
Abstract
Plaque psoriasis is a systemic immune-mediated disease driven by interleukin-17 producing cells under the regulation of interleukin-23. Interleukin-23 signaling is mediated by the intracellular kinase tyrosine kinase 2, a Janus kinase family member. Tyrosine kinase 2 is a potential target for oral small-molecule therapies to treat psoriasis and psoriatic arthritis. A number of tyrosine kinase 2 inhibitors are in development or approved for the treatment of psoriasis or psoriatic arthritis. Deucravacitinib, an oral, selective, allosteric tyrosine kinase 2 inhibitor, is approved by the US Food and Drug Administration as a first-in-class treatment for adults with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy, and is approved by Pharmaceuticals and Medical Devices Agency (PDMA) in Japan for patients with plaque psoriasis, generalized pustular psoriasis, and erythrodermic psoriasis who have had an inadequate response to conventional therapies. Deucravacitinib selectively binds to the unique tyrosine kinase 2 regulatory pseudokinase domain in an allosteric fashion, preventing a conformational change in the catalytic domain required for ATP substrate binding, thus effectively locking tyrosine kinase 2 in an inactive state. Two other tyrosine kinase 2 inhibitors in later stage clinical development, brepocitinib (PF-06700841) and ropsacitinib (PF-06826647), are orthosteric inhibitors that target the highly conserved catalytic domain. This selective allosteric tyrosine kinase 2 inhibition may explain the improved safety profile of deucravacitinib versus orthosteric Janus kinase and tyrosine kinase 2 inhibitors. Two phase 3 psoriasis trials demonstrated deucravacitinib was efficacious and not associated with safety concerns characteristic of Janus kinase inhibitors, hence the new class designation (TYK2 inhibitor) by health authorities in the USA and Japan. Allosteric tyrosine kinase 2 inhibitors represent a promising new class of molecules for the treatment of psoriasis and psoriatic arthritis, and longer-term trials will establish their place in therapy.
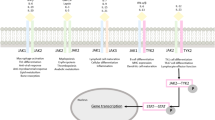
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
There are four kinases in the Janus kinase (JAK) family: JAK1, JAK2, JAK3, and tyrosine kinase 2 (TYK2). |
JAK inhibitors are associated with toxicities at efficacious doses, including infections, hematologic abnormalities, increases in high- and low-density lipoproteins and triglycerides, and other effects (reduced kidney function, increased creatinine phosphokinase levels, and liver toxicities). |
TYK2 inhibition is an alternate strategy for targeting the critical signal linkage between interleukin (IL)-23 and IL-17, thereby providing an improved benefit–risk profile. |
An allosteric TYK2 inhibitor, deucravacitinib, is approved for psoriasis and also in development for psoriatic arthritis, while two orthosteric TYK2 inhibitors are in development for psoriasis (brepocitinib and ropsacitinib) and psoriatic arthritis (brepocitinib). |
Deucravacitinib binds in an allosteric fashion to selectively inhibit TYK2, while ropsacitinib and brepocitinib are orthosteric inhibitors that bind to the catalytic domain and inhibit TYK2 and at least one other JAK. |
Digital Features
This article is published with digital features, including a graphical abstract, to facilitate understanding of the article. To view digital features for this article, go to https://doi.org/10.6084/m9.figshare.21716786.
Introduction
Plaque psoriasis (PsO), a chronic, systemic immune-mediated inflammatory skin disorder, affects patients’ physical and mental health, work productivity, and overall quality of life (QoL) [1,2,3]. Up to one-third of patients with PsO will develop psoriatic arthritis (PsA), and 10–40% of patients with PsO may have undiagnosed PsA [4, 5].
Current therapeutic options for PsO and PsA include biologics and small-molecule oral systemic agents. Biologics are effective but need intravenous or subcutaneous administration, and some are associated with loss of efficacy over time, safety issues, risk of immunogenicity, and relative expense [6, 7]. In contrast, small-molecule systemic agents are administered orally or topically, which can improve patient adherence and health-related QoL, and reduce healthcare costs [6]. However, conventional oral non-targeted therapies (methotrexate, cyclosporine, acitretin) are associated with adverse effects and long-term safety issues [6]. Apremilast, an oral targeted small molecule, provides limited therapeutic benefit in moderate-to-severe PsO or PsA [8, 9]. An unmet need exists for novel targeted oral therapies that are safe and highly efficacious in patients with PsO or PsA.
Chronic inflammation in PsO is maintained largely by interleukin (IL)-23, which maintains the differentiation of naive T cells into T-helper type 17 (Th17) cells, characterized by secretion of proinflammatory IL-17, and promotes their expansion [10]. IL-23 signal transduction is mediated by tyrosine kinase 2 (TYK2), which provides the critical link between IL-23 and IL-17, making TYK2 a potential target for regulating Th17 cell pathways [7]. Genetic studies showed a TYK2 coding variant that prevents receptor-mediated activation of TYK2 provided protection from several autoimmune diseases, including PsO [11].
There are three TYK2 inhibitors either approved or in later stage clinical development: deucravacitinib and brepocitinib, for both PsO and PsA, and ropsacitinib, only for PsO. Deucravacitinib is approved as a first-in-class TYK2 inhibitor drug by the US Food and Drug Administration (FDA) for the treatment of adults with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy and by the Pharmaceuticals and Medical Devices Agency (PDMA) in Japan for the treatment of patients with plaque psoriasis, generalized pustular psoriasis, and erythrodermic psoriasis who have had an inadequate response to conventional therapies [12, 13]. This review provides an overview of this new class of TYK2 inhibitors and how they differentiate from currently available treatments.
