
Abstract
Among invertebrates, cellular innate immunity is critical for wound healing and defense against parasites and pathogens. While the study of cellular immunity has received much attention in model insects, the study of hemocytes, including immune cells, in honey bees has received little attention. Much of our understanding of honey bee hemocytes is derived from a limited set of methodologies, predominately utilizing bright-field microscopy, which makes broad conclusions about honey bee cellular immunity difficult to infer. We build upon existing methodologies using differential cell staining, in vitro phagocytosis assays, and an analysis of respiratory burst activity as measured through reactive oxygen species (ROS) production. Further, we characterize the morphological diversity and functional capacity of honey bee hemocytes in both adult workers and young queen bees as well as the ontogeny of the hemocyte population across larval and adult stages of the worker caste. Our findings suggest that granulocytes are the major phagocytic cells in honey bees and that circulating larval granulocytes undergo mitotic cell division. Additionally, we demonstrate that ROS production in larval hemocytes can be stimulated with the protein kinase C activator, phorbol 12-myristate 13-acetate. This indicates the presence of a functional protein kinase C-dependent phagocyte oxidase system, though further experimentation is needed to confirm phagocyte oxidase as the source of ROS. Overall, this work expands our knowledge of honey bee hemocytes and provides additional methodological tools for studying immune mechanisms in insects.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
In recent years, notable advances in our understanding of honey bee immunology, pathology, and epidemiology have been made (Evans et al. 2006; Brutscher et al. 2015; Schwarz et al. 2015). Despite this progress, much of our current knowledge about honey bee host-pathogen interactions is derived from sequence-based genetic, transcriptomic, and metagenomic investigations (Weinstock et al. 2006; Runckel et al. 2011; Chen et al. 2014). As of yet, we know relatively little about honey bee immunity at the proteomic and cellular levels. While estimates have been made of the total abundance of circulating hemocytes over the honey bee life span (Schmid et al. 2008; Wilson-Rich et al. 2008), surprisingly little is known about the distinct immune cell subpopulations within honey bees and how these subpopulations differ between castes or how they change throughout development. Given the agricultural and ecological importance of honey bees, filling these knowledge gaps is an important goal for researchers seeking to understand the factors affecting bee health.
Recent studies on honey bee hemocytes have focused on characterizing cells using bright-field microscopy alone or alongside a limited number of complimentary functional assays (Schmid et al. 2008; Negri et al. 2013, 2014a, b). This approach is limited, however, as distinct hemocyte types may be difficult to distinguish through microscopy. Further, the nomenclatural conventions for describing hemocytes vary greatly, depending on the author and field of study. Within insect hemocyte literature at large, there are two major nomenclatural conventions, one for the description of Drosophila hemocytes and another for the description of hemocytes from non-drosopholine lineages. In both conventions, plasmatocytes are a commonly described cell type (Strand 2008). Importantly however, the respective function of plasmatocytes varies across drosopholine and non-drosopholine literature. Here, we utilize the conventions of non-drosopholine literature, wherein granulocytes are described as exhibiting a strong propensity for phagocytosis and plasmatocytes are predominately involved in the encapsulation response (Russo et al. 1996; Ribeiro and Brehélin 2006).
A limited number of studies have used functional assays to examine hemocyte immune activity in honey bees. A nitric oxide (NO) sensitive dye has been used to estimate NO production in honey bee hemocytes (Negri et al. 2013, Negri et al. 2014b); however, given the variety of oxidative species produced by immune cells, this method is not readily applicable to experiments concerning reactive oxygen species (ROS) such as superoxide (Fang 2004). While phagocytosis assays have been conducted using hemocytes from a breadth of arthropods (Castillo et al. 2006; Fallon et al. 2011; Qi et al. 2016), we know of only one assay examining honey bee hemocyte phagocytosis published over 50 years ago (Gilliam and Shimanuki 1967). Given the critical immune functions of ROS production and phagocytosis in pathogen killing, cell signaling, and pathogen clearance (Haas 2007; Nunes et al. 2013), further development of methods for interrogating immune function is warranted.
In this study, we investigate the morphological and functional characteristics of hemocytes collected from the circulating hemolymph of larval workers of 6 and 8 days of age after egg laying; adult workers at 0, 5, and 10 days after eclosion; and young unmated adult queen honey bees 2 to 5 days after eclosion. Using microscopy and differential staining, we identified two dominant hemocyte types in bees, plasmatocytes and granulocytes, and quantified their relative abundance across age and caste. Fluorescence-based techniques were used to characterize phagocytosis and ROS production, two important functions of the insect immune system (Lavine and Strand 2002), in granulocytes. We also employed a pH indicator dye, Neutral Red, to characterize hemocyte lysosomal area, a potential indicator of phagocytic activity.
2 Methods
2.1 Obtaining synchronously aged honey bee cohorts
We sampled hemolymph from synchronously aged cohorts of honey bees taken from three five-frame nucleus colonies headed by non-sister queens. To obtain uniformly aged larvae, an empty drawn frame was introduced into the colony and monitored daily until eggs were laid within the cells. Upon egg laying, the queen was caged for 3 days to ensure that any additional eggs laid in the frame would be recognizably younger. To obtain uniformly aged adult honey bees, frames of capped pupae were collected and placed in an incubator (34 °C, 60–80% relative humidity). Newly eclosed adult bees were brushed from the frame daily, painted on the thoracic notum using a spot of acrylic paint (DecoArt, Stanford, KY, USA), and reintroduced into their respective colonies to develop under natural conditions. For the experiments in this study, we utilized 6- and 8-day-old larval worker bees, as measured in days post egg laying; 0-, 5-, and 10-day-old adult worker bees; and 2- to 5-day-old unmated virgin queens. These age points were chosen pragmatically. In our experience, it was difficult to consistently obtain hemolymph samples of sufficient volume from larvae younger than 6 days of age and adults older than 10 days of age. Further, the hemolymph of pupating bees was found to contain large numbers of cells and tissue particles, likely due to the developmental rearrangements occurring during morphogenesis, impeding our ability to enumerate and characterize pupal hemocytes. To ensure sufficient nutrition and consistent and adequate levels of brood rearing, 100 to 200 g of protein supplement (AP23 Pollen Substitute, Dadant & Sons, Inc., Hamilton, IL, USA) was added to colonies as needed based on observations of underfed frames of developing larvae.
