
Abstract
One new eudesmane sesquiterpenoid (1) named ecdysantherol A and two new benzene derivatives ecdysantherols B (2) and C (3), together with five known benzene derivatives (4–8) were isolated from the stems of Ecdysanthera rosea. The structures of the new compounds were elucidated by extensive spectroscopic methods and X-ray diffraction. The known compounds were identified by the comparison of their spectroscopic data with reported literature data. Compound 1 showed moderate antibacterial activity against the Providensia smartii with MIC value of 12.5 μg/mL.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The Ecdysanthera comprises 15 species. Of which Ecdysanthera rosea is mainly distributed in tropical and subtropical areas of Asia and used as a traditional Chinese medicinal plant for the treatment of sore throat, chronic nephritis and trauma in China [1]. Terpenoids, benzene derivatives, steroids and their glycosides have been previously reported in this plant, they include three terpenoids and one steroid saponin with cytotoxic activities [2–12].
Given that the chemical constituents isolated from E.rosea are still limited and the existing bioactivity research of them are not related to its medicinal use directly. This attracted our attention to searching for more novel natural products from it. The present chemical investigation led to the isolation of three new compounds (1–3) (Fig. 1), and five known compounds: manglieside D (4) [13] erythro-guaiacylglycerol-β-O-4′-coniferyl alcohol (5) [14], (+)-(7S,8R)-guaiacylglycerol (6) [15], isocopoletin (7) [16], evofolin-B (8) [17] from this plant. In addition, preliminary test showed that compound 1 was a moderate antibacterial constituent against Providensia smartii with MIC value of 12.5 μg/mL, but a weak antibacterial constituent against Enterococcus faecalis and Staphylococcus aureus with MIC value of 50 μg/mL and 50 μg/mL respectively. In this paper, we report the isolation and structure elucidation of the new compounds.

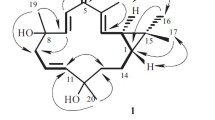
Chemical structures of compounds 1–4 and the standard chemicals
2 Results and Discussion
The molecular formula of compound 1 was determined to be C15H22O3 by the HREIMS at m/z 250.1579 [M]+ (calcd. 250.1569), indicating five degrees of unsaturation. The 13C NMR and DEPT spectroscopic data exhibited 15 carbon signals for three methyls at δC 26.8 (q), 27.6 (q), 21.4 (q), four methylenes at δC 22.9 (t), 22.6 (t), 32.7 (t), 117.0 (t), four methines at δC 57.8 (d), 59.4 (d), 37.7 (d), 43.2 (d), and four quaternary carbons at δC 143.2 (s), 46.9 (s), 71.7 (s), δC 209.7 (s). Except for one ketone and a pair of double bond, it’s suggested that there should be a tricyclic structure in compound 1 to fit the three degrees of unsaturation. The 1H NMR spectrum displayed two olefinic protons at δH 5.68 (1H, d, J = 2.1 Hz, H-15a), 5.36 (1H, d, J = 2.1 Hz, H-15b), in accordance with a terminal double bond at δC 117.0 (t) in 13C NMR spectrum which was supported by the HSQC experiment. The protons at δH 3.42 (1H, d, J = 3.9 Hz, H-2) and δH 3.99 (1H, d, J = 3.9 Hz, H-3) with the same coupling constant indicated an epoxy moiety which was also supported by the 1H-1H COSY correlation between them (Fig. 2). The HMBC correlations from the singlet methyl signal at δH 1.21 (3H, s, H-14) to δC 209.7 (C-1), δC 37.7 (C-5), δC 32.7 (C-9), δC 46.9 (C-10); from the proton at δH 2.73 (H-5) to δC 143.2 (C-4), δC 117.0 (C-15), δC 43.2 (C-7), δC 22.9 (C-6) and from the terminal methyl signals at δH 1.15 (3H, s, H-12) and δH 1.15 (3H, s, H-13) to δC 71.7 (C-11) and δC 43.2 (C-7), together with the 1H-1H COSY correlations between δH 1.63 (H-6a) and δH 2.73 (H-5), δH 1.66 (H-7) proposed an eudesmane sesquiterpenoid skeleton of compound 1. In addition, single-crystal X-ray diffraction (Fig. 2) using anomalous scattering of CuKα radiation (CCDC 1006467) revealed the absolute configuration of 1 as (2R,3S,5S,7S,10S)-2,3-epoxy-eudesm-4(15)-en-11-ol-1-one. Thus, compound 1 was elucidated as shown in Fig. 1, and named ecdysantherol A.