Literature Search Strategy
Randomized clinical trials of TYK2 inhibitors were identified via PubMed and limited to English language articles published up to 31 October 2022. To capture recent unpublished clinical data on these three compounds, we included recent congress presentations. Studies were included if the primary focus was a targeted TYK2 inhibitor being studied for PsO or PsA. It is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Current Targeted Systemic and Biologic Treatment Landscape
Therapies that target inflammatory pathways upregulated in PsO and PsA pathogenesis can be grouped by whether they target extracellular pathways (biologics) or intracellular pathways (small molecules) (Table 1) [8, 14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40].
Targeting Extracellular Cell Signaling Pathways
Tumor necrosis factor alpha (TNF-α) is a proinflammatory cytokine upregulated in the skin of patients with PsO [41]. TNF-α antagonists likely have an indirect effect on IL-17 through regulation of IL-23 production by dendritic cells [3]. All TNF-α antagonists have a boxed warning for serious infections and malignancies, and the most common adverse events (AE) are injection-site reactions, headache, and rash (Table 1) [14,15,16,17].
IL-12 and IL-23 inhibitors block the differentiation and activation of Th1 and Th17 cells, respectively (Table 1) [3, 42,43,44]. The IL-23 inhibitors bind the p19 subunit of IL-23, while ustekinumab targets both IL-12 and IL-23 through binding their shared p40 subunit [45]. The most common AEs associated with IL-23 and IL-12/23 inhibitors include infections, injection-site reactions, and headache (Table 1) [19,20,21,22].
IL-17 inhibitors act downstream of IL-23 through binding the IL-17 ligand or its receptors (Table 1) [46,47,48]. The most common AEs associated with IL-17 inhibitors are injection-site reactions, fungal infections (eg, candidiasis), and upper respiratory tract infections (URTIs) [23,24,25,26, 40]. In rare cases, IL-17 inhibitors have been associated with inflammatory bowel disease (IBD) onset [49], while brodalumab has a boxed warning for suicidal ideation and behavior [40].
Targeting Intracellular Cell Signaling Pathways
Apremilast, a small-molecule inhibitor of the phosphodiesterase-4 enzyme, is the only approved targeted oral therapy for PsO, and is also approved for PsA [8]. Apremilast reduces inflammation by inhibiting expression of proinflammatory cytokines and promoting expression of antiinflammatory molecules [8]. The most common AEs associated with apremilast include nausea, diarrhea, headache, and URTI (Table 1) [39].
Many cytokines linked to immune-mediated disease pathogenesis activate intracellular signaling pathways mediated by the Janus kinase-signal transducer and activator of transcription (JAK-STAT) signaling pathway [6, 50, 51]. There are four kinases in the JAK family: JAK1, JAK2, JAK3, and TYK2 [50]. JAK-STAT signaling inhibition has recently been reviewed elsewhere [6, 52]; an overview of the available JAK inhibitors (JAKinibs) for PsO and PsA is given here. Tofacitinib, a JAK1/3 inhibitor, was rejected by the US Food and Drug Administration for the treatment of PsO due to safety concerns, but is approved for PsA treatment (Table 1) [6, 34]. Upadacitinib, a JAK1 inhibitor, was not pursued in PsO, but is approved for PsA [6, 36]. JAKinibs are associated with toxicities at efficacious doses, including infections, hematologic abnormalities, increases in high- and low-density lipoproteins (HDL and LDL) and triglycerides, and other effects [reduced kidney function, increased creatinine phosphokinase (CPK) levels, and liver toxicities] [35, 53]. These safety concerns led the FDA to require boxed warnings about increased risk of serious heart-related events, cancer, blood clots, and death for JAKinibs, including tofacitinib, baricitinib, and upadacitinib [54].
However, TYK2-mediated signaling is limited to select inflammatory cytokine pathways [55]. The emerging class of TYK2 inhibitors (TYK2inibs) targets a similar IL-12/IL-23 pathway to many of the extracellular acting drugs, particularly ustekinumab, which also targets both of these pathways. Allosteric and orthosteric classes of TYK2inibs and how they differ from JAKinibs are further explored below.
Differentiating Between JAK1/2/3 and TYK2
Overview of JAK-STAT Signaling Pathway
The JAK-STAT pathway mediates signaling downstream of type I and type II cytokines (grouped by the shared structural elements in their receptors), which include many proinflammatory mediators such as IL-4, IL-5, IL-6, IL-12, IL-22, IL-23, and type I and II interferons (IFN) [35, 51, 53, 55, 56]. JAK enzymes have two domains: the catalytic domain that contains the adenosine triphosphate (ATP) binding site, and the regulatory pseudokinase domain (Fig. 1a) [28, 57, 58]. In its resting state, the enzyme is inactive because ATP is unable to bind; however, when a cytokine binds its receptor on the cell surface, the JAK enzyme undergoes a conformational change, allowing ATP access to its binding site on the catalytic domain (Fig. 1a).

The variety of inflammatory mediators that signal through JAK-STAT signaling pathways make JAKs an intriguing target for immunomodulatory drugs. However, cytokine signaling through JAK1/2/3 pathways is also important for cellular functions that are disrupted with JAKinibs (Fig. 2) [35, 51, 53, 55, 56, 59,60,61]. Increased HDL, LDL, and triglycerides as well as liver toxicities seen with JAKinibs are likely due to inhibition of IL-6 signaling, as IL-6 antagonists have similar effects [35, 53]. Natural killer (NK) cells, which are critical for antiviral defense, depend on JAK3-mediated cytokine signaling for their development and function [55]. Hematopoietic events such as the formation of erythrocytes, platelets, neutrophils, and lymphocytes are dependent on JAK2-mediated erythropoietin and thrombopoietin signaling [53, 55].