2.2 Honey bee hemocyte cytospin preparations
For morphological characterizations of hemocytes, hemolymph was collected. Larval hemolymph was obtained by piercing the left or right lateral cuticle of the posterior region with a sterilized 0.45-mm-diameter insect pin (BioQuip Products, Rancho Dominguez, CA, USA). Adult bee hemolymph was collected by anesthetizing individuals on ice, removing the abdomen and head with sterilized dissection scissors and collecting hemolymph from the anterior opening of the thorax. Preliminary experiments demonstrated that hemolymph collection was similar for un-anesthetized and cold-anesthetized bees. For both larval and adult hemolymph collection, a 20-μL micropipette was used.
Cells were centrifuged onto glass microscope slides using a Shandon CytoSpin II Cytocentrifuge (GMI, Ramsey, MN, USA). The cytospin technique is a method in which cells are concentrated, adhered to a designated area of a microscope slide surface, and dried through centrifugation. For all cytospin preparations, 4 μL of hemolymph was collected into 40 μL of phosphate-buffered saline (PBS) (Gibco, pH 7.2, Invitrogen, Waltham, MA, USA) on ice and centrifuged within 1 h. For the majority of samples, the 4-μL hemolymph sample was taken from one individual; however, for 10-day-old worker bees, a pooled 4-μL sample of hemolymph was taken from two individuals. Prior to cytospin centrifugation, the absorbent membrane in each cytospin chamber was wetted with 400 μL of PBS. Then, 200 μL of PBS was added along with 44 μL of hemolymph in PBS. Loaded cytospin chambers were centrifuged at 448 rcf for 3 min. Following centrifugation, slide-adhered cells were allowed to dry for 30 min at room temperature and stained with hematoxylin and eosin using the PROTOCOL Hema 3 Stat Pack (Fisher Scientific, Hampton, NH, USA).
2.3 Characterization of hemocyte communities
Hemocyte cytospin preparations from worker larvae, adult workers, and young adult queens were visualized using light microscopy (400×, AmScope T340B), and the relative abundance of each cell type was determined by following a transect across each cytospin sample along the diameter of the circle of adhered cells. Representative images were taken at 1000× magnification (AmScope MU900 9MP USB digital camera). Count data were log-transformed before a one-way ANOVA was used to determine whether significant differences in abundance existed across ages and castes for cell populations identified as plasmatocytes, granulocytes, or permeabilized cells. Tukey’s honest significance test was used to determine differences among pairwise comparisons of cell populations. Mitotic indices were estimated, as in Beals et al. (1999), for 6- and 8-day-old larvae, and a two-tailed Wilcoxon rank-sum test was used to test if rates of mitosis varied significantly between these two age points.
2.4 Assessing in vitro phagocytosis
To investigate the phagocytic activity of honey bee granulocytes, we used fluorophore-labeled K-12 strain Escherichia coli BioParticles (Alexa Fluor 488 conjugate, Thermo Fisher Scientific, Waltham, MA, USA) with a Trypan Blue quenched in vitro assay, similar to the methods of Qi et al. (2016). For this, 40 μL of HyClone TNM-FH Insect Cell Culture Medium (Fisher Scientific, Hampton, NH, USA) was mixed with 10 μL of hemolymph and placed on the surface of a microscope slide. We then mixed 50 μL of insect medium containing 3 μL of 0.5 mg/mL fluorophore-labeled E. coli with 50 μL of hemocyte suspension and incubated the slide at room temperature for 45 min. Following incubation, 50 μL of insect medium containing 15 μL of 0.4% Trypan Blue staining solution (Thermo Fisher Scientific, Waltham, MA, USA) was added and the slide was incubated for 5 min before being washed three times with 200 μL of PBS and fixed with 200 μL of PBS containing 4% paraformaldehyde and 1 μL of 143 μM 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Sigma-Aldrich, St. Louis, MO, USA). The fixing solution was removed, and a cover slip was placed over the sample using 15 μL of 80% glycerol in PBS. Cells were visualized using an inverted fluorescence microscope (Zeiss Axio Observer Z1 with AxioCam ICc 3 camera). The Trypan Blue was used to quench the fluorophore label on E. coli adhered to the cell surface and ensure that phagocytosed E. coli were internalized within honey bee hemocytes. We used ImageJ (Schneider et al. 2012) to estimate the average fluorescence intensity of labeled E. coli to compare the relative fluorescence of internal and external particles with a one-tailed Welch’s two-sample t test. These methods were effective for granulocytes, but the small size and low refractive index of plasmatocytes made them unsuitable for interrogation with this assay.
A Neutral Red assay was also used to compare lysosomal area, a potential indicator of phagocytic activity, between granulocytes and plasmatocytes. For this, 100 μL of insect medium containing 10 μL of hemolymph, 1 μL of 4 mg/mL Neutral Red dye (Sigma-Aldrich, St. Louis, MO, USA), and 5 μL of heat-killed E. coli were placed on the surface of a microscope slide. To prepare heat-killed bacteria, DH5 alpha E. coli were cultured in liquid LB medium for 12 h, pelleted, and washed two times in PBS by centrifugation at 5000g for 3 min, then diluted to an optical density of 0.2 at 600 nm. Diluted bacteria were then incubated for 60 min at 70 °C before a final 1000-fold dilution in PBS. After addition of heat-killed bacteria, the slides were incubated for 1 h before 80 μL of the solution was removed from the slide and a cover slip was added. Cells were visualized at 1000× magnification, and representative images were captured. To evaluate differences in phagocytic activity between cell types, we used color thresholding in ImageJ to estimate the total lysosomal area of each cell by converting each image to 8-bit color format and using a pixel color threshold of 120 across all images. A one-way ANOVA was then used with a Tukey’s honest significance test to determine differences in lysosome area between cell types.