Selected 1H-1H COSY ( ), HMBC correlations (→) and the X-ray structure of 1
), HMBC correlations (→) and the X-ray structure of 1
Compound 2 was isolated as a yellow powder. Its molecular formula C25H32O11 was deduced by the positive HR-ESIMS m/z 531.1841 [M + Na] +. The NMR of 2 were very similar to the known compound manglieside D [13]. By comparison of the NMR data in literatures, the same structure segments of a 1,3,5-trisubstituted aromatic ring, a disubstituted E-configuration double bond and a sugar unit were confirmed [18, 19]. The major difference was that compound 2 possessed a different 1′,3′,4′, 5′-tetrasubstituted aromatic ring, in which a methoxy group at C-3′ of manglieside D was replaced by a hydroxy group and this could be confirmed by its molecular formula and different proton signals at δH 6.81 (1H, d, J = 1.8 Hz, H-2′), δH 6.83 (1H, d, J = 1.8 Hz, H-6′). Therefore, compound 2 was elucidated as shown in Fig. 1, and named ecdysantherol B Fig. 1.
Confusingly, one literature neglected coupling constant and assigned same coupling pattern as 3-OCH3 and 4-OH substituted aromatic ring, because NOE correlation of methoxyl proton with only one aromatic proton (C-2) was observed in ROESY spectrum, which deduced a substituted aromatic carbon (C-4) [20]. Interestingly, same NOE correlation pattern was observed in ROESY spectrum of 2. Then, 1H NMR spectral data of compound 2 were further collected in different solvent, and the result indicated same coupling pattern without large coupling constant to meet Ortho-proton in aromatic ring. To further confirmed our assignment, we ordered standard chemicals of 3-methoxy-5-methylpehnol (CAS NO. 3209-13-0), and 4-ethyl-2-methoxyphenol (CAS NO. 2785-89-9), and 1H NMR spectral data of two compounds were record in DMSO-d6. The same coupling pattern of compound 2 and 3-methoxy-5-methylpehnol unambiguously confirmed 1,3,5-trisubstituted aromatic ring and proposed that no observation of NOE correlation between –OCH3 with both aromatic protons (H-2 and H-4) was not sufficient reason for 1,3,4-trisubstituted assignment. Besides, chemical shift of –OH in DMSO-d6 might be a characteristic for 3-methoxy-4-hydroxy substituted (ca δH 8.7) and 3-methoxy-5-hydroxy substituted (ca δH 9.3) in aromatic ring.
Compound 3 was isolated as a yellow powder. The molecular formula C27H34O16 was deduced by HRESIMS at m/z 614 [M + Na]+. Detailed analysis of NMR data indicated that 3 had a similar structure to that of the known compound previously reported, [21] except for the substituent groups on the aromatic ring. By comparison of its NMR data with those reported in literature, the signals at δH 7.34 (1H, s, H-3′′′), 7.34 (1H, s, H-7′′′) showed that 3 had a different 1,3,4,5-tetra-substituted aromatic ring. In addition, the HMBC correlations from δH 3.77 (3H, s, -OMe) to 149.1 (C-3) suggested it had another different 1,2,4-tri-substituted aromatic ring with the known compound. The above observations indicated that compound 3 was an analogue of the known compound. Furthermore, the detailed 2D NMR spectroscopic data revealed the position of the hydroxy groups and methoxy groups in compound 3. Thus, compound 3 was elucidated as shown in Fig. 1, and named ecdysantherol C.
3 Experimental Section
3.1 General Experimental Procedures
Optical rotations were obtained with a Jasco P-1020 Automatic Digital Polariscope. UV spectrum was measured with a Shimadzu UV2401PC in MeOH solution. IR spectra (KBr) were obtained on a Bruker tensor-27 infrared spectrophotometer. 1H, 13C, and 2D NMR spectra were recorded on a Bruker AM-400, a DRX-500 NMR and an Avance III 600 spectrometer with TMS as internal standard. MS data were obtained on a Waters Autospec Premier P776 for HREI. An APEX DUO (Bruker) instrument was used for the single crystal X-ray diffraction. Column chromatography (CC) was performed on Silica gel (200–300 mesh, Qingdao Marine Chemical Ltd., Qingdao, People’s Republic of China) and RP-18 gel (20–45 µm, Fuji Silysia Chemical Ltd., Tokyo, Japan). Fractions were monitored by TLC (GF 254, Qingdao Haiyang Chemical Co., Ltd., Qingdao, People’s Republic of China), and spots were visualized by 10 % H2SO4-ethanol reagent.
