
Abstract
Introduction
Receptor-interacting protein kinase 1 (RIPK1), a key mediator of inflammation through necroptosis and proinflammatory cytokine production, may play a role in the pathogenesis of immune-mediated inflammatory diseases such as chronic plaque psoriasis. An experimental medicine study of RIPK1 inhibition with GSK2982772 immediate-release formulation at doses up to 60 mg three times daily in mild to moderate plaque psoriasis indicated that efficacy may be improved with higher trough concentrations of GSK2982772.
Methods
This multicenter, randomized, double-blind, placebo-controlled, repeat-dose study (NCT04316585) assessed the efficacy, safety, pharmacokinetics, and pharmacodynamics of 960 mg GSK2982772 (once-daily modified-release formulation) in patients with moderate to severe plaque psoriasis. Twenty-nine patients were randomized 2:1 to GSK2982772 (N = 19) or placebo (N = 10) for 12 weeks.
Results
GSK2982772 was well tolerated with trough concentrations greater than tenfold higher than the previous phase 1 study with immediate release. Despite near complete RIPK1 target engagement in blood and modest reduction in circulating inflammatory cytokines, the proportion of patients achieving 75% improvement from baseline in Psoriasis Area Severity Index score at week 12 was similar between GSK2982772 and placebo (posterior median 1.8% vs 4.9%, respectively), with an estimated median treatment difference of − 2.3%. This analysis incorporated historical placebo data through the use of an informative prior distribution on the placebo arm. Week 4 changes in skin biopsy gene expression suggested sufficient local drug exposure to elicit a pharmacodynamic response.
Conclusion
Administration of the RIPK1 inhibitor GSK2982772 to patients with moderate to severe plaque psoriasis did not translate into meaningful clinical improvements.
Psoriasis is thought to be caused by problems with the immune system, including possibly receptor-interacting protein kinase 1 (RIPK1), which plays an important role in the development of inflammation. A previous study suggested that the drug, GSK2982772, which interferes with RIPK1, might improve symptoms in patients with psoriasis. This study examined whether higher doses of GSK2982772 than previously studied would be beneficial for patients with psoriasis. The study found that the severity of psoriasis was similar in patients treated with GSK2982772 for 12 weeks as in those who did not receive the drug, indicating that GSK298772 did not improve psoriasis.
AbstractSection Graphical Abstract
Similar content being viewed by others

Avoid common mistakes on your manuscript.
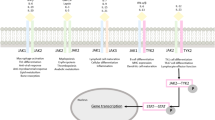
The pathogenesis of plaque psoriasis involves tumor necrosis factor (TNF) receptor 1 engagement that triggers receptor-interacting protein 1 kinase (RIPK1) activity; therefore, RIPK1 may be a potential therapeutic target in plaque psoriasis and in other immune-mediated inflammatory diseases. |
In a previous study of the RIPK1 inhibitor GSK298772 in patients with mild to moderate psoriasis, doses of 60 mg twice daily and 60 mg three times daily administered as an immediate-release formulation resulted in a decrease in plaque lesion severity score with increasing tertiles of GSK2982772 trough concentrations. |
In this study we evaluated whether the efficacy of GSK298772 may be improved with higher trough concentrations. |
Despite near complete RIPK1 target engagement in blood and modest reduction in circulating inflammatory cytokines, the proportion of patients achieving 75% improvement from baseline in Psoriasis Area Severity Index score at week 12 was similar between GSK2982772 and placebo. |
Although administration of the GSK2982772 to patients with moderate to severe plaque psoriasis did not translate into meaningful clinical improvements, these findings are an important addition to the literature on the RIPK1 pathway as a target in immune inflammatory diseases. |
Digital Features
This article is published with digital features, including a graphical abstract to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.24893724.
Introduction
Plaque psoriasis is a chronic relapsing inflammatory skin disease characterized by keratinocyte hyperproliferation and epidermal hyperplasia [1]. The pathogenesis of plaque psoriasis is complex, involving tumor necrosis factor (TNF) receptor 1 engagement that triggers receptor-interacting protein 1 kinase (RIPK1) activity. RIPK1 is a key mediator of TNF-mediated apoptosis, NF-κB signaling, necroptosis, and a regulator of proinflammatory cytokine production. Dysregulation of these processes results in sustained inflammation and profound effects on tissue homeostasis [2, 3].
