
Abstract
Honey bees (Apis mellifera) are an important social pollinator, crucial in maintaining ecological balance and biodiversity. The haploid-diploid sex determination system determines their sexes where males are haploids formed from unfertilized eggs while females are diploids formed from fertilized eggs. To better understand the gonad differentiation mechanism in honey bees, here the transcriptome difference between the honey bee ovary and testis were analyzed using the Illumina sequencing technology. A total of 4380 differentially expressed genes (DEGs) were identified, with 2255 up-regulated and 2125 down-regulated in the ovary compared to the testis. Among these, many candidate DEGs related to sex determination, spermatogenesis, and oogenesis were identified, such as csd, fem, and dsx. Moreover, 3092 differentially expressed alternative splicing events (ASEs) related to 1497 genes, such as tra2, ovo, and squid, were identified between the ovary and testis. The findings of this study provide valuable information on gene expression for understanding the underlying molecular mechanisms of gonad differentiation and gametogenesis in A. mellifera.
Similar content being viewed by others
1 Introduction
As important social-economic insects, honey bees (Apis mellifera L.) are a source of several products for human consumption while serving as important pollinators in nature. In colonies, the queens and worker bees are females, while drones are males. The sex of honey bees is determined by the haploid-diploid system, where males are haploids formed from unfertilized eggs while females are diploids resulting from fertilized eggs. The queens and drones have fully developed gonads, and their sexual maturity is asynchronous; after emergence, the queens need 5–10 days to reach sexual maturity (Chen, 2001), and the drones need 9–12 days (Bishop, 1920).
In animals, the sexual development process entails sex determination and differentiation, which not only leads to morphological differences in reproductive organs between male and female, but also affects many other aspects of the two sexes, such as behavior and physiology. In honey bees, sex is determined by the heterozygosity of a single complementary sex determiner (csd) which was identified by linkage analysis of genetic map (Beye et al., 2003). When the embryo is heterozygous at the csd locus, it develops into a fertile female; when the embryo is hemizygous at the csd locus, it develops into a fertile males; when the embryo contains two identical cds alleles, it develops into a diploid male (Beye et al., 2003). csd directs the female-specific splicing of feminizer (fem) gene, which then controls the female-specific splicing of Amdoublesex (Amdsx) gene, a conserved sex-determining gene at the end of the sex determination cascade in multicellular animals (Whiting, 1943). Therefore, the csd > fem > Amdsx genetic cascade controls the development of honey bee sexes, subsequently leading to sex-specific phenotypes (Beye et al., 2003; Cho et al., 2007; Hasselmann et al., 2008; Gempe et al., 2009).
Genes in the main sex determination cascade of honey bee have been identified (Beye et al., 2003; Hasselmann et al., 2008; Gempe et al., 2009; Cho et al., 2007). However, the molecular aspects of their reproductive activity and gonad development remain unknown. Therefore, in the present study, we performed comparative transcriptome sequencing analysis on the A. mellifera ovary and testis. The findings revealed major expression differences between the male and female gonads. These findings will increase the understanding of the underlying molecular mechanism in gonad differentiation, which provides valuable knowledge on gene expression for future research.
2 Material and methods
2.1 Insects
The honey bees (A. mellifera ligustica) were reared under natural conditions at the Honeybee Research Institute of Jiangxi Agricultural University (28°46′N, 115°49′E), Nanchang, Jiangxi, China.
The queens used for collecting ovaries were bred from two A. mellifera colonies. Briefly, the queens of the two colonies were respectively restricted to a worker cell comb for 12 h to lay fertilized eggs, after which the comb was transferred to the super box of the same colony until the eggs hatched. The 1-day old larvae of similar size were grafted to the queen cells using a grafting needle and then were placed in the super box. After emergence, the newly emerged queens were kept in queen cages and then transferred to their original colonies. The queen’s ovaries were dissected under a stereomicroscope (Guiguang, GL-99TI, Guilin, China) after 12 days, then frozen immediately in liquid nitrogen. Ovaries from two queens in each colony were collected as a sample, and two biological replicates were sampled.
At the same time, testis samples were collected from drones bred by the same two colonies. For this, the queen in each colony was restricted in a drone foundation to lay unfertilized eggs for 12 h. The queen was then released, and the comb was transferred to the super box in the same colony until the eggs hatched. A day before drone emergence, the comb was transferred to an incubator regulated at 34.5 °C and relative humidity of 70%. After emergence, the drones were marked by coloring the back of their thorax and returned to their original colony. The drones reached sexual maturity after 12 days (Graham, 2015), and they were captured. Their testes were dissected under a stereomicroscope and then were immediately frozen in liquid nitrogen. Testes collected from ten drones in each colony were pooled as a sample, and two biological replicates were sampled.