3.2 Plant Material
The dried stems of E. rosea were collected from Xishuangbanna Autonomous Prefecture, Yunnan Province, People’s Republic of China, and identified by Jingyun Cui of Xishuangbanna Botanic Garden. A voucher specimen (Cui 200811-03) has been deposited at the Herbarium of Kunming Institute of Botany, Chinese Academy of Sciences.
3.3 Extraction and Isolation
The air-dried and smashed stems of E. rosea (10 kg) were extracted with MeOH three times at room temperature. After in vacuum pump evaporation of the solvent, the combined crude extract was suspended in H2O and extracted with ethyl acetate three times. The EtOAc fraction (129.0 g) was eluted with gradient mixtures of CHCl3–MeOH (100:1 → 1:1) on silica gel column to yield 5 fractions [Fr.A (38.5 g), Fr.B (13 g), Fr.C (14 g), Fr.D (9 g), Fr.E (18 g)]. Fraction B (13 g) was isolated by Sephadex LH-20 and repeated silica gel column to yield compound 1 (11 mg). Fraction D (9 g) was chromatographed over Sephadex LH-20 (MeOH), MPLC (MeOH–H2O) and HPLC (MeCN–H2O) to provide compound 5 (7 mg). Fraction E (18 g) was subjected to the Sephadex LH-20, eluted with MeOH–H2O (1:1) and chromatographed over RP-C18 gel (MeOH–H2O ) to afford compounds 2 (6 mg), 3 (5 mg), 4 (17 mg), 6 (14 mg), 7 (14 mg) and 8 (8 mg).
Compound 1; colorless needle crystal; [α] 21.5D + 67.0 (c 0.1, MeOH); IR (KBr) νmax 3360, 1635 cm−1; 1H (400 MHz) and 13C NMR (100 MHz) data (MeOH), see Table 1, HREIMS m/z 250.1579 (calcd for C15H22O3, 250.1569).
Compound 2; yellow, amorphous powder; [α] 21.3D + 28.3 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 245.6 (4.12), 203.0 (4.65) nm; IR (KBr) νmax 3425, 2985, 1029 cm−1; 1H (400 MHz) and 13C NMR (100 MHz) data, see Table 1, HREIMS m/z 531.1851 (calcd for C25H32O11Na, 531.1842).
Compound 3; yellow, amorphous powder; [α] 22.3D – 64.1 (c 0.15, MeOH); UV (MeOH) λmax (log ε) 284.2 (4.05), 202.8 (4.57) nm; IR (KBr) νmax 3431, 1049 cm−1; 1H (400 MHz) and 13C NMR (100 MHz) data, see Table 1, HREIMS m/z 637.1754 (calcd for C27H34O16Na, 637.1745).
3.4 Antimicrobial assays
The microorganisms used in the antimicrobial assay were obtained from the American Type Culture Collection (ATCC). They included three bacteria strains: E. faecalis ATCC 10541, S. aureus ATCC 25922 and Providensia smartii ATCC29916. The MIC values of the compounds were determined by the broth microdilution method in 96-well microtitre. The 96-well plates were prepared by dispensing into each well 100 μL of Mueller–Hinton broth for bacteria. The test substances were initially prepared in 10 % DMSO in broth medium at 400 μg/mL for compounds or 50 μg/mL for the reference antibiotics, gentamycin. A volume of 100 μL of each test sample was added into the first wells of the microtitre plate (whose wells were previously loaded with 100 μL of broth medium). Serial two-fold dilutions of the test samples were made and 100 μL of bacterial inoculum standardized at 106 CFU/mL were added. This gave final concentration ranges from 100 to 0.781 μg/mL for the compounds and 12.5 to 0.097 μg/mL for reference substance. The plates were sealed with parafilm, then agitated with a plate shaker to mix their contents and incubated at 35 °C for 24 h.
MICs were determined upon addition of 50 μL (0.2 mg/mL) p-iodonitrotetrazolium chloride (INT, Sigma-Aldrich, South Africa). Viable bacteria reduced the yellow dye to a pink color. The MIC corresponded to the lowest well concentration where no color turbidity change was observed, indicating no growth of microorganism. All tests were performed in triplicates.
