23.8, 29.1, 29.3, 36.8, 39.1, 51.8, 54.2, 58.5, 70.5, 71.9, 139.0, 164.7, 170.3, 192.6; HRESIMS: m/z 317.1728 (calcd for C17H26O4Na, [M+H]+, 317.1723); spectroscopic and spectrometric data are in good agreement with the literature [17].
(+)-Ramulosin (9): Colorless amorphous powder, [α] 25D +12 (c 0.1 CH3OH); 13C NMR (chloroform-d, 125 MHz) δ 21.0, 21.8, 29.1, 29.6, 33.0, 37.5, 76.6, 96.8, 171.9, 174.8; HRESIMS: m/z 183.1026 (calcd for C10H15O3, [M+H]+, 183.1016); spectroscopic and spectrometric data are in good agreement with the literature [21].
3.6 Biological Assays
Antibacterial, antifungal and cytotoxic assays were performed as described by Surup et al. [35].
References
M. Stadler, D.N. Quang, A. Tomita, T. Hashimoto, Y. Asakawa, Mycol. Res. 110, 811–820 (2006)
F. Surup, K.I. Mohr, R. Jansen, M. Stadler, Phytochemistry 95, 252–258 (2013)
X.N. Wang, W.Y. Huang, J.C. Ju, C.Y. Li, J.K. Liu, Biochem. Syst. Ecol. 54, 157–159 (2014)
E. Kuhnert, S. Heitkämper, J. Fournier, F. Surup, M. Stadler, Fungal Biol. 118, 242–252 (2014)
E. Kuhnert, F. Surup, E.B. Sir, C. Lambert, K.D. Hyde, A.I. Hladki, A.I. Romero, M. Stadler, Fungal Divers. 71, 165–184 (2015)
E.B. Sir, E. Kuhnert, F. Surup, K.D. Hyde, M. Stadler, Mycol. Prog. 14, 28 (2015)
M. Stadler, V. Hellwig, Recent Res. Dev. Phytochem. 9, 41–93 (2005)
F. Surup, E. Kuhnert, E. Lehmann, S. Heitkämper, K.D. Hyde, J. Fournier, M. Stadler, Mycology 5, 110–119 (2014)
M. Daferner, S. Mensch, T. Anke, O. Sterner, Z. Naturforsch, C 54, 474–480 (1999)
C.J. Smith, N.R. Morin, G.F. Bills, A.W. Dombrowski, G.M. Salituro, S.K. Smith, A. Zhao, D.J. MacNeil, J. Org. Chem. 67, 5001–5004 (2002)
J. Pinon, P.D. Manion, Eur. J. For. Pathol. 21, 202–209 (1991)
T. Ando, K. Yoshida, M. Okuhara, J. Antibiot. 41, 31–35 (1988)
D.R. Jayanetti, Q. Yue, G.F. Bills, J.B. Gloer, J. Nat. Prod. 78, 396–401 (2015)
X.D. Qin, H.J. Shao, Z.J. Dong, J.K. Liu, J. Antibiot. 61, 556–562 (2008)
E. Kuhnert, F. Surup, V. Wiebach, S. Bernecker, M. Stadler, Phytochemistry 117, 116–122 (2015)
L.A. Paquette, R.A. Roberts, G.J. Drtina, J. Am. Chem. Soc. 106, 6690–6693 (1984)
I.G. Collado, R. Hernández-Galán, V. Prieto, J.R. Hanson, L.G. Rebordinos, Phytochemistry 41, 513–517 (1996)
R. Durán-Patrón, R. Hernández-Galán, I. Collado, J. Nat. Prod. 63, 182–184 (2000)
M. Tullberg, M. Grotli, K. Luthman, Tetrahedron 62, 7484–7491 (2006)
M. Soledade, C. Pedras, Y. Yu, J. Liu, Y.A. Tandron-Moya, Z. Naturforsch. C 60, 717–722 (2005)
D.B. Stierle, A.A. Stierle, A. Kunz, J. Nat. Prod. 61, 1277–1278 (1998)
Y. Amakura, K. Kondo, H. Akiyama, H. Ito, T. Hatano, T. Yoshida, T. Maitani, J. Food Hyg. Soc. Jpn. 47, 178–181 (2006)
F. Bohlmann, J. Jakupovic, Phytochemistry 19, 259–265 (1980)
P. Weyerstahl, H. Marschall, I. Seelmann, J. Jakupovic, Eur. J. Org. Chem. 6, 1205–1212 (1998)
B. Tudzynski, Appl. Microbiol. Biotechnol. 66, 597–611 (2005)
R.M. Coates, Z. Ho, M. Klobus, S.R. Wilson, J. Am. Chem. Soc. 118, 9249–9254 (1996)
I.G. Collado, A.J.M. Sánchez, J.R. Hanson, Nat. Prod. Rep. 24, 674–686 (2007)
K. Krohn, J. Dai, U. Flörke, H.J. Aust, S. Dräger, B. Schulz, J. Nat. Prod. 68, 400–405 (2005)
J.R. Anderson, L.E. Edwards, J. Chem. Soc. Perkin Trans. 1, 2185–2192 (1983)
W.F. Hendershot, C.W. Hesseltine, T.G. Pridham, R.G. Benedict, R.W. Jackson, Arch. Biochem. Biophys. 96, 166–170 (1962)
Y.S. Chen, Bull. Chem. Soc. Jpn 24, 372–381 (1960)
S. Halecker, F. Surup, E. Kuhnert, K.I. Mohr, N.L. Brock, J.S. Dickschat, C. Junker, B. Schulz, M. Stadler, Phytochemistry 100, 86–91 (2014)
E. Kuhnert, J. Fournier, D. Peršoh, J.J. Luangsa-ard, M. Stadler, Fungal Divers. 64, 181–203 (2014)
M. Stadler, T. Læssøe, J. Fournier, C. Decock, B. Schmieschek, H.V. Tichy, D. Peršoh, Stud. Mycol. 77, 1–143 (2014)
F. Surup, B. Thongbai, E. Kuhnert, E. Sudarman, K.D. Hyde, M. Stadler, J. Nat. Prod. 78, 934–938 (2015)
Acknowledgments
We thank Andreas Glatt for his preliminary work on the optimization of secondary metabolite production with talcum powder, Wera Collisi for conducting the bioassays, Philine Wotsch and Anke Skiba for assistance in the mycological lab, and Christel Kakoschke for recording NMR spectra. For HRESIMS measurements we are grateful to Aileen Teichmann and Heinrich Steinmetz. Jacques Fournier is gratefully acknowledged for sending us the fungal material. His collection trip in Martinique has benefited from “Investissements d’Avenir” Grants of the ANR (CEBA ANR-10-LABX-0025, CNRS Cayenne and Toulouse).
Conflict of interest
The authors declare no conflicts of interests.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Surup, F., Kuhnert, E., Liscinskij, E. et al. Silphiperfolene-Type Terpenoids and Other Metabolites from Cultures of the Tropical Ascomycete Hypoxylon rickii (Xylariaceae). Nat. Prod. Bioprospect. 5, 167–173 (2015). https://doi.org/10.1007/s13659-015-0065-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-015-0065-3