2.2 cDNA library construction and sequencing
Total RNAs were extracted from the ovary and testis samples using Trizol Reagent (US Patent No.5,346,994). The extracted total RNA purity (OD260/280), concentration, nucleic acid absorption peak, and RNA integrity were detected using NanoDrop 2000 (Thermo Fisher Scientific, Wilmington, DE) and Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). The mRNAs were then enriched using magnetic beads with oligo (dT) while being randomly interrupted in fragmentation buffer to construct the sequencing libraries. The first cDNA strands were synthesized using random hexamers and M-MuLVreverse transcriptase, while the second was synthesized using enzyme buffer, dNTPs, RNase H, and DNA polymerase I. The synthesized cDNAs were purified using AMPure XP beads (Beckman Coulter, USA) then subjected to end repairing, poly (A) tailing, and addition of sequencing adaptors. cDNA fragments of 240 bp in length were preferentially selected using AMPure XP beads, and the cDNA libraries were constructed by PCR amplification, enriched, and sequenced using the Illumina Hiseq 4000 platform (Illumina, CA, USA).
2.3 Mapping of sequencing data to the reference genome
Raw sequences were filtered using the FastQC software to remove reads containing just adaptors and low-quality reads (reads containing “N” more than 10%; reads containing bases with mass value Q ≤ 10 accounted for more than 50% of the whole read). The clean reads were submitted to the NCBI SRA database under the accession numbers SRR15276287, SRR15276288, SRR15276289, and SRR15276290.
Cleaned reads were aligned to the A. mellifera genome version Amel_HAv3.1 (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/003/254/395/GCA_003254395.2_Amel_HAv3.1/) using the HISAT2 software (Kim et al., 2015).
2.4 Gene expression level determination and standardization
The fragments per kilobase of script per million fragments mapped (FPKM) was used to measure gene expression using the cuffquant and cuffnorm components of the Cufflinks software (Trapnell et al., 2012). The FPKM was calculated as follows:
where the cDNA fragments represent the number of reads mapped to a transcript, mapped fragments (Millions) represent the total number of reads mapped to the transcripts, and transcript length (kb) represents the length of each transcript.
2.5 Screening of DEGs and ASEs
The DESeq software was used to identify differentially expressed genes between the ovary and testis. The Benjamini–Hochberg method was adopted to correct the p values, which is indicated as FDR (corrected p values). The |log2(fold change)|≥ 1 and FDR < 0.01 were used as the DEG screening criteria. Gene Ontology (GO) enrichment analysis of functional significance was then performed by mapping all DEGs to the GO database (Harris et al., 2004). Finally, pathway analysis was performed in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Kanehisa and Goto, 2000).
The cleaned reads were assembled into longer transcripts using the StringTie software (Pertea et al., 2015). The ASprofile software (Florea et al., 2013) determined the alternative splicing types in each gene and their corresponding expression levels. Differentially expressed ASEs were identified using rMATS software (Shen et al., 2014) following the |Δψ|> c (c = 0.0001) and FDR < 0.05 criteria.
2.6 Quantitative real-time PCR (qRT-PCR)
To perform quantitative PCR experiment, we bred many queens and drones. Then, ovary and testis samples were collected from 12-day-old queens and drones, respectively. Their total RNAs were extracted, and the RNA integrity was detected by agarose gel electrophoresis. In addition, the RNA concentration and purity (OD260/280 value) were detected using an ultraviolet spectrophotometer. The RNA samples (1 μg/μL) were reverse transcribed into cDNA using the MLV reverse transcriptase kit (Invitrogen, CA, USA). Nine DEGs (squid, rbp1, tra2, vgr, vasa, dicer1, fem, stat92e, dpp) were selected for qRT-PCR analysis using A. mellifera gapdh gene as an internal reference. The primer sequences (Table S1) were designed based on the mRNA sequences from the GenBank database using the Prime Primer 5.0 software. The qRT-PCR reaction system consisted of 5 μL of SYBR®Premix Ex Taq™ II, 0.2 μL of ROX correction fluid, 0.4 μL each of forward and reverse primers, 1 μL of cDNA, and 3 μL of H2O. The PCR conditions were as follows: 95 °C, 5 min; 94 °C, 2 min; 40 cycles (95 °C, 10 s, Tm, 15 s, 72 °C, 15 s); 72 °C, 10 min. To establish the melting curve of the qRT-PCR product, the primers were heated slowly with a gradual increase of 1 °C every 5 s from 72 to 99 °C. The specificity of the PCR products was analyzed by dissolution curve. For each gene, eight biological replicates were adopted, with four technical repeats each. The data were analyzed by 2−△△CT (Schmittgen and Livak, 2008). Significant differences were analyzed by t test with a cutoff value of 0.05 using the SPSS17.0 software (SPSS Inc., 2008).
3 Results and discussion
3.1 Transcriptome sequencing of ovary and testis
Four RNA-seq libraries of A. mellifera ovary and testis were generated from two biological replicates, each. A total of 35,310,578–40,246,018 high-quality reads were obtained after removing the sequencing adaptors and low-quality reads (Table I). The total length of reads in each sample was 4,443,325,050–5,062,141,958nt, with a percentage of Q30 bases more than 94.29%, implying that the base sequencing and identification were reliable and accurate. Approximately 30,660,123–34,952,594 (82.58–89.47%) of the cleaned reads mapped to the A. mellifera genome version Amel_HAv3.1.