Differentiating TYK2 and JAK signaling pathways [35, 51, 53, 55, 56, 59,60,61]. EPO erythropoietin, GH growth hormone, GM-CSF granulocyte macrophage colony-stimulating factor, IFN interferon, IL interleukin, JAK Janus kinase, MHC major histocompatibility complex, Th helper T cell, TNFα tumor necrosis factor alpha, TPO thrombopoietin, Treg regulatory T cell, TYK2 tyrosine kinase 2
TYK2 Signaling Pathway
TYK2 plays a central role in PsO and PsA pathophysiology via regulation of signaling downstream of the IL-12, IL-23, and type I IFN-α and IFN-β receptors (Fig. 2) [28, 51]. IL-12 is involved in the development of Th1 cells, which release TNF-α and IFN-γ. IL-23 controls the expansion and survival of Th17 cells and Th22 cells, which produce IL-17 and IL-22, respectively [56]. Th1, Th17, and Th22 cells mediate keratinocyte activation, epidermal hyperplasia, and tissue inflammation inherent in PsO and PsA [50, 56]. IFN-α and IFN-β mediate inflammatory events, including dendritic cell activation, B-cell antibody production, Th1 and Th17 cell polarization, and reduction in regulatory T-cell function, and may also play a role in the initiation of psoriatic plaques [51, 62]. While all these pathways play a role in PsO and PsA pathogenesis, IL-23 and IL-17 are considered the key cytokines in initiating and maintaining chronic inflammation and TYK2 is the critical intracellular signal transduction link between them (Fig. 3) [3, 51].

TYK2 connects IL-23 and IL-17 [3, 51]. TYK2 is the critical intracellular transduction link between interleukin (IL)-23 and the production of IL-17. Inhibition of TYK2 signaling by allosteric (alloTYK2inib) or orthosteric (orthoTYK2inib) inhibitors breaks the link between IL-23 and IL-17 production. JAK Janus kinase, STAT signal transducer and activator of transcription, Th17 T-helper cell type 17, TYK2 tyrosine kinase 2, TYK2inib tyrosine kinase 2 inhibitor
Inhibition of TYK2 Signaling
There are two types of TYK2 inhibitors: allosteric inhibitors indirectly change the conformation of an enzyme by binding a site other than the active site and thus render the enzyme inactive, and orthosteric competitive inhibitors compete for the active binding site (Fig. 1b). Inhibition of TYK2 signaling breaks the link between IL-23 and IL-17 (Fig. 3), thereby inhibiting critical PsO and PsA signaling and producing a significant clinical response. Three oral inhibitors targeting TYK2 are approved or in clinical development: deucravacitinib (FDA and PDMA approved), ropsacitinib, and brepocitinib. Deucravacitinib is a selective TYK2 inhibitor designed to bind in an allosteric fashion to the regulatory pseudokinase domain rather than the highly conserved catalytic domain where ropsacitinib and brepocitinib bind (Fig. 1) [28, 57, 58]. This is an important distinction because there is far greater structural differentiation between TYK2 and JAK1/2/3 in the regulatory pseudokinase domain than in the catalytic domain [57, 58]. Allosteric binding to the regulatory pseudokinase domain blocks the receptor-mediated conformational change of the catalytic domain required for ATP binding, locking TYK2 in an inactive state and preventing downstream signaling of IL-23, IL-12, or Type 1 interferons, which are involved in PsO and PsA pathogenesis (Fig. 1) [28, 57, 58]. Ropsacitinib, a dual TYK2 and JAK2 inhibitor, is an orthosteric inhibitor that competes with ATP for binding to the active site on the catalytic domains of TYK2 and JAK2 [32]. Brepocitinib, which is also an orthosteric competitive inhibitor, binds TYK2, JAK1, and JAK2 [30].
In human cellular assays, deucravacitinib, ropsacitinib, and brepocitinib potently inhibited TYK2-dependent IL-12, IL-23, and IFN-α signaling [28, 30, 32]. Brepocitinib is a potent inhibitor of JAK1/3-dependent IL-2 signaling, and brepocitinib and ropsacitinib potently inhibit JAK2-dependent erythropoietin signaling [28, 30]. At clinically relevant concentrations, simulated daily average TYK2 inhibition by deucravacitinib was > 50%, whereas minimal inhibition (≤ 1%) of JAK1/3 and JAK2/2 occurred [63]. The orthosteric competitive JAKinibs tofacitinib, upadacitinib, and baricitinib inhibited JAK1/3 and JAK2/2 to varying degrees (23–94%) but produced no meaningful TYK2 inhibition (≤ 2%). Compared with JAK1/2/3 inhibitors, deucravacitinib inhibition of JAK1/3-dependent IL-2 signaling and JAK2/2-dependent thrombopoietin signaling was more than 97- and 46-fold less potent, respectively, in whole blood assays, consistent with its profile as a selective TYK2 inhibitor [63]. As tofacitinib, upadacitinib, and baricitinib do not inhibit TYK2 at therapeutic concentrations, this would suggest AEs typical of JAKinibs are not due to TYK2 inhibition.
Taken together, these data show that TYK2 inhibition targets similar pathways, i.e., IL-23/IL-17, to many of the currently available biologic treatment options, but has the advantage of being orally administered, which may increase patient adherence. In comparison to currently available oral treatments such as apremilast (the only other approved oral treatment in PsO besides deucravacitinib, and also approved in PsA) and JAKinibs (none approved in PsO but some approved in PsA), TYK2 inhibition provides greater selectivity for immune pathways, potentially reducing safety issues.