Preclinical studies have shown that TNF-dependent inflammation can be prevented through the inhibition of RIPK1 [3,4,5,6]. Thus, RIPK1 may be a potential therapeutic target in immune-mediated inflammatory diseases in which these pathways are implicated, including plaque psoriasis. GSK2982772 selectively binds to the allosteric pocket of the RIPK1 domain and has been previously studied in mild to moderate plaque psoriasis, rheumatoid arthritis, and ulcerative colitis [7,8,9,10].
In patients with mild to moderate psoriasis, doses of 60 mg twice daily and 60 mg three times daily administered as an immediate-release (IR) formulation resulted in a decrease in plaque lesion severity score (PLSS) with increasing tertiles of GSK2982772 trough concentrations, suggesting that efficacy may be improved with higher trough concentrations of GSK2982772 [7]. A once-daily modified-release (MR) formulation was developed to provide a flatter GSK2982772 concentration–time profile and to provide higher trough concentrations. Modified-release doses up to 960 mg were well tolerated in healthy volunteers and provided more than tenfold higher trough concentrations compared to the 60-mg IR three-times-daily regimen used in the previous study in mild to moderate plaque psoriasis. This magnitude of increase in trough concentration should result in a significant shift in the pharmacological response curve to fully test the utility of the RIPK1 inhibition mechanism for the treatment of psoriasis [11].
The objective of this study was to assess the efficacy, safety, tolerability, pharmacokinetic (PK), and pharmacodynamic (PD) profiles of 960 mg GSK2982772 administered as a once-daily MR tablet in patients with moderate to severe plaque psoriasis.
Methods
Study Design
This was a multicenter, randomized, double-blind, placebo-controlled, repeat-dose study conducted at 10 centers in Canada and one in Poland between September 28, 2020 and October 12, 2021. Following a screening period of up to 35 days , patients were randomized 2:1 to either 960-mg GSK2982772 MR once-daily formulation [11] or placebo (Fig. 1), and stratified by prior use of biologic therapy.

Study design. Following a screening and washout period of up to 35 days, patients were randomized 2:1 to GSK2982772 960 mg MR or placebo for 12 weeks. The primary endpoint was PASI75 at week 12. Patients were followed for 28 days after the final dose. MR modified release; PASI75 75% improvement from baseline in Psoriasis Area Severity Index response
On July 14, 2020, in response to the COVID-19 pandemic, additional safety monitoring was instituted in a protocol amendment. In a second amendment on January 6, 2021, the number of skin biopsies required was reduced to aid in study recruitment (see “Study Assessments”).
The study was approved by the main ethics committee, Institutional Review Board Services Advarra, Ontario Canada and at every participating institution and was conducted according to the recommendations of Good Clinical Practice and the Declaration of Helsinki. The study protocol, amendments, informed consent, and other information requiring preapproval were reviewed and approved by applicable institutional review boards (Table S1). All patients provided written informed consent to participate in the study.
Patients
Male and female patients aged 18 to 75 years with moderate to severe plaque psoriasis for at least 6 months prior to the screening visit and with a Psoriasis Area Severity Index (PASI) score of 12 or more, psoriatic body surface area (BSA) of at least 10%, and Static Investigator Global Assessment (sIGA) of disease activity score of at least 3 were eligible. Patients agreed to avoid prolonged exposure to natural or artificial sources of ultraviolet radiation during the study.
Patients with nonplaque forms of psoriasis (e.g., erythrodermic, guttate, or pustular), drug-induced psoriasis, psoriatic arthritis, uveitis, inflammatory bowel disease, or other immune-mediated conditions requiring immunosuppressive therapy were excluded. Patients were also excluded if they had previously been exposed to at least three biologic therapies or any type of prescribed anti-TNFα biologic therapies for at least 3 months with no response. Patients with an active infection (including SARS-CoV-2), history of liver or kidney disease, unstable or uncontrolled cardiovascular disease (including hypertension), hereditary or acquired immunodeficiency disorders, history of malignant neoplasm (except for adequately treated basal or squamous cell skin cancer or carcinoma in situ of the cervix), progressive neurologic disorders (e.g., multiple sclerosis, amyotrophic lateral sclerosis, Alzheimer disease), or a history of suicidal ideation behavior were also ineligible.