3.2 Repeated correlation assessment
The correlation heat map between gene expression of the paired biological replicates of ovary and testis revealed that the r2 values between two different biological replicates were more than 0.88 and between ovary and testis were less than 0.1, suggesting high reliability and repeatability of the RNA-seq data (Figure S1).
3.3 Differentially expressed genes between the ovary and testis
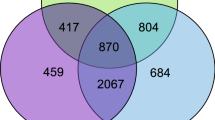
A total of 4380 DEGs between the ovary and testis, including 2255 up-regulated genes in the ovary and 2125 in the testis, were identified (Figure 1). The top three up-regulated genes in the ovary were odorant-binding protein 7 (LOC677668), two-pore potassium channel protein sup-9 (LOC413020), and cilia- and flagella-associated protein 58 (LOC412109). In the testis, hypothetical protein WN51_08187 (LOC113219380), carboxypeptidase B-like (LOC551327), and galactose-1-phosphate uridylyltransferase (LOC724540) were the top three up-regulated genes (Table S2).

Up- and down-regulated DEGs in the ovary compared to the testis in 12-day-old honey bees (Apis mellifera). Both ovary and testis contain two biological replicates
In addition, GO enrichment of the DEGs showed that eight terms were significantly enriched, including transmembrane transport (GO:0,055,085), oxidation–reduction process (GO:0,055,114), potassium ion transmembrane transport (GO:0,071,805), regulation of transcription, DNA-templated (GO:0,006,355), integral component of membrane (GO:0,016,021), and transcription factor activity, sequence-specific DNA binding (GO:0,003,700) (q < 0.05; Table S3). The KEGG pathway analysis revealed that three signaling pathways, namely ECM-receptor interaction (KO04512), lysosome (KO04142), and glycosaminoglycan degradation (KO00531), were significantly enriched (q < 0.05; Table S4). The ECM-receptor interaction pathway is implicated in cell migration, proliferation, follicle growth, and oocyte maturation (Berkholtz et al., 2006).
Among the up-regulated genes in the ovary, ten GO terms and two KEGG pathways were significantly enriched (q < 0.05; Tables S3 and S4). However, the testis up-regulated genes significantly enriched eight GO terms and 17 KEGG pathways (q < 0.05; Tables S3 and S4); of them, the ascorbate and aldarate metabolism (ko00053) is the most relevant pathway to the caput epididymis and may play important roles in sperm maturation (Hu et al., 2018).
3.4 DEGs involved in sex determination
Five DEGs related to sex determination in honey bee or Drosophila were identified, including csd (log2(testis/ovary) = 4.74), fem (log2(testis/ovary) = 4.92), dsx (log2(testis/ovary) = 2.44), tra2(log2(testis/ovary) = -2.07), and rbp1 (log2(testis/ovary) = − 1.06), with GenBank accession numbers LOC406074, LOC724970, LOC725126, LOC725195, and LOC413835. The csd, fem, and dsx were up-regulated in the testis, while tra2 and rbp1 were up-regulated in the ovary (Figure 2).

Heatmap of eighteen candidate DEGs involved in sex determination and development of gonads and germ cells. X-axis indicates the names of the samples. Y-axis shows the names of the 18 genes and their functional classifications
The csd gene is the initial signal for honey bee sex determination (Beye et al., 2003), which is realized through heterozygosity of csd gene. When csd is heterozygous, the embryo develops into a female; when csd is homozygous or hemizygous, the embryo develops into a male (Beye et al., 2003). The csd gene has many alleles among the bee populations hence subjected to balance selection (Hasselmann and Beye, 2004; Liu et al., 2011, 2012; Wang et al., 2012). Coming second in the honey bee sex determination cascade is fem, which maintains and stabilizes the female-specific development of embryos while regulating the female-specific splicing of its downstream gene dsx (Hasselmann et al., 2008; Gempe et al., 2009). The fem expression is also necessary for female gonad differentiation (Gempe et al., 2009). However, in this study these two genes are up-regulated in the testis; it is unknown whether they have function in honey bee testis.
dsx is a conservative gene expressed at the end of the sex determination cascade in insects. It controls the somatic cells’ sex identity and the germ cells’ development (Steinmann-Zwicky, 1994). Besides, in A. mellifera, dsx produces sex-specific proteins (DSX-F and DSX-M) through alternative splicing to regulate sex development (Cho et al., 2007). In addition, tra2 and rbp1 are important splicing factors regulating alternative splicing of dsx pre-mRNA in the sex determination pathway in Drosophila melanogaster (Burtis and Baker, 1989; Hoshijima et al., 1991). Moreover, the tra2 homologs have been successively isolated and linked to sex determination in other insects, such as A. mellifera (Nissen et al., 2012), Musca domestica (Burghardt et al., 2005), Ceratitis capitata (Marco et al., 2009), Bombyx mori (Suzuki et al., 2012), Tribolium castaneum (Shukla and Palli, 2013), and Sclerotia aquatilis (Nguantad et al., 2020). tra2 also regulates ovariole number and ovary size in Aedes albopictus (Li et al., 2019). In the present study tra2and rbp1 were significantly expressed higher in ovary than in testis, implying that they might be involved in ovary development or oogenesis in honey bees.