Other TYK2 inhibitors in early stage (phase 1) development include NDI-034858 (Nimbus Therapeutics), VTX-958 (Ventyx Biosciences), ESK-001 (Esker/Alumis Therapeutics), and ICP-332 (Innocare). Top-line results from a phase 1b trial of another TYK2 inhibitor, GLPG3667 (Galapagos, NV, USA), in 31 patients with moderate-to-severe PsO indicate it was safe and well tolerated with positive efficacy signals observed with a high dose (NCT04594928) [64].
Clinical Profile of TYK2 Inhibitors in PsO
Deucravacitinib
Deucravacitinib, an oral, once-daily, selective, allosteric TYK2 inhibitor (alloTYKinib), is a first-in-class TYK2 inhibitor approved by the US FDA for the treatment of adults with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy [12]. In a phase 2 trial (NCT02931838) of deucravacitinib in patients with moderate-to-severe PsO, the proportion of patients who achieved a 75% or greater improvement from baseline in Psoriasis Area and Severity Index (PASI 75) at week 12 (primary endpoint) was significantly higher, with deucravacitinib 3 mg twice daily (BID; 69%), 6 mg BID (67%), and 12 mg daily (QD; 75%) compared with placebo (7%; P < 0.001) [27].
Results from two phase 3 trials, POETYK PSO-1 (NCT03624127) and POETYK PSO-2 (NCT03611751), showed deucravacitinib 6 mg QD treatment was superior to apremilast and placebo in PASI 75 response at week 16 (53.0% to 58.4% versus 35.1% to 39.8% versus 9.4% to 12.7%, respectively; P < 0.001 for all) [65, 66]. Of patients who had achieved PASI 75 at week 24 with deucravacitinib treatment and continued treatment, 81.3% (PSO-1) and 81.8% (PSO-2) maintained PASI 75 response at week 52. In PSO-1, of patients treated with deucravacitinib who achieved PASI 75 at week 16, 91.0% maintained a PASI 75 response at 2 years [67]. In PSO-1, a greater proportion of patients treated with deucravacitinib achieved PASI 90 compared with patients in the placebo and apremilast groups at week 16 (35.5% versus 4.2% versus 19.6%, respectively; P ≤ 0.0002 for all) and compared with the apremilast group at week 24 (42.2% versus 22.0%; P < 0.0001). Furthermore, 62.6% of patients who achieved PASI 75 at week 16 also achieved PASI 90 at week 16, and this PASI 90 response rate was maintained for up to 2 years (63.0%). A greater percentage of patients in the deucravacitinib group compared with the apremilast group achieved complete skin clearance, as assessed by PASI 100 response, at week 16 and at week 24 in the PSO-1 trial (week 16, deucravacitinib 14.2% versus apremilast 3.0%, P < 0.0001; week 24, deucravacitinib 17.5% versus apremilast 6.5%, P = 0.0007). In PSO-1 and PSO-2, the percentages of patients achieving a static Physician’s Global Assessment (sPGA) score of 0 or 1 at week 16 were 49.5% to 53.6% with deucravacitinib treatment compared with 32.1% to 33.9% in the apremilast group and 7.2% to 8.6% in placebo (P < 0.0001 for all). In patients with a scalp-specific Physician’s Global Assessment (ss-PGA) score greater than 3 (moderate to severe) at baseline, 59.7% to 70.3% of those treated with deucravacitinib achieved an ss-PGA of 0/1 (clear/almost clear) at week 16 compared with 36.7% to 39.1% in the apremilast group and 17.3% to 17.4% in the placebo group (P < 0.0001 for all). In the pooled PSO-1 and PSO-2 population of patients with moderate-to-severe fingernail psoriasis, defined as PGA-Fingernails (PGA-F) ≥ 3 at baseline (moderate to severe), the proportion of patients who achieved PGA-F 0/1 (clear/minimal) at week 16 was significantly greater with deucravacitinib versus placebo (20.5% versus 8.3%, P = 0.0272) [68]. In patients with moderate-to-severe palmoplantar psoriasis (palmoplantar PGA [pp-PGA] ≥ 3 at baseline) in the pooled population, a greater proportion of patients achieved pp-PGA 0/1 at week 16 with deucravacitinib compared with placebo (49.1% versus 16.0%, P = 0.0052). Patients treated with deucravacitinib also experienced significant patient-reported QoL improvements at week 16 in the PSO-1 and PSO-2 trials, as measured by greater reduction from baseline in Psoriasis Symptoms and Signs Diary scores [adjusted mean change from baseline (SE), PSO-1 −26.7 (1.8) versus −17.8 (2.2) versus −3.6 (2.1) and PSO-2 −28.3 (1.1) versus −21.1 (1.4) versus −4.7 (1.4) for deucravacitinib versus apremilast versus placebo, respectively; P < 0.0001 for all] and a score of 0 or 1 on the Dermatology Life Quality Index (DLQI; PSO-1 41.0% versus 28.6% versus 10.6% and PSO-2 in patients with DLQI score ≥ 2 at baseline 37.6% versus 23.1% versus 9.8%; P < 0.01 for all). Serious AEs and treatment discontinuations due to AEs were infrequent in deucravacitinib-treated patients.
Deucravacitinib is approved as a first-line systemic therapy at a dosage of 6 mg QD and is not recommended for use in combination with other potent immunosuppressants. The most common AEs (≥ 1%) in the PSO-1 and PSO-2 trials through week 16 with deucravacitinib treatment, and observed at a higher rate than in the placebo group, were URTI, blood CPK increased, herpes simplex, mouth ulcers, folliculitis, and acne (Table 1) [12].