3.5 Crystallographic Data of 1
C15H22O3, M = 250.33, orthorhombic, a = 5.85670(10) Å, b = 11.1899(2) Å, c = 19.6555(3) Å, α = 90.00°, β = 90.00°, γ = 90.00°, V = 1288.14(4) Å3, T = 100(2) K, space group P212121, Z = 4, μ(CuKα) = 0.706 mm−1, 7204 reflections measured, 2162 independent reflections (R int = 0.0345). The final R 1 values were 0.0346 (I > 2σ(I)). The final wR(F2) values were 0.1028 (I > 2σ(I)). The final R 1 values were 0.0347 (all data). The final wR(F2) values were 0.1029 (all data). The goodness of fit on F2 was 1.130. Flack parameter = 0.1(2). The Hooft parameter is 0.06(6) for 852 Bijvoet pairs. The crystal structure of compound 1 was solved by direct method SHELXS-97 and expanded using the difference Fourier techniques, refined by the program SHLXL-97 and the full-matrix least-squares calculations. Crystallographic data for the structure of compound 1 have been deposited with the Cambridge Crystallographic data centre (deposition no. CCDC 1006467). Copies of these data can be obtained free of charge via www.ccdc.cam.ac.uk.
References
Y. Jiang, B.T. Li, Flora of China, vol. 63 (Science Press, Beijing, 1977), p. 234
P. Luger, M. Weber, N.X. Dung, P.T. Ky, L. Chinh, Acta. Crystallogr. 52, 1574–1576 (1996)
D.K. Chu, T.C. Le, T.K. Pham, Tap. Chi. Duoc. Hoc. 4, 13–16 (1997)
P. Luger, M. Weber, X.D. Nguyen, T.K. Pham, Cryst. Res. Technol. 33, 325–332 (1998)
T.K. Pham, T.C. Le, Tap. Chi. Duoc. Hoc. 5, 14–16 (1997)
C. Le, T. Nguyen, T.K. Pham, H. Trinh, X.D. Nguyen, P. Luger, Tap. Chi. Duoc. Hoc. 4, 16–17 (1995)
X.D. Zhu, Q.H. Zhang, F. Wang, Y.J. Ye, Zhongcaoyao 42, 237–240 (2011)
K.F. Huang, M.L. Sy, J.S. Lai, J. Chin. Chem. Soc. 37, 187–189 (1990)
X.D. Zhu, Q.H. Zhang, L. Kong, F. Wang, S.D. Luo, Fitoterapia 81, 906–909 (2010)
F.Q. Xu, H.Y. Liu, C.X. Chen, H.M. Zhong, Chem. Nat. Compd. 44, 308–316 (2008)
F.Q. Xu, H.Y. Liu, F. Teng, C.X. Chen, H.M. Zhong, Tianran Chanwu Yanjiu Yu Kaifa 19, 365–368 (2007)
X.D. Zhu, G.S. Wu, J.Y. Xiang, H.R. Luo, S.D. Luo, H.M. Zhu, Fitoterapia 82, 632–636 (2011)
V.K. Phan, D.T. Mai, V.D.T. Le, H.T. Nguyen, N.H. Nguyen, Chem. Pharm. Bull. 56, 1270–1275 (2008)
L. Lyia, P.S. Navindra, J. Agric. Food. Chem. 58, 11673–11679 (2010)
D.G. Marina, F. Antonio, M. Pietro, P. Lucio, Synth. Commun. 28, 3693–3700 (1998)
W.J. Liang, Q.Y. Ma, H.Z. Jiang, J. Zhou, J. Pang, Y.X. Zhao, Chin. Tradit. Herb. Drugs 42, 1271–1275 (2011)
T.S. Wu, J.H. Yeh, P.L. Wu, Phytochemistry 40, 121–124 (2011)
G. Fang, C.P. Tang, Y. Yang, Helv. Chim. Acta 91, 1023–1030 (2008)
Q.A. Zheng, M. Xu, C.R. Yang, D. Wang, H.Z. Li, H.T. Zhu, Y.J. Zhang, Nat. Prod. Bioprospect. 2, 111–116 (2012)
S. Britta, S. Silke, H. Josef, K. Nasser, H. Sonja, M. Christa, J. Nat. Prod. 65, 1479–1485 (2002)
W. Tsutomu, N. Yoshimi, N. Tadataka, Chem. Pharm. Bull. 54, 14–20 (2006)
Acknowledgments
This project was supported by the National Natural Science Foundation of China (81225024), the National Science and Technology Support Program of China (2013BAI11B02).
Conflict of Interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
This article is published under license to BioMed Central Ltd. Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Song, CW., Lunga, PK., Qin, XJ. et al. Chemical Constituents from the Stems of Ecdysanthera rosea. Nat. Prod. Bioprospect. 4, 319–323 (2014). https://doi.org/10.1007/s13659-014-0041-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-014-0041-3