2.5 Assessing reactive oxygen species production
To observe the production of ROS by hemocytes in vitro, we used an oxidant sensitive dye, 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) (Cabiscol et al. 2000; Görlach et al. 2000). For these observations, 10 μL of hemolymph was extracted from 6- to 8-day-old worker larvae or newly eclosed adult workers and placed in 8-well chambered slides (Nunc Lab-Tek II, Thermo Fisher Scientific, Waltham, MA, USA) with 190 μL of insect medium. To this, 50 μL of HyClone insect medium containing 2.5 μL of 1000 μM H2DCFDA (Thermo Fisher Scientific, Waltham, MA, USA) and 1 μL of 143 μM DAPI were added to each well. Cells were imaged at 200× magnification following 5 min of incubation with the dye.
To test for protein kinase C (PKC)-dependent induction of ROS production, phorbol 12-myristate 13-acetate (PMA), a model ROS inducer, was added to cells in a 96-well plate fluorescence assay. For paired experimental and control wells, 140 μL of insect medium was added to each well. A pool of hemolymph (80 μL) was collected from 5- to 8-day-old larvae and divided evenly between the two wells. On a 60-s time stagger, cells were stimulated through addition of 20 μL of insect medium containing 2 μL of 1000 μM H2DCFDA dye and 2 μL of 2 mg/mL PMA (Alfa Aesar, Fisher Scientific, Hampton, NH, USA) in dimethyl sulfoxide (DMSO) (ACS Reagent Grade, Fisher Scientific, Hampton, NH, USA). For control wells, PMA was substituted with DMSO control solvent. The fluorescence intensity, using excitation and emission wavelengths of 495 and 520, respectively, of each well was then measured at 60-s intervals (PerkinElmer EnSpire Multimode Plate Reader). Following data collection, we removed initial fluorescence measurements from each well according to their position in the time series. Further, the fluorescence intensities were baseline corrected by subtracting the initial fluorescence measurement. Significant differences in relative fluorescence between the DMSO-treated control and PMA-treated experimental cells were inferred using repeated measures ANOVA.
3 Results
3.1 Differentiation and characterization of honey bee hemocyte communities
Across the age points investigated, we found that hemocyte communities in bees were predominately composed to two cell types, granulocytes (Figure 1a, e) and plasmatocytes (Figure 1d). After differential staining, adult and larval granulocytes were relatively large and rounded with cytoplasm which stained moderate to dark purple and contained many vacuolar bodies. While granulocytes from adult worker and queen hemolymph comprised only a small fraction of the circulating hemocytes (0.6 to 5.9% in workers and 8.0% in queens on average); this cell type was dominant in larval hemolymph (33.6 to 67.1% in 6- and 8-day-old larvae, respectively). In larvae, we also noted some cells that appeared similar to granulocytes in terms of staining and nucleus to cytoplasm ratio, but lacked vacuolar bodies and exhibited plasmatocyte-like asymmetric spreading and an affinity for sample debris, such as setae (Figure 1b). These cells resembled those described as L5-2 by Negri et al. (2014b) but appeared quite rarely and were never observed in slide transects when relative hemocyte abundances were estimated.

The major circulating particulate types of honey bee hemolymph imaged at 1000× magnification. Representative images show larval granulocytes (a); rare larval hemocytes resembling those described as L5-2 by Negri et al. (2014b) (b); larval large permeable nuclei (c) differentially stained (left), incubated in Trypan Blue (middle), and a merger of bright-field and DAPI-stained images (right); adult plasmatocytes (d); adult granulocytes (e); and granulocytes (f) and plasmatocytes (g) exhibiting signs of deterioration. Scale bars represent 10 μm.
Adult hemolymph was primarily composed of smaller cells resembling plasmatocytes that exhibited a high nucleus to cytoplasm ratio and spread asymmetrically on a glass surface (Marringa et al. 2014; Feitosa et al. 2015). In newly eclosed workers, this cell type comprised 87.5% of total hemocytes. While adult plasmatocytes stained purple with hematoxylin and eosin in samples of younger bees, these cells often appeared more neutrophilic, exhibiting only light or minimal staining.
In addition, cells or particles which were difficult to characterize were also observed. For example, particles which we refer to as permeabilized cells resembled granulocytes and plasmatocytes undergoing cell lysis, cell death, or degranulation and were consistently observed in both adults and larvae (Figure 1f, g). Such cellular deterioration could be the product of our methodological handling of samples, though permeabilized cells have similarly been found in flow cytometric studies (Marringa et al. 2014; Burritt et al. 2016). Hemolymph particles resembling large permeable nuclei were also observed in larvae (Figure 1c). These notably large particles, as large as 50 μm in diameter, appeared only in 6-day-old larvae and resembled a non-viable nucleus. The membrane surrounding these particles was consistently permeable, as evidenced through uptake of Trypan Blue (Figure 1c, middle panel). Further, DAPI staining resulted in the entire area of the particle exhibiting fluorescence (Figure 1c, right panel), suggesting that they are either nuclei with no cytoplasm or nuclei with an exceedingly thin and permeable cytoplasm.
We found significant differences in the relative abundance of granulocytes, plasmatocytes, and permeabilized cells across life stages and castes (Figure 2; P < 0.001 for all tests). Large permeable nuclei were not included in this comparison since these particles were only seen in hemolymph of 6-day-old larvae and were not abundant.

Mean and standard error of transect counts of granulocytes, plasmatocytes, and permeabilized cells measured across honey bee ages and castes. Permeabilized cells correspond to the cell types shown in Figure 1f, g. Adult queen hemocyte communities are representative of three unmated individuals between 2 and 5 days of age. Worker bee counts are representative of at least five individuals. Different letters indicate statistical differences between groups. Numbers in parentheses indicate the age of the individual, in days, following egg laying or eclosion. Light gray bars represent worker bees while queens are represented with dark gray bars.
Granulocyte abundance in workers peaked in 8-day-old larvae, then significantly decreased in newly eclosed adults (P < 0.01) and further decreased in 5-day-old workers (P < 0.001) to a level that was statistically indistinguishable from 10-day-old workers. Observed plasmatocyte abundance was inverse to granulocyte abundance. Plasmatocytes were not observed in the larval life stage, were most abundant in newly eclosed adults, then became much less abundant in 5- and 10-day-old workers (P < 0.01 for both comparisons). The only significant difference observed in permeabilized cell abundance was observed when comparing 6-day-old larvae to newly eclosed workers, in which newly eclosed workers had significantly fewer permeabilized cells (P < 0.005).