Ropsacitinib
Ropsacitinib, an orthosteric competitive catalytic-site inhibitor that binds TYK2 and JAK2, has been studied in patients with moderate-to-severe PsO in a phase 1 trial and a phase 2b trial. In the phase 1, double-blind, placebo-controlled trial, 40 patients with moderate-to-severe PsO were randomized to ropsacitinib (100 mg or 400 mg) or placebo QD for 28 days (NCT03210961) [33]. Ropsacitinib was well tolerated and efficacious in reducing disease activity at 28 days, as measured by PASI 75, target plaque severity score, and body surface area [33]. In the phase 2b, double-blind, placebo-controlled trial, 179 patients were randomized 1:1:2:2:2 to ropsacitinib (50 mg, 100 mg, 200 mg, or 400 mg) or placebo QD for 16 weeks, after which patients on the 200 mg or 400 mg dose continued this dose and patients randomized to other doses were rerandomized to 200 mg or 400 mg QD (NCT03895372) [69]. At week 16, a significantly greater proportion of patients achieved PASI 90, the primary endpoint, with ropsacitinib at a dose of 200 mg (33.0%, P = 0.0004) and 400 mg (46.5%, P < 0.0001) compared with placebo. Similarly significant results were reported for PASI 75 at week 16 for ropsacitinib 200 mg and 400 mg versus placebo. A Physician’s Global Assessment (PGA) response, defined as a PGA score of 0/1 (clear/almost clear) with a decrease from baseline ≥ 2 points, was achieved by a numerically greater proportion of patients across all treatment groups compared with placebo from weeks 6 to 16. The most common treatment-emergent AEs in patients treated with ropsacitinib up to week 16 were nasopharyngitis, URTI, and increased blood pressure (Table 1) [69]. AEs due to hematology, chemistry, and lipid laboratory abnormalities were observed in a dose-dependent manner. There were no deaths, adjudicated cardiovascular events, or treatment-related clinically detectable findings in electrocardiogram (ECG) [33, 69, 70].
Brepocitinib
Brepocitinib, an orthosteric competitive catalytic-site inhibitor that binds TYK2, JAK1, and JAK2, has completed phase 1 and phase 2 trials in patients with moderate-to-severe PsO. Oral brepocitinib is not being further pursued in PsO; phase 2 trials will evaluate the topical formulation in mild-to-moderate PsO (NCT03850483). In a phase 1 trial in healthy subjects and patients with moderate-to-severe PsO (NCT02310750), brepocitinib was generally safe and well tolerated [31]. In a phase 2a trial in patients with moderate-to-severe PsO, 212 patients were randomized to brepocitinib (30 mg or 60 mg) or placebo QD for 4 weeks (induction period), followed by 10 mg QD, 30 mg QD, 100 mg once weekly, or placebo (8-week maintenance) (NCT02969018). Significantly greater changes in PASI at week 12 were reported in brepocitinib-treated patients compared with placebo (least square mean change −17.3 with 30 mg QD continuous treatment; P < 0.05) [29]. At week 12, the proportion of patients achieving PASI 75 and PASI 90 was greater with all brepocitinib treatment groups compared with placebo, with the highest achievement observed in the 30 mg QD continuous treatment group (PASI 75: 25/29, 86.2%; PASI 90: 15/29, 51.7%). The most common AEs in brepocitinib-treated patients were nasopharyngitis, URTI, and headache (Table 1) [29]. Biomarker analyses indicated brepocitinib treatment reduced inflammatory gene expression in PsO lesions to levels observed in nonlesional skin; reductions in inflammatory gene expression correlated with improvements in histologic and clinical outcomes [71].
Clinical Profile of TYK2 Inhibitors in PsA and Other Indications
A phase 2 trial of deucravacitinib in patients with active PsA (NCT03881059) demonstrated efficacy of 52.9% to 62.7% in patients treated with deucravacitinib 6 mg QD and 12 mg QD, respectively, compared with 31.8% in placebo-treated patients, in American College of Rheumatology 20 response at week 16 (P < 0.05) [72]. Most AEs were mild to moderate, and the most common AEs (≥ 5%) in patients treated with deucravacitinib were nasopharyngitis, URTI, sinusitis, bronchitis, rash, headache, and diarrhea. There were no serious AEs and no occurrence of herpes zoster infections, major cardiovascular events, or significant changes in laboratory parameters with deucravacitinib treatment. Phase 3 trials of deucravacitinib in patients with PsA are currently ongoing. Topline results from a phase 2 trial in patients with active systemic lupus erythematosus (SLE) showed that deucravacitinib was significantly more efficacious than placebo in multiple endpoints and was well tolerated [73]. Additional phase 2 trials are evaluating the efficacy of deucravacitinib in moderate-to-severe UC, CD, and cutaneous lupus erythematosus.
While ropsacitinib is not currently being pursued in PsA, a phase 2 trial will evaluate ropsacitinib and brepocitinib in moderate-to-severe hidradenitis suppurativa. Phase 2 trials are exploring the efficacy of oral brepocitinib in active PsA, active non-segmental vitiligo, and moderate-to-severe SLE, CD, and UC, and as a topical cream in mild-to-moderate atopic dermatitis.
Conclusions
An improved understanding of PsO and PsA pathophysiology has resulted in therapies that target inflammatory mediators upregulated in these diseases, in particular IL-23 and its downstream signaling pathways, including IL-17. While many of these biologic therapies are efficacious, there is still an unmet need for effective and safe oral therapies targeting these same pathways.