Study Assessments
Study visits occurred at screening, baseline (day 1), and weeks 2, 4, 8, 12, and 16. Clinical efficacy (PASI, BSA, and sIGA) and safety assessments were made at each visit. Predose plasma concentrations of GSK2982772 were obtained at weeks 2, 4, 8, and 12 using a validated analytical method based on protein precipitation followed by high-performance liquid chromatography with tandem mass spectrometry (HPLC/MS/MS) analysis. For assessment of target engagement, blood samples from each time point were collected by venipuncture into an ethylenediaminetetraacetic acid (EDTA) tube. A 1-mL aliquot of whole blood was transferred to a Nunc cryovial, flash frozen in liquid nitrogen, then shipped on dry ice to the central laboratory (Q2 Solutions, Valencia, CA) and stored at − 70 °C until analysis. Target engagement (%) was measured by immunoassay (TEAR1) as previously described [12]. For proteomic analyses, blood samples were collected by venipuncture into an EDTA tube. A 1-mL aliquot of plasma was transferred to a 1.8-mL Nunc cryovial and flash frozen in liquid nitrogen. Frozen samples were shipped on dry ice to Olink, Inc (Waltham, MA) for analysis using their proximity extension assay (PEA) technology and stored at − 70 °C. For metabolic analyses, blood samples were collected by venipuncture into serum separation tubes (SST). Serum samples were shipped to the central laboratory (Q2 Solutions) on day of collection either at ambient temperature (lipid panel, direct low-density lipoprotein cholesterol, creatinine clearance) or on dry ice (insulin and homeostatic model assessment for insulin resistance [HOMA-IR]).
Baseline skin biopsies were obtained from a target lesion of at least 3 cm2 on the trunk or extremities and from unaffected skin close to the target lesion. At week 12, biopsies were collected from the outer one-third of the target lesion. All biopsies were divided and processed for histological and gene expression analysis.
Full-thickness, 4-mm punch biopsies were collected by qualified personnel using standard methods. After removal of subcutaneous fat, biopsies were bisected so that each half comprised epidermis and dermis. For histology, one-half biopsy was placed in 50 mL of buffered formalin for a minimum of 24 h, then shipped at ambient temperature for preparation of formalin-fixed paraffin-embedded blocks. For PK analysis and TEAR, half biopsies were snap frozen in liquid nitrogen. Unfortunately, a protocol amendment that adjusted the number of samples taken, and sample quality issues limited PK analysis and TEAR analysis (protocol amendment changed the biopsy requirement from two to one lesional biopsy to aid recruitment) and precluded histopathological scoring of these biopsies (approximately 20% of specimens were deemed inadequate because of improper embedding and/or too little tissue). For transcriptomic analysis, one-half biopsy was placed in 5 mL of RNAlater®, shipped at ambient temperature for 24 h, then stored at − 20 °C. Frozen samples were shipped to Epistem, Ltd (Manchester, UK) for processing and analysis by RNAseq.
Endpoints
The primary endpoint was PASI75 response at week 12, defined as achieving at least 75% improvement in PASI score from baseline. Secondary efficacy endpoints included PASI50, PASI90, and PASI100 response rates (achieving at least 50%, at least 90%, and 100% improvement in score from baseline, respectively), change from baseline in PASI scores, sIGA response (achieving a sIGA score of “clear” [0] or “almost clear” [1]), and change from baseline in BSA, all evaluated at week 12, with additional time points as exploratory endpoints.
Additional secondary endpoints included trough plasma concentrations of GSK2982772, and safety and tolerability measures including adverse events, clinical laboratory values (hematology, chemistry, and urinalysis), vital signs, and 12-lead electrocardiogram monitoring assessed at each study visit.