Young queens did not have significantly more granulocytes or plasmatocytes relative to newly eclosed workers, but did have more granulocytes and plasmatocytes relative to 5-day-old workers (P < 0.001 for both comparisons). Young queens had more permeabilized cells relative to both newly emerged (P < 0.001) and 5-day-old workers (P < 0.01).
3.2 Observation of larval granulocyte mitosis
While observing larval granulocytes, we noticed clear instances of cells undergoing all stages of mitotic cell division (Figure 3a). Mitotic indices were not significantly different between 6- and 8-day-old larvae (P = 0.41, Figure 3c). After observing mitosis in fixed samples, we recorded a video of a live larval granulocyte undergoing mitosis in HyClone insect medium. Representative images from this recording are shown in Figure 3b. The full recording is provided as supplementary material (Appendix A).

Representative images of larval granulocyte mitosis (a). Image frames extracted from a video recording of a live larval granulocyte undergoing mitosis (b). All images were taken at 1000× magnification. Scale bars represent 10 μm. Mitotic indices for 6- and 8-day-old larvae show estimated fraction of mitotic cells (c). Distributions of mitotic indices are representative of at least seven individuals.
3.3 Characterization of phagocytic activity in honey bee granulocytes and plasmatocytes
In our initial fluorophore-labeled E. coli phagocytosis assay, we observed phagocytosis in granulocytes (Figure 4). Particles internalized within the granulocyte cytoplasm exhibited significantly greater fluorescence intensity relative to particles which were external to the cytoplasm (P < 0.0001, Figure 4c).

Larval granulocyte phagocytosis of fluorophore-labeled E. coli bioparticles. Images showing DAPI nuclear stain, green fluorescent bioparticles (EC), a bright-field view (BF), and a merger of all three frames at 400× (a) and 1000× (b). Scale bars represent 10 μm. Relative fluorescence intensities of internalized and externalized bioparticles demonstrate the effect of Trypan Blue quenching on particle fluorescence (c). Estimations of relative fluorescence are representative of 26 E. coli particles each.
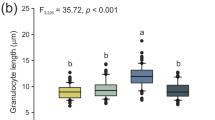
We adapted a Neutral Red assay to measure the potential for phagocytic activity in granulocytes and plasmatocytes and found that all cell types formed Neutral Red-stained endocytic bodies following incubation in medium containing Neutral Red and heat killed E. coli (Fig. 5). We found that the area of the lysosome varied significantly across cell types (P < 0.0001). Furthermore, we found that all pairwise comparisons of lysosome area resulted in significant differences (P < 0.001 for all comparisons, Figure 5d).

Neutral Red phagocytosis assays of larval granulocytes (a), adult plasmatocytes (b), and adult granulocytes (c). All images were taken at 1000× magnification. Scale bars represent 10 μm. Significant differences in relative lysosomal area across all pairwise comparisons of cell types, inferred by color thresholding, suggest differences in potential phagocytic capacity across cell types (d). Estimates of lysosomal area are shown in arbitrary units and are representative of at least 15 cells from three individuals.
3.4 Reactive oxygen species production in honey bee granulocytes and plasmatocytes
We observed basal levels of ROS production for both larval hemocytes and newly eclosed adult plasmatocytes and granulocytes in cell culture using fluorescence microscopy (Figure 6a, b). Notably, most larval hemocytes observed in this assay appeared symmetrically rounded and granulocyte-like; however, the sample shown in Figure 6b shows atypical cells that exhibit asymmetric spreading similar to the cell type L5-2 described by Negri et al. (2014b). Given that we did not perform tests of the source or intensity of basal ROS production in individual cell types, further work is needed to understand the mechanisms of ROS release as well as its intensity and response to immunostimulation across cell types. Using the 96-well plate reader assay, we observed a significant increase in relative fluorescence in PMA-exposed cells (P < 0.0001, Figure 6c), indicating that ROS production was elevated in these cells.

Basal honey bee adult granulocyte and plasmatocyte ROS production (a) and larval hemocyte ROS production (b) taken at 200× magnification. Notably, b shows larval cells with atypical asymmetric morphology resembling those described as L5-2 by Negri et al. (2014b). DAPI staining was used to distinguish dead and live cells and a bright-field view of cells is shown (BF). Scale bars represent 10 μm. A mean and standard error time series plot shows relative ROS production in PMA-exposed and control larval cells (c). ROS production measurements are representative of five replicates, each performed on a separate date with larvae from one of three colonies, for both experimental and control treatments.
4 Discussion
Negri et al. (2014b) made considerable progress toward understanding and elucidating the types of hemocytes present in honey bees using traditional approaches based on bright-field microscopy. However, the characterization of honey bee hemocytes is a nascent field and technological advancements, including the use of immunohistochemistry (Gardiner and Strand 1999; Bryant and Michel 2016), RNA sequencing (Irving et al. 2005; Pinto et al. 2009), and flow cytometry (Marringa et al. 2014; Burritt et al. 2016; Hystad et al. 2017), will ultimately be needed to more conclusively distinguish, separate, and interrogate honey bee hemocyte types. The past hemocyte-specific monoclonal antibody works of Strand and Johnson (1996) and Pech and Strand (2000) provide clear evidence of the potential utility of such efforts. Further, the recent immunohistochemistry panel-based flow cytometric methods of Yu et al. (2016) illustrate the utility of applying such high-throughput techniques to the differentiation of cell populations. In addition to methodological differences, there has been a lack of consensus in the way hemocyte types are named between studies (Strand 2008; Negri et al. 2014b). Continued collaborations among researchers is needed to harmonize the categorization of different hemocyte types using both morphological and functional characteristics.