3.5 DEGs involved in oocyte development and oogenesis
Several genes involved in ovary development or oogenesis were up-regulated in the ovary, such as vgr, vasa, squid, ago3, aub, ovo, and otu with GenBank accession numbers LOC725920, LOC410692, LOC408936, LOC725111, LOC412427, LOC552100, and LOC100576989, respectively (Figure 2).
In insects, vgr codes for an ovary-specific protein that mediates the absorption of vitellogenin (vg) from the hemolymph by the developing oocytes in the ovaries (Tufail and Takeda, 2009). vgr expression is essential for reproductive development. For example, knocking down the vgr expression by RNAi in Nilaparvata lugens (Lu et al., 2015) and Haemaphysalis longicornis Neumann (Boldbaatar et al., 2008) led to abnormal development of oocytes and abnormal eggs. The vasa gene is essential for polar cell formation where the VASA protein binds to mRNAs of target genes involved in germ cell establishment and oogenesis, promoting their translation (Gavis et al., 1996; Tomancak et al., 1998; Styhler et al., 1998). For example, RNAi-based knockdown of vasa in Schistosoma mansoni led to a reduction in the ovary volume and a lower number of dividing cells in immature ovary (Skinner et al., 2020). The squid gene functions in nuclear export, cytoplasmic transport, and translational control of the gurken mRNA during oogenesis (Norvell et al., 1999). The aub and ago3 are germ cell PIWI-interacting RNA (piRNA) pathway components, essential in germline development, stem cell self-renewal, epigenetic regulation, and transposon silencing (Kennerdell et al., 2002; Malone and Hannon, 2009; Thomson and Lin, 2009). They form a “ping-pong” feed-forward piRNA amplification loop with PIWI in the Drosophila ovary, in which ago3 binds to a sense piRNA and guides cleavage of antisense transposon mRNAs to produce antisense piRNAs and in turn aub binds to an antisense piRNA and guides cleavage of sense transposon mRNAs to produce a new sense piRNA (Brennecke et al., 2007; Gunawardane et al., 2007). Therefore, the up-regulation of these genes in the ovary implied that they might be involved in ovary development or oogenesis, but it needs further experimental verification.
In Drosophila, ovo and otu genes are crucial in oogenesis’ early and late stages (Kennerdell et al., 2002; Oliver et al., 1987; Steinhauer and Kalfayan, 1992; Tirronen et al., 1995; Hinson et al., 1999). The ovo mutation leads to the early arrest of oogenesis and complete absence of germline (Oliver et al., 1987), while out mutation causes abnormal ovarian development (Steinhauer and Kalfayan, 1992; Tirronen et al., 1995). A higher expression of both in the ovary suggests a common function in honey bees and Drosophila.
3.6 DEGs involved in testis development and spermatogenesis
Several genes involved in testis development or spermatogenesis were up-regulated in the testis, including dpp, stat92e, dicer1, spata5, spata17, and muc1 with GenBank accession numbers LOC727101, LOC413742, LOC726766, LOC411003, LOC107964258, and LOC551924, respectively (Figure 2).
The dpp signaling regulates germline stem cell (GSC) self-renewal and differentiation by interacting with Birt-Hogg-Dubé syndrome (BHD) in fruit fly testis (Singh et al., 2006). The dicer1 enzyme is required for germ cell development and spermatogenesis in mammals (Björkgren and Sipilä, 2015). For example, in male mice, the absence of dicer1 results in infertility prolongs the time required for germ cells to enter the first prophase during meiosis from interphase and increases the apoptosis of primary spermatocytes (Yannick et al., 2011). stat92e activation promotes the male GSC establishment and maintenance in adult testes (Kiger et al., 2001; Tulina and Matunis, 2001) and male germ cell sex determination during gonad coalescence (Wawersik et al., 2005). SPATA5 and SPATA17 are members of the AAA-protein family, which consists of ATPase associated with diverse activities. spata5 is expressed at the early stages of spermatogenesis in mice (Liu et al., 2000). spata5 knockdown in male Nilaparvata lugens decreases the male accessory gland protein content and causes the malformation of vas deferens and seminal vesicle (Ge et al., 2016). Besides, SPATA17 is important in testis development and testis-specific apoptosis (Nie et al., 2011, 2013). MUC1 is a nonglobular transmembrane glycoprotein expressed in male germ cell line and ejaculated sperms (Martínez-Conejero et al., 2008), facilitating the separation of delayed cell maturation and splitting of spermatozoa into the lumen of the seminiferous tubules (Franke et al., 2001). Therefore, the up-regulation of these genes in the testis implied that they are involved in testis development or spermatogenesis in honey bees.