TYK2 regulates cytokine signaling central in PsO pathogenesis (e.g., IL-23, IL-12, and type 1 interferons) and has been confirmed by genetic studies as a target in PsO. TYK2 inhibition is an alternate strategy for targeting the critical signal linkage between IL-23 and IL-17. Three TYK2 inhibitors are approved or in later stage clinical development. Deucravacitinib, an allosteric inhibitor of TYK2, is approved in the USA for treatment of patients with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy and in Japan for treatment of patients with plaque psoriasis, generalized pustular psoriasis, and erythrodermic psoriasis who have had an inadequate response to conventional therapies. Deucravacitinib binds to the regulatory pseudokinase domain, resulting in selective allosteric inhibition of TYK2 [28]. In contrast, the orthosteric TYK2 inhibitors, brepocitinib and ropsacitinib, target the active site in the catalytic domain and are potent inhibitors of 2 or more JAK family kinases [30, 32]. Allosteric inhibition using therapeutic doses of deucravacitinib results in high selectivity for TYK2 versus JAK1/2/3, thereby avoiding the safety implications associated with JAK1/2/3 inhibition as well as orthosteric TYK2 inhibitors [28]. Deucravacitinib, based on the safety profile observed in the two large phase 3 PsO trials, did not receive a boxed warning similar to what is found on JAK inhibitors (e.g., tofacitinib and upadacitinib) for serious infections, malignancies, thrombosis, and death. The FDA approval of deucravacitinib notes that it is not yet known whether TYK2 inhibition will result in the potential adverse reactions associated with JAK inhibition [12]. This suggests that the FDA views TYK2 inhibitors as a separate class of drugs from JAK inhibitors and places deucravacitinib as the first approved therapy in this new class. Long-term data will add to the current clinical evidence. Because of the unique mechanism of action of deucravacitinib, it should be regarded as the first in a new class of TYK2 inhibitor, an allosteric TYK2 inhibitor (alloTYKinib), compared with orthosteric nonselective TYK2 inhibitors (orthoTYKinib). Longer-term efficacy and safety studies will help to establish and reinforce the optimal positioning of TYK2 inhibitors in the PsO and PsA therapeutic armamentarium.
References
Global Report on Psoriasis. Switzerland: World Health Organization, 2016.
Armstrong AW, Read C. Pathophysiology, clinical presentation, and treatment of psoriasis: a review. JAMA. 2020;323(19):1945–60.
Hawkes JE, Yan BY, Chan TC, Krueger JG. Discovery of the IL-23/IL-17 signaling pathway and the treatment of psoriasis. J Immunol. 2018;201(6):1605–13.
Ritchlin CT, Colbert RA, Gladman DD. Psoriatic arthritis. N Engl J Med. 2017;376(10):957–70.
Mease PJ, Palmer JB, Hur P, Strober BE, Lebwohl M, Karki C, et al. Utilization of the validated Psoriasis Epidemiology Screening Tool to identify signs and symptoms of psoriatic arthritis among those with psoriasis: a cross-sectional analysis from the US-based Corrona Psoriasis Registry. J Eur Acad Dermatol Venereol. 2019;33(5):886–92.
Nogueira M, Puig L, Torres T. JAK inhibitors for treatment of psoriasis: focus on selective TYK2 inhibitors. Drugs. 2020;80(4):341–51.
Kvist-Hansen A, Hansen PR, Skov L. Systemic treatment of psoriasis with JAK inhibitors: a review. Dermatol Ther (Heidelb). 2020;10(1):29–42.
Vangipuram R, Alikhan A. Apremilast for the management of moderate to severe plaque psoriasis. Expert Rev Clin Pharmacol. 2017;10(4):349–60.
Torres T, Puig L. Apremilast: a novel oral treatment for psoriasis and psoriatic arthritis. Am J Clin Dermatol. 2018;19(1):23–32.
Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278(3):1910–4.
Dendrou CA, Cortes A, Shipman L, Evans HG, Attfield KE, Jostins L, et al. Resolving TYK2 locus genotype-to-phenotype differences in autoimmunity. Sci Transl Med. 2016;8(363):363ra149.
Sotyktu [package insert]. Princeton, NJ, USA: Bristol-Myers Squibb Company, 2022.
Sotyktu Japan [package insert]. Tokyo, Japan: Bristol-Meyers Squibb K.K., 2022.
Humira [package insert]. North Chicago, IL: Abbott Laboratories (AbbVie), 2021.
Enbrel [package insert]. Thousand Oaks, CA: Immunex Corporation, 2021.
Cimzia [package insert]. Smyrna, GA: UCB, Inc., 2019.
Avsola [package insert]. Thousand Oaks, CA: Amgen, Inc., 2019.
Simponi Aria [package insert]. Horsham, PA, USA: Janssen Biotech, Inc., 2021.
Skyrizi [package insert]. North Chicago, IL, USA: AbbVie Inc., 2022.
Ilumya [package insert]. Cranbury, NJ: Sun Pharmaceutical Industries, Inc., 2020.
Tremfya [package insert]. Horsham, PA: Janssen Biotech, Inc., 2020.
Stelara [package insert]. Horsham, PA: Janssen Biotech, Inc, 2020.
Taltz [package insert]. Indianapolis, IN: Eli Lilly and Company, 2021.
Cosentyx [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation, 2021.
Reis J, Vender R, Torres T. Bimekizumab: the first dual inhibitor of interleukin (IL)-17A and IL-17F for the treatment of psoriatic disease and ankylosing spondylitis. BioDrugs. 2019;33(4):391–9.
Freitas E, Torres T. Bimekizumab: the new drug in the biologics armamentarium for psoriasis. Drugs Context. 2021;10(2021–4–1):1–4.
Papp K, Gordon K, Thaci D, Morita A, Gooderham M, Foley P, et al. Phase 2 trial of selective tyrosine kinase 2 inhibition in psoriasis. N Engl J Med. 2018;379(14):1313–21.