Our observations of larval hemocyte communities revealed that granulocytes were the predominant cell type. After differential staining, these cells appeared similar to accounts of granulocytes from other organisms (Inoue et al. 2001; Kadota et al. 2003). However, a second larval cell type, resembling those described as L5-2 by Negri et al. (2014b), were also observed though they were not encountered during cytospin transect counting, indicating that they are either rare or not easily distinguished using the methods described here. It is possible that the cytospin method we employed resulted in the loss or degradation of cell types observed in other works (e.g., López et al. 2014, 2017). Alternatively, additional cell types may be inducible upon infection or wounding. In Drosophila, cells known as lamellocytes are found occasionally in the larval stage and are inducible with infection and involved in the encapsulation response (Lanot et al. 2001). Given the appearance and rarity of the second larval cell type we observed, future research into their behavior and abundance in response to pathogenesis is warranted.
With respect to the ontogeny of hemocytes within worker caste honey bee hemolymph, our results differ from those of Wilson-Rich et al. (2008) in that we observed hemocyte abundance to be greatest in young worker bees relative to larvae. However, this may be due to differences in sampling and methodology or the reliance on bright-field microscopy by Wilson-Rich et al. (2008), which can make the observation of the small plasmatocytes quite challenging.
Our observation of mitotic cells in larval hemolymph suggests that some portion of hematopoiesis occurs through the division of circulating hemocytes. The granulocyte-like appearance of these mitotic cells suggests that granulocyte differentiation does not coincide with a loss of mitotic capacity. Further, this finding is in agreement with previous accounts of plasmatocyte-derived hematopoiesis in the circulatory hemolymph of Drosophila, in which plasmatocytes appear to be the dominant phagocytes (Holz et al. 2003). This observation is also congruent with previous accounts of hemocyte mitosis in mosquitoes (King and Hillyer 2013).
Development and improvement of phagocytosis assays is an important step toward understanding the basic immune biology of honey bee hemocytes (Qi et al. 2016). In addition to better understanding basic immune biology, phagocytosis assays can also be used to investigate the effects of environmental factors, such as toxic exposure, on insect immunity (Baines et al. 1992; Fallon et al. 2011). We were able to implement a commonly used in vitro fluorescence-based phagocytosis assay on honey bee granulocytes, but we failed to implement this using adult plasmatocytes due to their small size and challenges in maintaining and manipulating them in cell culture. However, we surveyed the potential for phagocytic activity in both granulocytes and plasmatocytes using a proxy measurement, Neutral Red lysosomal staining. In the Neutral Red assay, the pH-dependent reduction of Neutral Red dye results in a color transition from yellow to red under decreasing pH conditions ranging from pH 8.0 to pH 6.8. As such, when membrane-permeable Neutral Red enters acidic lysosomal compartments within the cell, it undergoes acid-catalyzed reduction to yield a product which is both membrane-impermeable and red (Guillermo et al. 2008). A key assumption of our use of this dye is that stained vesicles are phagolysosomes as opposed to non-immune-related lysosomes (Antal et al. 1995; Hermann et al. 2004; Pinsino et al. 2015). Regardless of the true function, lysosomal compartments are a pre-requisite for phagocytic activity and measuring their presence enables researchers to measure the phagocytic potential of a cell using this straightforward assay.
In addition to phagocytosis, Neutral Red has been used to investigate lysosomal membrane stability (Harreus et al. 1997; Svendsen et al. 2004). When lysosomal membranes become permeable, such as during apoptosis or following toxic exposure, the chemically reduced dye permeates from the lysosome into the cell cytoplasm. Permeable lysosomes appear less structured relative to stable, impermeable lysosomes. Further, as dye diffuses into the cytoplasm, the cell cytoplasm becomes visibly stained with light red to orange coloration.
In Neutral Red assays, we observed that granulocytes exhibited a significantly greater lysosomal area relative to plasmatocytes when incubated with heat-killed bacteria. However, we also observed notable differences between larval and adult granulocytes, with larval granulocytes exhibiting significantly less lysosome area and appearing more structured than adult granulocytes. Some adult granulocytes exhibited distinct lysosome compartments within the cell, but most had non-discrete lysosomal compartments in which the cytoplasm appeared diffusely stained light red or orange. This suggests that, while both larval and adult granulocytes actively participate in phagocytosis, adult granulocyte lysosomes exhibit membrane permeability. This may be related to the significant loss of granulocytes as adult bees age from 0 to 5 days post eclosion. Overall, our results suggest that phagocytosis is primarily accomplished by granulocytes and that the immunity provided by granulocytes decreases with age in worker bees. Young queens, however, maintained a large population of granulocytes. Thus, queens appear to undergo a slower rate of senescence in immune function, just as they age more slowly relative to workers in other aspects of their physiology (Remolina and Hughes 2008).
The targeted production of ROS is a critical component of the immune response. Through the use of an oxidant sensitive dye, H2DCFDA, we show that ROS production can be investigated in honey bee primary hemocyte cultures. Upon reacting with ROS, H2DCFDA is oxidized to yield a fluorescent dichlorofluorescein derivative that can be measured through fluorometric methods. We observed basal levels of ROS production in honey bee granulocytes and plasmatocytes in vitro using fluorescence microscopy. Additionally, we quantified in vitro larval granulocyte ROS production in response to the protein kinase C (PKC)-activating phorbol compound, PMA. PMA activates ROS induction through its allosteric effects on PKC isoforms (Zavoico et al. 1985; Inoguchi et al. 2003; Leonard et al. 2011). In mammals, induction of the superoxide-forming phagocyte oxidase system requires PKC-mediated p47phox phosphorylation, resulting in the formation of a trimeric complex of p67phox, p40phox, and p47phox. Upon translocation to the plasma or endosomal membrane, this trimeric complex associates with flavocytochrome b558, a complex of gp91phox, small G protein Rac, and p22phox, to produce active phagocyte oxidase (Nunes et al. 2013). However, the superoxide-forming catalytic core of mammalian oxidase, gp91phox, is a Nox2 NADPH oxidase and the honey bee genome lacks a Nox2 orthologue. In some organisms, phagocyte oxidase function is instead achieved via a dual oxidase system (Duox) and the honey bee genome does encode such an oxidase (Kawahara et al. 2007). Despite this, our finding that PMA stimulates a respiratory burst in honey bee granulocytes suggests that a similar cascade results in the activation of honey bee phagocyte oxidase, though further experiments are needed to confirm this mechanism more directly.