Exploratory endpoints included ex vivo GSK2982772 target engagement in blood, change from baseline in inflammatory biomarkers in blood, and change from baseline in metabolic biomarkers in fasting blood (HOMA [glucose and insulin], hemoglobin A1C [HbA1c], and lipid panel), all assessed at weeks 0, 4, and 12. Transcriptomic profiling of mRNA isolated from skin at weeks 0, 4, and 12 was conducted and gene set variation analysis (GSVA) [13] enrichment scores and specific pathway improvement scores derived. Psoriasis, TNFα, and interferon gamma (IFNγ) pathway improvement scores were calculated at weeks 4 and 12. GSVA methods and lists of gene sets analyzed are shown in Table S2.
Statistical Analyses
It was determined that a sample size of 21 patients (GSK2982772, 14; placebo, 7) would ensure approximately a 99% probability of achieving a posterior probability of at least 0.975 that the true treatment difference in PASI75 response rate over placebo was greater than zero. This assumed a true response rate of 7% with prior distribution beta (3.5, 46.5) and 55% with prior distribution of beta (0.33, 0.33) for placebo and GSK2982772 arms, respectively.
All patients who were randomized and who had received at least one dose of study intervention (intention-to-treat population) were included in the efficacy analyses; the safety population (used for safety and PD analyses) consists of the same participants. Pharmacokinetic population required at least one nonquantifiable postbaseline assessment.
The primary analysis was a Bayesian logistic regression of the PASI75 response rate at week 12, adjusting for covariates, including prior biologic use and baseline PASI score. Equivalent analyses for the other categorical endpoints (PASI50, PASI90, PASI100, and sIGA response) were not conducted because of the observed low response rates (see “Results”). Summary tables of observed responses rates were reported. Missing response data were imputed as nonresponders.
To leverage historical placebo PASI75 response data into the primary analysis, a meta-analytic prior [14] was derived from historical data from similar clinical trials; the effective sample size was approximately 63. The final analysis prior used for the placebo response rate was of the following form: 90% weight on beta (5.39, 69) and 10% on beta (0.33, 0.33). All other priors across the primary and secondary analyses were chosen to be noninformative.
A Bayesian mixed-model repeated measures (MMRM) approach was used for continuous secondary PASI and BSA end points, adjusting for visit, baseline scores, prior biologic use, and baseline-by-visit and treatment-by-visit interactions. For target engagement, the ratio of free to total RIP1 kinase was analyzed on the log scale using an MMRM approach adjusted for baseline.
Descriptive summaries were provided for all safety endpoints, including frequency of adverse events and serious adverse events. Descriptive statistics and graphics of PK and exploratory biomarker endpoints were presented.
Results
A total of 29 patients were randomized (GSK2982772, 19; placebo, 10) and 18 completed the study (Fig. S1). The majority of patients were male (72%) and white/Caucasian (90%), with a mean age of 48.8 years (Table 1) and weight of 92.4 kg. At baseline, mean PASI score was 18.3 and most patients (76%) had a moderate sIGA score. Patients with previous biologic therapy use were evenly distributed between the treatment groups.
Clinical Efficacy
One (5%) patient treated with GSK2982772 achieved PASI75 at week 12, compared with none in the placebo group (Table 2). Among patients treated with GSK2982772, PASI50 was achieved as early as week 4 in 2 (11%) patients and 8 (42%) patients at week 12, compared to no patients at week 4 and 2 (20%) patients at week 12 in the placebo group. No patient from either treatment group achieved PASI90 or PASI100.
From the analysis of the PASI75 response at week 12 (primary endpoint), incorporating historical placebo data, posterior median estimates were 1.8% for GSK2982772 and 4.9% for placebo, with a difference of − 2.3% (95% credible interval [CrI] − 9.2, 8.3) (Table 3), with a corresponding 28% posterior probability of the true difference greater than zero. The corresponding analysis based on trial data alone (using noninformative placebo prior) also suggested no difference between treatment groups in PASI75 response rates at week 12.
The posterior mean change from baseline in PASI score at week 12 was − 7.6 in the GSK2982772 group and − 6.4 for placebo (Fig. 2), a difference between groups of − 1.2 (95% CrI − 5.3, 3.2), with a corresponding 72.1% posterior probability that the true difference is less than zero (Table S3). The posterior mean change from baseline in BSA was also similar between treatment groups at week 12 (GSK2982772 − 3.9%, placebo − 4.0%; difference between groups, 0.1% [95% CrI − 6.2%, 6.8%], Fig. 2 and Table S4). The posterior probability of a true difference of less than zero was 50.4%.