3.7 Differentially expressed alternative splicing events (ASEs)
Alternative splicing is essential in the sex determination of many insects, such as Drosophila where a regulatory cascade of X:A > sxl > tra > dsx determines sex through alternative splicing (Sánchez, 2008). This study analyzed 12 types of ASEs present in the ovary and testis. A total of 35,486 and 37,774 ASEs were detected in the ovary and testis, respectively. The alternative 5′ first exon (transcription start site) and alternative 3′ last exons (transcription terminal site) were the two most abundant ASE types in the ovary and testis (Figure 3). Additionally, 3092 differentially expressed ASEs between the ovary and testis related to 1497 genes were identified (Tables II and S5). We found that multiple DEGs mentioned above contain ASEs, such as tra2, ovo, squid, aub, muc1, and dicer1.

Statistical bar chart of ASEs detected in the ovary and testis. X-axis indicates the number of ASEs. Y-axis shows the 12 types of ASEs
The GO analysis of the differentially expressed ASEs related genes revealed that 341 GO terms were significantly enriched (q < 0.05, Table S6). Among them, oocyte anterior/posterior axis specification (GO:0,007,314) and oocyte microtubule cytoskeleton organization (GO:0,016,325) were related to oogenesis. However, only the WNT signaling pathway (ko04310) was significantly enriched (q < 0.05, Table S7) in the KEGG analysis. The WNT signaling pathway is implicated in gonad differentiation and development (Harwood et al., 2008; Golestaneh et al., 2009; Nicol and Guigen 2011). These findings imply that alternative splicing is important in A. mellifera gonad and germ cell development.
3.8 Validation of DEGs by qRT-PCR analysis
Nine DEGs were selected for the validation of the reliability of the RNA-seq results using qRT-PCR. All the nine genes showed significant expression differences between the ovary and testis (t test, p < 0.05, Table S8), with five being up-regulated in the ovary and four in the testis (Figure 4). This is consistent with the RNA-seq results, indicating that the transcriptome analysis findings were highly reliable.

Quantitative RT-PCR verification of nine DEGs between the ovary and testis. X-axis indicates the gene names. Y-axis shows the relative expression levels of genes.*represents significant difference
In summary, this study analyzed the transcriptome difference between the A. mellifera ovary and testis using RNA-seq. A large amount of DEGs and ASEs were identified, and many of them are related to sex determination, oogenesis, and spermatogenesis. These findings provide valuable information for understanding reproductive biology of honey bees.
Data availability
The clean reads for this study are available from the NCBI SRA database. Other data that support the results of this study are available from the corresponding author upon request.
References
Berkholtz C.B., Shea L.D. & Woodruff T.K. (2006) Extracellular matrix functions in follicle maturation. Seminars In Reproductive Medicine, 24(4), 262-269. https://doi.org/10.1055/s-2006-948555.
Beye M., Hasselmann M., Fondrk M.K., Page R.E. & Omholt S.W. (2003) The gene csd is the primary signal for sexual development in the honeybee and encodes an SR-type protein. Cell, 114(4), 419-429. https://doi.org/10.1016/s0092-8674(03)00606-8.
Bishop G.H. (1920) Fertilization in the honeybee, I. The male sexual organs: their histological structure and physiological functioning. Journal of Experimental Zoology, 31(2), 225–265.
Björkgren I. & Sipilä P. (2015) The role of Dicer1 in the male reproductive tract. Asian Journal Of Andrology, 17(5), 737-741. https://doi.org/10.4103/1008-682X.155542.
Boldbaatar D., Battsetseg B., Matsuo T., Hatta T., Umemiya-Shirafuji R., et al. (2008) Tick vitellogenin receptor reveals critical role in oocyte development and transovarial transmission of Babesia parasite. Biochemistry And Cell Biology, 86(4), 331-344. https://doi.org/10.1139/O08-071.
Brennecke J., Alexei A.A., Stark A., Dus M., Kellis M., et al. (2007) Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell, 128(6), 1089-1103. https://doi.org/10.1016/j.cell.2007.01.043.
Burghardt G., Hediger M., Siegenthaler C., Moser M., Dübendorfer A., et al. (2005) The transformer2 gene in Musca domestica is required for selecting and maintaining the female pathway of development. Development Genes And Evolution, 215(4), 165-176. https://doi.org/10.1007/s00427-004-0464-7.
Burtis K.C. & Baker B.S. (1989) Drosophila doublesex gene controls somatic sexual differentiation by producing alternatively spliced mRNAs encoding related sex-specific polypeptides. Cell, 56(6), 997-1010. https://doi.org/10.1016/0092-8674(89)90633-8.
Chen S. (2001) Honeybee biology in: The Apicultural Science in China. China Agricultural Press, 1st, p154–155.
Cho S., Huang Z.Y. & Zhang J. (2007) Sex-specific splicing of the honeybee doublesex gene reveals 300 million years of evolution at the bottom of the insect sex-determination pathway. Genetics, 177(3), 1733-1741. https://doi.org/10.1534/genetics.107.078980.
Florea L., Song L. &Salzberg, S.L. (2013) Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Research, 2, 188. 10.5281/zenodo.7068.
Franke F.E., Kraus S., Eiermann C., Pauls K., Lalani E.N., et al. (2001) MUC1 in normal and impaired spermatogenesis. Molecular Human Reproduction, 7(6), 505-512. https://doi.org/10.1093/molehr/7.6.505.