Recent research suggests that plasma membrane translocation of p47phox is required for ROS induction in the wax moth, Galleria mellonella (Fallon et al. 2011), providing further support for a model of insect ROS induction that is similar to the mammalian model. It is important to note, however, that while the data of Fallon et al. (2011) and this work provide convergent evidence in support of a PKC-mediated mechanism of ROS induction in insects, the H2DCFDA assay we employed has some caveats. Particularly, H2DCFDA can be oxidized by non-immune-related ROS and redox factors and can undergo a superoxide-mediated redox-cycling mechanism which can lead to artificially high fluorescence measurements (Kalyanaraman et al. 2012). Even so, our data clearly indicate a relative difference in ROS production as a result of PMA exposure. In summary, while we are confident about the effect of PMA on insect hemocyte ROS production, our fluorescence measurements are only useful for interpreting relative, not absolute, ROS production. Additionally, our experiments are not sufficient for determining the source of ROS production and further research is needed to test the proposed phagocyte oxidase-dependent mechanism.
In addition to the functional assays we have performed for this work, phenoloxidase (PO) activity is another important component of the insect immune system worth interrogating in future studies. To date, researchers have characterized PO activity across honey bee development (Wilson-Rich et al. 2008; Schmid et al. 2008), finding that PO activity is generally low in larval honey bees and increases as bees age from adult nurses to foragers. Given these findings, we speculate that the larval granulocytes described in this work likely exhibit minimal PO expression. Conversely, the adult plasmatocytes we describe represent a potential source of PO expression in adults, given the dominance of plasmatocytes in adult honey bees. However, additional work is required to test these hypotheses.
In summary, our investigations provide new evidence concerning the morphology, ontogeny, and function of honey bee hemocytes. Efforts to understand how environmental insults, such as xenobiotic exposure, nutritional status, and parasitism, impact hemocyte-derived immunity represent an emerging area of honey bee research (Szymaś and Jędruszuk 2003; Amdam et al. 2004; Brandt et al. 2016; Marringa et al. 2014). However, such studies are dependent upon a clear understanding of the composition and function of hemocyte communities.
References
Amdam, G.V., Hartfelder, K., Norberg, K., Hagen, A., Omholt, S.W. (2004) Altered physiology in worker honey bees (Hymenoptera: Apidae) infested with the mite Varroa destructor (Acari: Varroidae): A factor in colony loss during overwintering? J. Econ. Entomol. 97, 741–747
Antal, P., Sipka, S., Surányi, Csipo, I., Seres, T., Maródi, L., Szegedi, G. (1995) Flow cytometric assay of phagocytic activity of human neutrophils and monocytes in whole blood by neutral red uptake. Ann. Hematol. 70, 259–265
Baines, D., DeSantis, T., Downer, R.G.H. (1992) Octopamine and 5-hydroxytryptamine enhance the phagocytic and nodule formation activities of cockroach (Periplaneta americana) haemocytes. J. Insect Physiol. 38, 905–914
Beals, M., Gross, L., Harrell, S. (1999) Mitotic index and cell division. Available at http://www.tiem.utk.edu/~gross/bioed/webmodules/mitoticindex.htm (Accessed 12/27/2016)
Brandt, A., Gorenflo, A., Siede, R., Meixner, M., Büchler, R. (2016) The neonicotinoids thiacloprid, imidacloprid, and clothianidin affect the immunocompetence of honey bees (Apis mellifera L.). J. Ins. Phys. 86, 40–47
Brutscher, L.M., Daughenbaugh, K.F., Flenniken, M.L. (2015) Antiviral defense mechanisms in honey bees. Curr. Opin. Insect Sci. 10, 71–82
Bryant, W.B., Michel, K. (2016) Anopheles gambiae hemocytes exhibit transient states of activation. Dev. Comp. Immunol. 55, 119–129
Burritt, N.L., Foss, N.J., Neeno-Eckwall, E.C., Church, J.O., Hilger, A.M., Hildebrand, J.A., et al. (2016) Sepsis and hemocyte loss in honey bees (Apis mellifera) infected with Serratia marcescens strain sicaria. PLoS ONE 11, e0167752
Cabiscol, E., Piulats, E., Echave, P., Herrero, E., Ros, J. (2000) Oxidative stress promotes specific protein damage in Saccharomyces cerevisiae. J. Biol. Chem. 275, 27393–27398
Castillo, J.C., Robertson, A.E., Strand, M.R. (2006) Characterization of hemocytes from the mosquitoes Anopheles gambiae and Aedes aegypti. Insect Biochem. Mol. Biol. 36, 891–903
Chen, Y.P., Pettis, J.S., Corona, M., Chen, W.P., Li, C.J., Spivak, M., et al. (2014) Israeli acute paralysis virus: Epidemiology, pathogenesis and implications for honey bee health. PLoS Pathog. 10, e1004261
Evans, J.D., Aronstein, K., Chen, Y.P., Hetru, C., Imler J.L., Jiang, H., Kanost, M., Thompson, G.J., Zou, Z., Hultmark, D. (2006) Immune pathways and defence mechanisms in honey bees Apis mellifera. Insect Mol. Bio. 15, 645–656
Fallon, J.P., Reeves, E.P., Kavanagh, K. (2011) The Aspergillus fumigatus toxin fumagillin suppresses the immune response of Galleria mellonella larvae by inhibiting the action of haemocytes. Microbiol. Read. Engl. 157, 1481–1488
Fang, F.C. (2004) Antimicrobial reactive oxygen and nitrogen species: Concepts and controversies. Nature Rev. Microbiol. 2, 820–832