Posterior mean change from baseline in a PASI score and b BSA by treatment group (ITT population). The posterior mean (95% CrI) for change from baseline in PASI score at 2, 4, 8, and 12 weeks for patients treated with placebo or GSK2982772 960 mg MR. BSA body surface area; CrI credible interval; ITT intent to treat; MR modified release; PASI Psoriasis Area Severity Index
Two (11%) patients treated with GSK2982772 achieved a sIGA score of almost clear at week 12 compared with none in the placebo group (Table S5). No patients in either group achieved a sIGA score of clear during the study.
Pharmacokinetics
GSK2982772 mean and median trough concentrations following administration of 960 mg MR once daily were more than tenfold higher than those observed in the previous study in mild to moderate plaque psoriasis at all visits with PK sampling (Table 4).
Relationship Between PK and PD Parameters
The relationship between exposure and target engagement in the skin could not be determined. There was no clear relationship between PASI score change from baseline and trough plasma drug levels of GSK2982772 at any time point during of the study.
Pharmacodynamics
Target Engagement and Biomarkers in Skin
The posterior mean percentage target engagement in blood for GSK2982772 was 96.1% at week 4 and 93.2% at week 12 (Fig. 3 and Table S6). All (16/16) patients had greater than 90% target engagement at week 4 and 11/14 achieved this threshold at week 12.

Target engagement over time by treatment for blood (safety population). The posterior mean (95% CrI) TEAR1 percentage target engagement at 0, 4, and 12 weeks for patients treated with placebo or GSK2982772 960 mg MR. CrI credible interval; MR modified release
GSK2982772 modestly reduced levels of proinflammatory cytokines in the blood relative to placebo, with average fold changes more apparent at week 4 than at week 12 (Fig. S2). There were no meaningful differences in metabolic biomarker levels between GSK2982772 and placebo.
Transcriptomic Profiling in Skin
In lesional skin, the mean change from baseline in GSVA enrichment scores for upregulated psoriasis genes was lower with GSK2982772 (week 4, − 0.21; week 12, − 0.21) than placebo (week 4, − 0.04; week 12, − 0.33) (Fig. 4a). For downregulated psoriasis genes, the mean change in GSVA enrichment scores was higher with GSK2982772 (0.12) than placebo (0.08) at week 4 and lower with GSK2982772 (0.17) than placebo (0.23) at week 12 (Fig. 4b). The improvement scores for the psoriasis, TNFα, and IFNγ pathway signatures were slightly higher at week 4 with GSK2982772 than with placebo; however, there was little difference between the two groups at week 12 (Fig. 4c).

Transcriptomic profiling scores over time by treatment. a Psoriasis upregulated gene pathway. b Psoriasis downregulated pathway. c Improvement score for gene pathways (safety population). Improvement score (based on Krueger et al. J Allergy Clin Immunol. 2012;130:145–54.e9) was defined as the proportion of the disease burden reversed posttreatment (calculated from the ratio of fold change in expression in lesional tissue posttreatment [compared to baseline] and at baseline [compared to nonlesional tissue]). The median across patients in each treatment group and visit was taken as the gene improvement score. The shaded box shows interquartile range. The horizontal line inside each faded box marks the median. The whiskers running from the box represent 1.5 × IQR or min/max (whichever is smaller) and the faded shape within each box represents the mean. GSVA, gene set variation analysis; L, lesional; NL, nonlesional
Safety
Adverse events were reported in 12 (63%) patients in the GSK2982772 group and 6 (60%) patients in the placebo group (Table 5). The most frequently reported adverse event (AE) was headache in both groups (GSK2982772, 3 [16%]; placebo, 3 [30%]). There was one serious AE reported in the study (GSK2982772, acute kidney injury). This serious adverse event was considered drug related and resolved after treatment discontinuation.