Gavis E.R., Lunsford L., Bergsten S.E. & Lehmann, R. (1996) A conserved 90 nucleotide element mediates translational repression of nanos RNA. Development, 122(9), 2791-2800. https://doi.org/10.1242/dev.122.9.2791.
Ge L.Q., Xia T., Huang B., Song Q.S., Zhang H.W., et al. (2016) Suppressing male spermatogenesis-associated protein 5-like gene expression reduces vitellogenin gene expression and fecundity in Nilaparvata lugens Stål. Scientific Reports, 6, 28111. https://doi.org/10.1038/srep28111.
Gempe T., Hasselmann M., Schiøtt M., Hause G., Otte M., et al. (2009) Sex determination in honeybees: two separate mechanisms induce and maintain the female pathway. PLos Biology, 7(10), e1000222. https://doi.org/10.1371/journal.pbio.1000222.
Golestaneh N., Beauchamp E., Fallen S., Kokkinaki M., Uren A., et al. (2009) Wnt signaling promotes proliferation and stemness regulation of spermatogonial stem/progenitor cells. Reproduction, 138(1), 151-162. https://doi.org/10.1530/REP-08-0510.
Graham B.M. (2015) The hive and the honey bee. Hamilton, Ilnois, USA: Dadant & Sons Press.
Gunawardane L.S., Saito K., Nishida K.M., Miyoshi K., Kawamura Y., et al. (2007) A slicer-mediated mechanism for repeat-associated siRNA 5' end formation in Drosophila. Science, 315(5818), 1587-1590. https://doi.org/10.1126/science.1140494.
Harris M.A., Clark J., Ireland A., Lomax J., Ashburner M., et al. (2004) The Gene Ontology (GO) database and informatics resource. Nucleic Acids Research, 32(Database issue), D258–261. https://doi.org/10.1093/nar/gkh036.
Harwood B.N., Cross S.K., Radford E.E., Haac B.E. & De Vries W.N. (2008) Members of the WNT signaling pathways are widely expressed in mouse ovaries, oocytes, and cleavage stage embryos. Developmental Dynamics, 237, 1099-1111. https://doi.org/10.1002/dvdy.21491.
Hasselmann M. & Beye M. (2004) Signatures of selection among sex-determining alleles of the honey bee. Proceedings of the National Academy of Sciences, 101(14), 4888-4893. https://doi.org/10.1073/pnas.0307147101.
Hasselmann M., Gempe T., Schiøtt M., Nunes-Silva C.G., Otte M., et al. (2008) Evidence for the evolutionary nascence of a novel sex determination pathway in honeybees. Nature, 454(7203), 519-522. https://doi.org/10.1038/nature07052.
Hinson S., Pettus J. & Nagoshi, R.N. (1999) Regulatory and functional interactions between ovarian tumor and ovo during Drosophila oogenesis. Mechanisms of Development, 88(1), 3-14. https://doi.org/10.1038/nature07052.
Hoshijima K., Inoue K., Higuchi I., Sakamoto H. & Shimura Y. (1991) Control of doublesex alternative splicing by transformer and transformer-2 in Drosophila. Science, 252(5007), 833-836. https://doi.org/10.1126/science.1902987.
Hu S.G., Liang A.J., Yao G.X., Li X.Q., Zou M., et al. (2018) The dynamic metabolomic changes throughout mouse epididymal lumen fluid potentially contribute to sperm maturation. Andrology, 6(1), 247-255. https://doi.org/10.1111/andr.12434..
Kanehisa M. & Goto S. (2000) Kegg: kyoto encyclopedia of genes and genomes. Nucleic acids research, 28(1), 27-30. https://doi.org/10.1093/nar/28.1.27.
Kennerdell J.R., Yamaguchi S. & Carthew R.W. (2002) RNAi is activated during Drosophila oocyte maturation in a manner dependent on aubergine and spindle-E. Genes & Development, 16(15):1884-9. https://doi.org/10.1101/gad.990802.
Kiger A.A., Jones D.L., Schulz C., Rogers M.B. & Fuller M.T. (2001) Stem cell self-renewal specified by JAK-STAT activation in response to a support cell cue. Science, 294(5551), 2542-2545. https://doi.org/10.1126/science.1066707.
Kim D., Langmead B. & Salzberg S.L. (2015) HISAT: a fast spliced aligner with low memory requirements. Nature Methods, 12(4) 357-360. https://doi.org/10.1038/nmeth.3317
Liu Y., Black J., Kisiel N. & Kulesz-Martin M.F. (2000) SPAF, a new AAA-protein specific to early spermatogenesis and malignant conversion. Oncogene, 19(12), 1579-1588. https://doi.org/10.1038/sj.onc.1203442.
Liu Z.Y., Wang Z.L., Wu X.B., Yan W.Y. & Zeng Z.J. (2011) csd alleles in the red dwarf honey bee (Apis florea, Hymenoptera: Apidae) show exceptionally high nucleotide diversity. Insect Science, 18, 645-651. https://doi.org/10.1111/j.1744-7917.2011.01437.x
Liu Z.Y., Wang Z.L., Yan W.Y., Wu X., Zeng Z.J. & Huang Z.Y. (2012) The sex determination gene shows no founder effect in the giant honey bee, Apis dorsata. PLoS One, 7(4), e34436. https://doi.org/10.1371/journal.pone.0034436.