Feitosa, A.P.S., Alves, L.C., Chaves, M.M., Veras, D.L., Silva, E.M., Alianca, A.S.S., Franca, I.R.S., Goncalves, G.G.A., Lima-Filho, J.L., Brayner, F.A. (2015) Hemocytes of Rhipicephalus sanguineus (Acari: Ixodidae): Characterization, population abundance, and ultrastructural changes following challenge with Leishmania infantum. J. Med. Entomol. 52, 1193–1202
Gardiner, E.M.M., Strand, M.R. (1999) Monoclonal antibodies bind distinct classes of hemocytes in the moth Pseudoplusia includes. J. Insect Physiol. 45, 113–126
Gilliam, M., Shimanuki, H. (1967) In vitro phagocytosis of Nosema apis spores by honey-bee hemocytes. J. Invertebr. Pathol. 9, 387–389
Görlach, A., Brandes, R.P., Nguyen, K., Amidi, M., Dehghani, F., Busse R. (2000) A gp91phox containing NADPH oxidase selectively expressed in endothelial cells is a major source of oxygen radical generation in the arterial wall. Circ. Res. 87, 26–32
Guillermo, R., del Peso, A., Zurita, J. (2008) Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 3, 1125–1131
Haas, A. (2007) The phagosome: Compartment with a license to kill. Traffic 8, 311–330
Harreus, D., Köhler, H.-R., Weeks, J.M. (1997) Combined non-invasive cell isolation and neutral-red retention assay for measuring the effects of copper on the Lumbricid Aporrectodea rosea (Savigny). Bull. Environ. Contam. Toxicol. 59, 44–49
Hermann, A.C., Millard, P.J., Blake, S.L., Kim, C.H. (2004) Development of a respiratory burst assay using zebrafish kidneys and embryos. J. Immunol. Methods 294, 119–129
Holz, A., Bossinger, B., Strasser, T., Janning, W., Klapper, S. (2003) The two origins of hemocytes in Drosophila. Development 130, 4955–4962
Hystad, E.M., Salmela, H., Amdam, G.V., Münch, D. (2017) Hemocyte-mediated phagocytosis differs between honey bee (Apis mellifera) worker castes. PLoS ONE 12, e0184108
Inoguchi, T., Sonta, T., Tsubouchi, H., Etoh, T., Kakimoto, M., Sonoda, N., Sato, N., Sekiguchi, N., Kobayashi, K., Sumimoto, H., Utsumi, H., Nawata, H. (2003) Protein Kinase C-dependent increase in reactive oxygen species (ROS) production in vascular tissues of diabetes: Role of vascular NAD(P)H oxidase. J. Am. Soc. Nephrol. 14, S227-S232
Inoue, N., Hanada, K., Tsuji, N., Igarashi, I., Nagasawa, H., Mikami, T., Fujisaki, K. (2001) Characterization of phagocytic hemocytes in Ornithodoros moubata (Acari: Ixodidae). J. Med. Entomol. 38, 514–519
Irving, P., Ubeda, J.M., Doucet, D., Troxler, L., Lagueux, M., Zachary, D., Hoffmann, J.A., Hetru, C., Meister, M. (2005) New insights into Drosophila larval haemocyte functions through genome-wide analysis. Cell Microbiol. 7, 335–350
Kadota, K., Walter, S., Claveria, F.G., Igarashi, I., Taylor, D., Fujisaki, K. (2003) Morphological and populational characteristics of hemocytes of Ornithodoros moubata nymphs during the ecdysial phase. J. Med. Entomol. 40, 770–776
Kalyanaraman, B., Darley-Usmar, V., Davies, K.J., Dennery, P.A., Forman, H.J., Grisham, M.B., Mann, G.E., Moore, K., Roberts, L.J., Ischiropoulos, H. (2012) Measuring reactive oxygen and nitrogen species with fluorescent probes: Challenges and limitations. Free Radic. Biol. Med. 52, 1–6
Kawahara, T., Quinn, M.T., Lambeth, J.D. (2007) Molecular evolution of the reactive oxygen-generating NADPH oxidase (Nox/Duox) family of enzymes. BMC Evol. Biol. 7, 109
King, J.G., Hillyer, J.F. (2013) Spatial and temporal in vivo analysis of circulating and sessile immune cells in mosquitoes: Hemocyte mitosis following infection. BMC Biol 11, 55
Lanot, R., Zachary, D., Holder, F., Meister, M. (2001) Postembryonic hematopoiesis in Drosophila. Dev. Biol. 230, 243–257
Lavine, M.D., Strand, M.R. (2002) Insect hemocytes and their role in immunity. Ins. Biochem. Mol. Biol. 32, 1295–1309
Leonard, T.A., Różycki, B., Saidi L.F., Hummer G., Hurley J.H. (2011) Crystal structure and allosteric activation of protein kinase C βII. Cell 144, 55–66
López, J.H., Schuehly, W., Crailsheim, K., Riessberger-Gallé, U. (2014) Trans-generational immune priming in honeybees. Proc. R. Soc. B 281, 20140454
López, J.H., Krainer, S., Engert, A., Schuely, W., Riessberger-Gallé, U., Crailsheim, K. (2017) Sublethal pesticide doses negatively affect survival and the cellular responses in American foulbrood-infected honeybee larvae. Sci. Rep. 7, 40853
Marringa, W.J., Krueger, M.L., Burritt, N.L., Burritt J.B. (2014) Honey bee hemocyte profiling by flow cytometry. PLoS ONE 9, e108486
Negri, P., Maggi, M., Correa-Aragunde, N., Brasesco, C., Eguaras, M., Lamattina, L. (2013) Nitric oxide participates at the first steps of Apis mellifera cellular immune activation in response to non-self recognition. Apidologie 44, 575–585
Negri, P., Quintana, S., Maggi, M., Szawarski, N., Lamattina, L., Eguaras, M. (2014a) Apis mellifera hemocytes generate increased amounts of nitric oxide in response to wounding/encapsulation. Apidologie 45, 610–617
Negri, P., Maggi, M, Szawarski, N, Lamattina, L., Eguaras, M. (2014b) Apis mellifera haemocytes in-vitro: What type of cells are they? Functional analysis before and after pupal metamorphosis. J. Apic. Res. 53, 576–589
Nunes, P., Demaurex, N., Dinauer, M.C. (2013) Regulation of the NADPH oxidase and associated ion fluxes during phagocytosis. Traffic 14, 1118–113