Burke JR, Cheng L, Gillooly KM, Strnad J, Zupa-Fernandez A, Catlett IM, et al. Autoimmune pathways in mice and humans are blocked by pharmacological stabilization of the TYK2 pseudokinase domain. Sci Transl Med. 2019;11(502):eaaw1736.
Forman SB, Pariser DM, Poulin Y, Vincent MS, Gilbert SA, Kieras EM, et al. TYK2/JAK1 inhibitor PF-06700841 in patients with plaque psoriasis: phase IIa, randomized, double-blind, placebo-controlled trial. J Invest Dermatol. 2020;140(12):2359-70.e5.
Fensome A, Ambler CM, Arnold E, Banker ME, Brown MF, Chrencik J, et al. Dual inhibition of TYK2 and JAK1 for the treatment of autoimmune diseases: discovery of (( S)-2,2-difluorocyclopropyl)((1 R,5 S)-3-(2-((1-methyl-1 H-pyrazol-4-yl)amino)pyrimidin-4-yl)-3,8-diazabicyclo[3.2.1]octan-8-yl)methanone (PF-06700841). J Med Chem. 2018;61(19):8597–612.
Banfield C, Scaramozza M, Zhang W, Kieras E, Page KM, Fensome A, et al. The safety, tolerability, pharmacokinetics, and pharmacodynamics of a TYK2/JAK1 inhibitor (PF-06700841) in healthy subjects and patients with plaque psoriasis. J Clin Pharmacol. 2018;58(4):434–47.
Gerstenberger BS, Ambler C, Arnold EP, Banker ME, Brown MF, Clark JD, et al. Discovery of tyrosine kinase 2 (TYK2) inhibitor (PF-06826647) for the treatment of autoimmune diseases. J Med Chem. 2020;63:13561–77.
Tehlirian C, Peeva E, Kieras E, Scaramozza M, Roberts ES, Singh RSP, et al. Safety, tolerability, efficacy, pharmacokinetics, and pharmacodynamics of the oral TYK2 inhibitor PF-06826647 in participants with plaque psoriasis: a phase 1, randomised, double-blind, placebo-controlled, parallel-group study. Lancet Rheumatol. 2021;3(3):e204–13.
Xeljanz [package insert]. New York, NY: Pfizer Inc., 2020.
Winthrop KL. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat Rev Rheumatol. 2017;13(4):234–43.
Rinvoq [package insert]. North Chicago, IL, USA: AbbVie Inc., 2022.
Cohen SB, van Vollenhoven RF, Winthrop KL, Zerbini CAF, Tanaka Y, Bessette L, et al. Safety profile of upadacitinib in rheumatoid arthritis: integrated analysis from the SELECT phase III clinical programme. Ann Rheum Dis. 2020;80(3):304–11.
McInnes IB, Byers NL, Higgs RE, Lee J, Macias WL, Na S, et al. Comparison of baricitinib, upadacitinib, and tofacitinib mediated regulation of cytokine signaling in human leukocyte subpopulations. Arthritis Res Ther. 2019;21(1):183.
Otezla [package insert]. Thousand Oaks, CA: Amgen Inc., 2021.
Siliq [package insert]. Bridgewater, NJ: Bausch Health US LLC, 2020.
Hawkes JE, Chan TC, Krueger JG. Psoriasis pathogenesis and the development of novel targeted immune therapies. J Allergy Clin Immunol. 2017;140(3):645–53.
Alwan W, Nestle FO. Pathogenesis and treatment of psoriasis: exploiting pathophysiological pathways for precision medicine. Clin Exp Rheumatol. 2015;33(5 Suppl 93):S2-6.
Nakamura M, Lee K, Jeon C, Sekhon S, Afifi L, Yan D, et al. Guselkumab for the treatment of psoriasis: a review of phase III trials. Dermatol Ther (Heidelb). 2017;7(3):281–92.
Chiricozzi A, Antonioli L, Panduri S, Fornai M, Romanelli M, Blandizzi C. Risankizumab for the treatment of moderate to severe psoriasis. Expert Opin Biol Ther. 2019;19(1):1–8.
Benson JM, Peritt D, Scallon BJ, Heavner GA, Shealy DJ, Giles-Komar JM, et al. Discovery and mechanism of ustekinumab: a human monoclonal antibody targeting interleukin-12 and interleukin-23 for treatment of immune-mediated disorders. MAbs. 2011;3(6):535–45.
Mease PJ, van der Heijde D, Ritchlin CT, Okada M, Cuchacovich RS, Shuler CL, et al. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase III trial SPIRIT-P1. Ann Rheum Dis. 2017;76(1):79–87.
Farahnik B, Beroukhim K, Abrouk M, Nakamura M, Zhu TH, Singh R, et al. Brodalumab for the treatment of psoriasis: a review of phase III trials. Dermatol Ther (Heidelb). 2016;6(2):111–24.
Fala L. Cosentyx (secukinumab): first IL-17A antagonist receives FDA approval for moderate-to-severe plaque psoriasis. Am Health Drug Benefits. 2016;9(Spec Feature):60–3.
Fieldhouse KA, Ukaibe S, Crowley EL, Khanna R, O'Toole A, Gooderham MJ. Inflammatory bowel disease in patients with psoriasis treated with interleukin-17 inhibitors. Drugs Context. 2020;9(2020–2–1):1–9.
Howell MD, Kuo FI, Smith PA. Targeting the Janus kinase family in autoimmune skin diseases. Front Immunol. 2019;10:2342.
Baker KF, Isaacs JD. Novel therapies for immune-mediated inflammatory diseases: what can we learn from their use in rheumatoid arthritis, spondyloarthritis, systemic lupus erythematosus, psoriasis, Crohn’s disease and ulcerative colitis? Ann Rheum Dis. 2018;77(2):175–87.