Four patients reported AEs that led to permanent discontinuation of the study intervention or withdrawal from the study (GSK2982772: creatinine renal clearance abnormal [n = 1], acute kidney injury [n = 1]; placebo: suicidal ideation [n = 1], alanine aminotransferase increased [n = 1]).
Discussion
Treatment with 960 mg GSK2982772 MR administered once daily during this 12-week study was well tolerated with a safety profile that was similar to what has been previously reported [7, 9]. The most common AE was headache, and one patient experienced a serious AE (acute kidney injury) that resolved after treatment discontinuation. GSK2982772 trough plasma concentrations were more than tenfold higher than what has previously been achieved with the 60 mg IR three-times-daily dosing in the previous psoriasis study.
The high systemic exposure achieved in most patients resulted in near complete ex vivo target engagement in the blood and a relative reduction in circulating inflammatory cytokines with GSK2982772 compared to placebo over the duration of the study. Additionally, psoriasis-associated gene expression and TNFα and IFNγ gene signatures were reduced in lesional biopsies at week 4, suggesting that sufficient local drug exposure was achieved to elicit a PD response. With only one patient treated with GSK2982772 achieving PASI75 at week 12, the apparent PD response did not translate into clinical efficacy; however, the limited PK sampling in the target organ makes robust interpretation of the lack drug exposure at the affected site challenging.
Differences in gene expression between patients treated with GSK2982772 and placebo were less well defined by week 12, which could be due to compensatory mechanisms after week 4 or increased variability at week 12. The proinflammatory and cell-death promoting functions of RIPK1 are complex and occur in a cellular, tissue, and disease context and are subject to cross-regulation via posttranslational modifications [15]. Thus, inhibition of the kinase activity of RIPK1 by GSK2982772 may initially reduce necroptotic proinflammatory cytokine production, whereas long-term inhibition of RIPK1 kinase activity may promote compensatory NF-κB signaling via the scaffolding function of RIPK1, increasing proinflammatory mediator production. In addition, psoriasis is a heterogeneous disease and many other cytokines and pathways (e.g., interleukin (IL)-12/23 pathway, Th17, etc.) are involved in its pathogenesis.
The modest systemic anti-inflammatory effect observed (i.e., reduction in circulating inflammatory cytokines) did not translate to a significant clinical benefit when compared to other available treatments [16]. One explanation for this lack of clinical efficacy may be the low contribution of necroptosis in driving inflammation in the skin in psoriasis. Compared to psoriasis, necroptotic signaling plays a greater role in autoimmune skin conditions such as lupus erythematosus, lichen planus, dermatomyositis, erythema multiforme, and toxic epidermal necrolysis, and, as such, RIPK1 may be a potential therapeutic target in these diseases [17, 18]. Defining a molecular signature for identifying diseases that will be the most responsive could help direct future clinical trials with RIPK1 inhibitors.
Although there is preclinical evidence that RIPK1 may be a potential therapeutic target in psoriasis, and a prior phase 2a study suggested higher dosing with GSK2982772 in patients with more severe disease may impact clinical response [7], clinical efficacy for patients treated daily with 960 mg GSK2982772 MR in this study was similar to placebo. Because there are already well-studied, efficacious treatment options for patients with psoriasis, such as anti-TNF, anti-IL-17, and anti-IL-23 biologics [19,20,21,22], no further studies are planned for GSK2982772 in psoriasis.
The study was designed to detect a large effect on PASI75 response. Therefore, this study was limited regarding drawing robust conclusions on more sensitive endpoints or detecting smaller effect sizes. In addition, PK sampling was limited, making interpretation of the data and drug exposure challenging.
Conclusions
Despite more than tenfold increased plasma trough concentrations of GSK2982772, high target engagement in the blood, evidence of a reduction in inflammatory cytokines, and a PD effect observed in skin at week 4, the clinical benefit of daily treatment with the 960-mg MR formulation of GSK2982772 was limited.
Data Availability
The datasets generated during and/or analyzed during the current study are available in the repository, https://www.gsk-studyregister.com/en/.
References
Lowes MA, Bowcock AM, Krueger JG. Pathogenesis and therapy of psoriasis. Nature. 2007;445(7130):866–73. https://doi.org/10.1038/nature056632.