Li X.C., Jin B.B., Dong Y.Q., Chen X.G., Tu Z.J. & Gu J.B. (2019) Two of the three Transformer-2 genes are required for ovarian development in Aedes albopictus. Insect Biochemistry and Molecular Biology, 109, 92-105. https://doi.org/10.1016/j.ibmb.2019.03.008.
Lu K., Shu Y.H., Zhou J.J., Zhang X.Y., Zhang X.Y., et al. (2015) Molecular characterization and RNA interference analysis of vitellogenin receptor from Nilaparvata lugens (Stål). Journal Of Insect Physiology, 73, 20-29. https://doi.org/10.1016/j.jinsphys.2015.01.007.
Malone C.D. & Hannon G.J. (2009) Small RNAs as guardians of the genome. Cell, 136(4), 656-668. https://doi.org/10.1016/j.cell.2009.01.045.
Marco S., Mark R., Benjamin A., Peter A., Lino C.P., et al. (2009) Ceratitis capitatatransformer-2 gene is required to establish and maintain the autoregulation of Cctra, the master gene for female sex determination. International Journal of Developmental Biology, 53(1), 109-120. https://doi.org/10.1387/ijdb.082681ms.
Martínez-Conejero J., Garrido N., Remohí J., Pellicer A., Simón C., et al. (2008) MUC1 in human testis and ejaculated spermatozoa and its relationship to male fertility status. Fertility & Sterility, 90(2), 450-452. https://doi.org/10.1016/j.fertnstert.2007.06.104.
Nicol B. & Guiguen Y. (2011) Expression profiling of Wnt signaling genes during gonadal differentiation and gametogenesis in rainbow trout. Sex Development, 5(6), 318-329. https://doi.org/10.1159/000334515.
Nie D., Liu Y. & Xiang Y. (2011) Overexpression a novel zebra fish spermatogenesis-associated gene 17 (SPATA17) induces apoptosis in GC-1 cells. Molecular Biology Reports, 38(6), 3945-3952. https://doi.org/10.1007/s11033-010-0511-6.
Nie D.S., Yu L., He J. & Xiang Y. (2013) Overexpression of human SPATA17 protein induces germ cell apoptosis in transgenic male mice. Molecular Biology Reports, 40(2), 1905-1910. https://doi.org/10.1007/s11033-012-2246-z.
Nissen I., Müller M. & Beye M. (2012) The Am-tra2 gene is an essential regulator of female splice regulation at two levels of the sex determination hierarchy of the honeybee. Genetics, 192(3), 1015-1026. https://doi.org/10.1534/genetics.112.143925.
Nguantad S., Chumnanpuen P., Thancharoen A., Vongsangnak W. & Sriboonlert A. (2020) Identification of potential candidate genes involved in the sex determination cascade in an aquatic firefly, Sclerotia aquatilis (Coleoptera, Lampyridae). Genomics, 112(3), 2590-2602. https://doi.org/10.1016/j.ygeno.2020.01.025.
Norvell A., Kelley R.L., Wehr K. & Schüpbach T. (1999) Specific isoforms of squid, a Drosophila hnRNP, perform distinct roles in Gurken localization during oogenesis. Genes & Development, 13(7), 864-876. https://doi.org/10.1101/gad.13.7.864.
Oliver B., Perrimon N. & Mahowald A.P. (1987) The ovo locus is required for sex-specific germ line maintenance in Drosophila. Genes & Development, 1(9), 913-923. https://doi.org/10.1101/gad.1.9.913.
Pertea M., Pertea G.M., Antonescu C.M., Chang T.C., Mendell J.T., et al. (2015) StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature biotechnology, 33(3), 290-295. https://doi.org/10.1038/nbt.3122.
Sánchez L. (2008) Sex-determining mechanisms in insects. International Journal of Developmental Biology, 52(7), 837-856. https://doi.org/10.1387/ijdb.072396ls.
Schmittgen T.D. & Livak K.J. (2008). Analyzing real-time PCR data by the comparative CT method. Nature Protocols, 3, 1101-1108. https://doi.org/10.1038/nprot.2008.73.
Shen S., Park J.W., Lu Z., Lin L., Henry M.D., et al. (2014) rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proceedings of the National Academy of Sciences of the United States of America, 111(51), E5593-5601. https://doi.org/10.1073/pnas.1419161111.
Shukla J.N. & Palli S.R. (2013) Tribolium castaneum Transformer-2 regulates sex determination and development in both males and females. Insect Biochemistry and Molecular Biology, 43(12), 1125-1132. https://doi.org/10.1016/j.ibmb.2013.08.010.