Pech, L.L., Strand, M.R. (2000) Plasmatocytes from the moth Pseudoplusia includes induce apoptosis of granular cells. J. Insect Physiol. 46, 1565–1573
Pinsino, A., Russo, R., Bonaventura, R., Brunelli, A., Marcomini, A., Matranga, V. (2015) Titanium dioxide nanoparticles stimulate sea urchin immune cell phagocytic activity involving TLR/p38 MAPK-mediated signalling pathway. Sci. Rep. 5, 14492
Pinto, S.B., Lombardo, F., Koutsos, A.C., Waterhouse, R.M., McKay, K., An, C., Ramakrishnan, C., Kafatos, F.C., Michel, K. (2009) Discovery of Plasmodium modulators by genome-wide analysis of circulating hemocytes in Anopheles gambiae. Proc. Natl. Acad. Sci. USA 106, 21270–21275
Qi, Y.-X., Huang, J., Li, M.-Q., Wu, Y.-S., Xia, R.-Y., Ye G.-Y. (2016) Serotonin modulates insect hemocyte phagocytosis via two different serotonin receptors. eLIFE 5, e12241
Remolina, S.C., Hughes, K.A. (2008) Evolution and mechanisms of long life and high fertility in queen honey bees. Age 30, 177–185
Ribeiro, C., Brehélin, M. (2006) Insect haemocytes: What type of cell is that? J. Insect Physiol. 52, 417–429
Runckel, C., Flenniken, M.L., Engel, J.C., Ruby, J.G., Ganem, D., Andino, R., DeRisi, J.L. (2011) Temporal analysis of the honey bee microbiome reveals four novel viruses and seasonal prevalence of known viruses, Nosema, and Crithidia. PLoS ONE 6, e20656
Russo, J., Dupas, S., Frey, F., Carton, Y., Brehelin, M. (1996) Insect immunity: Early events in the encapsulation process of parasitoid (Leptopilina boulardi) eggs in resistant and susceptible strains of Drosophila. Parasitology 112, 135–142
Schmid, M.R., Brockmann, A., Pirk, C.W.W., Stanley, D.W., Tautz, J. (2008) Adult honeybees (Apis mellifera L.) abandon hemocytic, but not phenoloxidase-based immunity. J. Insect Physiol. 54, 439–444
Schneider, C.A., Rasband, W.S., Eliceiri, K.W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nature Methods 9, 671–675
Schwarz, R.S., Huang, Q., Evans, J.D. (2015) Hologenome theory and the honey bee pathosphere. Curr. Opin. Insect Sci. 10, 1–7
Strand, M.R. (2008) Insect hemocytes and their role in immunity. In Beckage, N.E. [Ed.] Insect Immunology, 25–47. Elsevier, San Diego
Strand, M.R., Johnson, J.A. (1996) Characterization of monoclonal antibodies to hemocytes of Pseudoplusia includens. J. Insect Physiol. 42, 21–31
Svendsen, C., Spurgeon, D.J., Hankard, P.K., Weeks, J.M. (2004) A review of lysosomal membrane stability measured by neutral red retention: Is it a workable earthworm biomarker? Ecotoxicol. Environ. Saf. 57, 20–29
Szymaś, B., Jędruszuk, A. (2003) The influence of different diets on haemocytes of adult worker honey bees, Apis mellifera. Apidologie 34, 97–102
Weinstock, G.M., Robinson, G.E., Gibbs, R.A. et al. (2006) Insights into social insects from the genome of the honeybee Apis mellifera. Nature 443, 931–949
Wilson-Rich, N., Dres, T.D., Starks, P.T. (2008) The ontogeny of immunity: Development of innate immune strength in the honey bee (Apis mellifera). J. Insect Physiol. 54, 1392–1399
Yu, Y.-R.A., Hotten, D.F., Malakhau, Y., Volker, E., Ghio, A.J., Noble, P.W., Kraft, M., Hollingsworth, J.W., Gunn, M.D., Tighe, R.M. (2016) Flow cytometric analysis of myeloid cells in human blood, bronchoalveolar lavage, and lung tissues. Am. J. Respir. Cell Mol. Biol. 54, 13–24
Zavoico, G.B., Halenda, S.P., Sha’afi, R.I., Feinstein, M.B. (1985) Phorbol myristate acetate inhibits thrombin-stimulated Ca2+ mobilization and phosphatidylinositol 4,5-bisphosphate hydrolysis in human platelets. Proc. Natl. Acad. Sci. USA 82, 3859–3862
Acknowledgements
The authors thank Jason Duncan for providing the E. coli culture and Manjula Karpurapu, Sangwoon Chung, Brenda Reader, and Luiza Ruso for helpful advice regarding laboratory equipment and techniques. The authors thank Karen Goodell for laboratory space. This work was supported by a North American Pollinator Protection Campaign Honey Bee Health Grant and Project Apis m.—Costco Honey Bee Biology Fellowship to RTR and support provided by state and federal funds appropriated to The Ohio State University, Ohio Agricultural Research and Development Center.
Author information
Authors and Affiliations
Contributions
All authors helped conceive and design experiments. RTR conducted experiments and wrote the manuscript. RTR, RMJ, MNB, and JWC helped analyze the data and edit the manuscript.
Corresponding author
Additional information
Handling editor: Monique Gauthier
Caractérisation morphologique et fonctionnelle des groupes de cellules hémocytes de l’abeille mellifère, Apis mellifera
immunité cellulaire / plasmatocytes / granulocytes / phagocytose / production de ROS
Morphologische und funktionelle Charakterisierung von Hämozyten-Zellgruppen der Honigbiene, Apis mellifera
zelluläre immunität / plasmatozyten / granulozyten / phagozytose / ROS produktion
Electronic supplementary material
ESM 1.
Supplementary video showing larval hemocyte cell division. (AVI 120770 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Richardson, R.T., Ballinger, M.N., Qian, F. et al. Morphological and functional characterization of honey bee, Apis mellifera, hemocyte cell communities. Apidologie 49, 397–410 (2018). https://doi.org/10.1007/s13592-018-0566-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13592-018-0566-2