Rendon A, Schäkel K. Psoriasis pathogenesis and treatment. Int J Mol Sci. 2019;20(6):1475. https://doi.org/10.3390/ijms20061475.
O’Donnell MA, Hase H, Legarda D, Ting AT. NEMO inhibits programmed necrosis in an NFκB-independent manner by restraining RIP1. PLoS ONE. 2012;7(7):e41238. https://doi.org/10.1371/journal.pone.0041238.
Berger SB, Harris P, Nagilla R, et al. Characterization of GSK’963: a structurally distinct, potent and selective inhibitor of RIP1 kinase. Cell Death Discov. 2015;1:15009. https://doi.org/10.1038/cddiscovery.2015.9.
Berger SB, Kasparcova V, Hoffman S, et al. Cutting edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J Immunol. 2014;192(12):5476–80. https://doi.org/10.4049/jimmunol.1400499.
Patel S, Webster JD, Varfolomeev E, et al. RIP1 inhibition blocks inflammatory diseases but not tumor growth or metastases. Cell Death Differ. 2020;27(1):161–75. https://doi.org/10.1038/s41418-019-0347-0.
Weisel K, Berger S, Papp K, et al. Response to inhibition of receptor-interacting protein kinase 1 (RIPK1) in active plaque psoriasis: a randomized placebo-controlled study. Clin Pharmacol Ther. 2020;108(4):808–16. https://doi.org/10.1002/cpt.1852.
Weisel K, Berger S, Thorn K, et al. A randomized, placebo-controlled experimental medicine study of RIPK1 inhibitor GSK2982772 in patients with moderate to severe rheumatoid arthritis. Arthritis Res Ther. 2021;23(1):85. https://doi.org/10.1186/s13075-021-02468-0.
Weisel K, Scott N, Berger S, et al. A randomised, placebo-controlled study of RIPK1 inhibitor GSK2982772 in patients with active ulcerative colitis. BMJ Open Gastroenterol. 2021;8(1):e000680. https://doi.org/10.1136/bmjgast-2021-000680.
Weisel K, Scott NE, Tompson DJ, et al. Randomized clinical study of safety, pharmacokinetics, and pharmacodynamics of RIPK1 inhibitor GSK2982772 in healthy volunteers. Pharmacol Res Perspect. 2017;5(6):e00365. https://doi.org/10.1002/prp2.365.
Tompson D, Whitaker M, Pan R, et al. Development of a once-daily modified-release formulation for the short half-life RIPK1 Inhibitor GSK2982772 using DiffCORE technology. Pharm Res. 2022;39(1):153–65. https://doi.org/10.1007/s11095-021-03124-7.
Finger JN, Brusq JM, Campobasso N, et al. Identification of an antibody-based immunoassay for measuring direct target binding of RIPK1 inhibitors in cells and tissues. Pharmacol Res Perspect. 2017;5(6):e00377. https://doi.org/10.1002/prp2.377.
Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. https://doi.org/10.1186/1471-2105-14-7.
Schmidli H, Gsteiger S, Roychoudhury S, O’Hagan A, Spiegelhalter D, Neuenschwander B. Robust meta-analytic-predictive priors in clinical trials with historical control information. Biometrics. 2014;70(4):1023–32. https://doi.org/10.1111/biom.12242.
Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol. 2013;14(11):727–36. https://doi.org/10.1038/nrm3683.
Papp K, Reich K, Leonardi CL, et al. Apremilast, an oral phosphodiesterase 4 (PDE4) inhibitor, in patients with moderate to severe plaque psoriasis: results of a phase III, randomized, controlled trial (efficacy and safety trial evaluating the effects of apremilast in psoriasis [ESTEEM] 1). J Am Acad Dermatol. 2015;73(1):37–49. https://doi.org/10.1016/j.jaad.2015.03.049.
Kim SK, Kim WJ, Yoon JH, et al. Upregulated RIP3 expression potentiates MLKL phosphorylation-mediated programmed necrosis in toxic epidermal necrolysis. J Invest Dermatol. 2015;135(8):2021–30. https://doi.org/10.1038/jid.2015.90.