Singh S.R., Zhen W., Zheng Z., Wang H., Oh S.W., et al. (2006) The Drosophila homolog of the human tumor suppressor gene BHD interacts with the JAK-STAT and Dpp signaling pathways in regulating male germline stem cell maintenance. Oncogene, 25(44), 5933-5941. https://doi.org/10.1038/sj.onc.1209593.
Skinner D.E., Popratiloff A., Alrefaei Y.N., Mann V.H., Rinaldi G., et al. (2020) Functional analysis of vasa/PL10-like genes in the ovary of Schistosoma mansoni. Molecular and biochemical parasitology, 236, 111259. https://doi.org/10.1016/j.molbiopara.2020.111259.
Steinhauer W.R. & Kalfayan L.J. (1992) A specific ovarian tumor protein isoform is required for efficient differentiation of germ cells in Drosophila oogenesis. Genes & Development, 6(2), 233-243. https://doi.org/10.1101/gad.6.2.233.
Steinmann-Zwicky M. (1994) Sex determination of the Drosophila germ line: tra and dsx control somatic inductive signals. Development, 120(3), 707-716. https://doi.org/10.1534/genetics.107.078980.
Styhler S., Nakamura A., Swan A., Suter B. & Lasko P. (1998) vasa is required for GURKEN accumulation in the oocyte, and is involved in oocyte differentiation and germline cyst development. Development, 125(9), 1569-1578. https://doi.org/10.1007/s004290050153.
Suzuki M.G., Suzuki K., Aoki F. & Ajimura M. (2012) Effect of RNAi-mediated knockdown of the Bombyx mori transformer-2 gene on the sex-specific splicing of Bmdsx pre-mRNA. International Journal of Developmental Biology, 56, 693-699. https://doi.org/10.1387/ijdb.120049ms.
Thomson T. & Lin H. (2009) The biogenesis and function of PIWI proteins and piRNAs: progress and prospect. Cell & Developmental Biology, 25(1), 355-376. https://doi.org/10.1146/annurev.cellbio.24.110707.175327.
Tirronen M., Lahti V.P., Heino T.I. & Roos C. (1995) Two otu transcripts are selectively localised in Drosophila oogenesis by a mechanism that requires a function of the otu protein. Mechanisms of Development, 52(1), 65-75. https://doi.org/10.1016/0925-4773(95)00390-M.
Tomancak P., Guichet A., Zavorszky P. & Ephrussi A. (1998) Oocyte polarity depends on regulation of gurken by Vasa. Development, 125(9), 1723-1732. https://doi.org/10.1242/dev.125.9.1723.
Trapnell C., Roberts A., Goff L., Pertea G., Kim D., et al. (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols, 7(3), 562-578. https://doi.org/10.1038/nprot.2012.016.
Tufail M. & Takeda M. (2009) Insect vitellogenin/lipophorin receptors: molecular structures, role in oogenesis, and regulatory mechanisms. Journal of Insect Physiology, 55(2), 87-103. https://doi.org/10.1016/j.jinsphys.2008.11.007.
Tulina N. & Matunis E. (2001) Control of stem cell self-renewal in Drosophila spermatogenesis by JAK-STAT signaling. Science, 294(5551), 2546-2549. https://doi.org/10.1126/science.1066700.
Wang Z.L., Liu Z.Y., Wu X.B., Yan W.Y. & Zeng Z.J. (2012) Polymorphism analysis of csd gene in six Apis mellifera subspecies. Molecular Biology Reports, 39(3), 3067-3071. https://doi.org/10.1007/s11033-011-1069-7.
Wawersik M., Milutinovich A., Casper A.L., Matunis E., Williams B., et al. (2005) Somatic control of germline sexual development is mediated by the JAK/STAT pathway. Nature, 436(7050), 563-567. https://doi.org/10.1038/nature03849.
Whiting P.W. (1943) Multiple Alleles in Complementary Sex Determination of Habrobracon. Genetics, 28(5):365-82. https://doi.org/10.1093/genetics/28.5.365.
Yannick R., Oliver M., Papaioannou M.D., Béatrice C., Corinne G., et al. (2011) Dicer1 depletion in male germ cells leads to infertility due to cumulative meiotic and spermiogenic defects. PLos One, 6(10), e25241. https://doi.org/10.1371/journal.pone.0025241.
Acknowledgements
We thank Zhen-Xiu Zeng for providing help in honey bee colony breeding and sample collection.
Funding
This work was supported by the National Natural Science Foundation (No. 31402147, 31860686) and the Natural Science Foundation of Jiangxi province (No. 2018ACB21028, 20202BABL205007), China.
Author information
Authors and Affiliations
Contributions
ZLW conceived and designed the experiments. LXP, WWH, FPC, and XFH performed the experiments and analyzed the data. LXP and ZLW wrote the paper.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent for publication
All authors have read and approved the final manuscript for publication.
Conflict of interest
The authors declare no competing interests.
Additional information
Communicated by Zachary Huang.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
PAN, LX., HU, WW., CHENG, FP. et al. Transcriptome analysis reveals differentially expressed genes between the ovary and testis of the honey bee Apis mellifera. Apidologie 53, 15 (2022). https://doi.org/10.1007/s13592-022-00926-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13592-022-00926-5