Lauffer F, Jargosch M, Krause L, et al. Type I immune response induces keratinocyte necroptosis and is associated with interface dermatitis. J Invest Dermatol. 2018;138(8):1785–94. https://doi.org/10.1016/j.jid.2018.02.034.
Pantano I, Mauro D, Romano F, et al. Real-life efficacy of guselkumab in patients with early psoriatic arthritis. Rheumatology (Oxford). 2022;61(3):1217–21.
Valenti M, Narcisi A, Pavia G, Gargiulo L, Costanzo A. What can IBD specialists learn from IL-23 trials in dermatology? J Crohns Colitis. 2022;16(Suppl_2):ii20–9.
Valenti M, Pavia G, Gargiulo L, et al. Biologic therapies for plaque type psoriasis in patients with previous malignant cancer: long-term safety in a single- center real-life population. J Dermatolog Treat. 2022;33(3):1638–42.
Yiu ZZN, Becher G, Kirby B, et al. Drug survival associated with effectiveness and safety of treatment with guselkumab, ixekizumab, secukinumab, ustekinumab, and adalimumab in patients with psoriasis. JAMA Dermatol. 2022;158(10):1131–41.
Acknowledgements
Medical Writing, Editorial, and Other Assistance
Medical editorial support, Allyson Lehrman, DPM (assembling tables and figures, collating author comments, copyediting, fact checking, and referencing) and graphic services were provided by AOIC, LLC and were funded by GSK.
Funding
Funding for this study (NCT04316585 available from www.clinicaltrials.gov) was provided by GSK, including the study, medical editorial, and graphics support, and journal fees (i.e., Rapid Service Fee). Artificial intelligence (AI) technologies such as Language Learning Models, chatbots, and image creators were not used in the production of this work.
Author information
Authors and Affiliations
Contributions
Conception and design of the study: Valerie J. Ludbrook, Katie Thorn, Debra Tompson, Bartholomew J. Votta, Amy Lee, and Amanda Peppercorn. Acquisition of data: Amy Lee and Xin Chen. Conducted analyses: Debra Tompson. Conducted statistical analyses: Katie Thorn. Contributed to the data interpretation: Valerie J. Ludbrook, David C. Budd, Katie Thorn, Debra Tompson, Bartholomew J. Votta, Lucy Walker, Xin Chen, and Wei Jing Loo. Drafted the work or revised it critically: Valerie J. Ludbrook, David C. Budd Katie Thorn, Debra Tompson, Bartholomew J. Votta, Lucy Walker, Amy Lee, Xin Chen, Amanda Peppercorn, and Wei Jing Loo. All listed authors meet the criteria for authorship set forth by the International Committee for Medical Journal Editors. The authors would like to thank the participants as well as the investigators and all the study staff for their contributions to the study.
Corresponding author
Ethics declarations
Conflict of Interest
Valerie J. Ludbrook, David C. Budd, Katie Thorn, Debra Tompson, Bartholomew J. Votta, Lucy Walker, Amy Lee, Xin Chen, and Amanda Peppercorn are employees of and stockholders in GSK. Wei Jing Loo has served as an investigator, speaker, advisor/consultant for and/or received grants/honoraria from AbbVie, Amgen, Arcutis, Bausch Health, BMS, Celgene, Eli Lilly, Galderma, GSK, Janssen, LEO Pharma, Meiji Seika Pharma, Pediapharm, Novartis, Pfizer, Sanofi Genzyme, Sun Pharma, UCB, and Valeant, and she and her institution received funding for this study from GSK.
Ethical Approval
The study was approved by the main ethics committee, Institutional Review Board Services Advarra, Ontario Canada every participating institution and was conducted according to the recommendations of Good Clinical Practice and the Declaration of Helsinki. The study protocol, amendments, informed consent, and other information requiring preapproval were reviewed and approved by an institutional review board. All patients provided written informed consent to participate in the study.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Ludbrook, V.J., Budd, D.C., Thorn, K. et al. Inhibition of Receptor-Interacting Protein Kinase 1 in Chronic Plaque Psoriasis: A Multicenter, Randomized, Double-Blind, Placebo-Controlled Study. Dermatol Ther (Heidelb) 14, 489–504 (2024). https://doi.org/10.1007/s13555-024-01097-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-024-01097-0