Krueger JG, McInnes IB, Blauvelt A. Tyrosine kinase 2 and Janus kinase-signal transducer and activator of transcription signaling and inhibition in plaque psoriasis. J Am Acad Dermatol. 2022;86:148–57.
Gadina M, Chisolm DA, Philips RL, McInness IB, Changelian PS, O’Shea JJ. Translating JAKs to Jakinibs. J Immunol. 2020;204(8):2011–20.
FDA requires warnings about increased risk of serious heart-related events, cancer, blood clots, and death for JAK inhibitors that treat certain chronic inflammatory conditions [press release]: U. S. Food and Drug Administration, 2021.
Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov. 2017;16(12):843–62.
Ergen EN, Yusuf N. Inhibition of interleukin-12 and/or interleukin-23 for the treatment of psoriasis: what is the evidence for an effect on malignancy? Exp Dermatol. 2018;27(7):737–47.
Tokarski JS, Zupa-Fernandez A, Tredup JA, Pike K, Chang C, Xie D, et al. Tyrosine kinase 2-mediated signal transduction in T lymphocytes is blocked by pharmacological stabilization of its pseudokinase domain. J Biol Chem. 2015;290(17):11061–74.
Wrobleski ST, Moslin R, Lin S, Zhang Y, Spergel S, Kempson J, et al. Highly selective inhibition of tyrosine kinase 2 (TYK2) for the treatment of autoimmune diseases: discovery of the allosteric inhibitor BMS-986165. J Med Chem. 2019;62(20):8973–95.
Dougan M, Dranoff G, Dougan SK. GM-CSF, IL-3, and IL-5 family of cytokines: regulators of inflammation. Immunity. 2019;50(4):796–811.
Duvallet E, Semerano L, Assier E, Falgarone G, Boissier MC. Interleukin-23: a key cytokine in inflammatory diseases. Ann Med. 2011;43(7):503–11.
Marrack P, Kappler J, Mitchell T. Type I interferons keep activated T cells alive. J Exp Med. 1999;189(3):521–30.
Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361(5):496–509.
Chimalakonda A, Burke J, Cheng L, Catlett I, Tagen M, Zhao Q, et al. Selectivity profile of the tyrosine kinase 2 inhibitor deucravacitinib compared with Janus kinase 1/2/3 inhibitors. Dermatol Ther (Heidelb). 2021;11(5):1763–76.
Galapagos reports positive topline results with selective TYK2 inhibitor GLPG3667 in Phase 1b psoriasis study [press release]. Mechelen, Belgium: Galapagos NV, 2021.
Armstrong AW, Gooderham M, Warren RB, Papp KA, Strober B, Thaçi D, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, placebo-controlled phase 3 POETYK PSO-1 trial. J Am Acad Dermatol. 2022;July 9 [online ahead of print].
Strober B, Thaçi D, Sofen H, Kircik L, Gordon KB, Foley P, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, phase 3 POETYK PSO-2 trial. J Am Acad Dermatol. 2022;September 14 [online ahead of print].
Lebwohl M, Warren RB, Sofen H, Imafuku S, Paul C, Szepietowski JC, et al. Deucravacitinib long-term efficacy with continuous treatment in plaque psoriasis: 2-year results from the phase 3 POETYK PSO study program [abstract] Annual Congress of the European Academy of Dermatology and Venereology. Milan, Italy, 2022.
Blauvelt A, Rich P, Sofen H, Lambert J, Merola JF, Lebwohl M, et al. Deucravacitinib, an oral, selective tyrosine kinase 2 (TYK2) inhibitor, versus placebo in scalp, nail, and palmoplantar psoriasis: subset analyses of the phase 3 POETYK PSO-1 and POETYK PSO-2 trials [poster] Annual Meeting of the Maui Derm for Dermatologists. Maui, HI, USA, 2022.
Tehlirian C, Singh RSP, Pradhan V, Roberts ES, Tarabar S, Peeva E, et al. Oral tyrosine kinase 2 inhibitor PF-06826647 demonstrates efficacy and an acceptable safety profile in participants with moderate-to-severe plaque psoriasis in a phase 2b, randomized, double-blind, placebo-controlled study. J Am Acad Dermatol. 2022;87(2):333–42.
Singh RSP, Pradhan V, Roberts ES, Scaramozza M, Kieras E, Gale JD, et al. Safety and pharmacokinetics of the oral TYK2 inhibitor PF-06826647: a phase I, randomized, double-blind, placebo-controlled, dose-escalation study. Clin Transl Sci. 2021;14:671–82.
Page KM, Suarez-Farinas M, Suprun M, Zhang W, Garcet S, Fuentes-Duculan J, et al. Molecular and cellular responses to the TYK2/JAK1 inhibitor PF-06700841 reveal reduction of skin inflammation in plaque psoriasis. J Invest Dermatol. 2020;140:1546–55.
Mease PJ, Deodhar AA, van der Heijde D, Behrens F, Kivitz AJ, Neal J, et al. Efficacy and safety of selective TYK2 inhibitor, deucravacitinib, in a phase II trial in psoriatic arthritis. Ann Rheum Dis. 2022;81(6):815–22.
Morand E, Pike M, Merrill JT, Van Vollenhoven R, Werth VP, Hobar C, et al. Efficacy and safety of deucravacitinib, an oral, selective, allosteric TYK2 inhibitor, in patients with active systemic lupus erythematosus: a phase 2, randomized, double-blind, placebo-controlled study [abstract LB0004]. Ann Rheum Dis. 2022;81(suppl 1):209.
Armstrong A, Gooderham M, Warren R, Papp K, Strober B, Thaçi D, et al. Efficacy and safety of deucravacitinib, an oral, selective tyrosine kinase 2 (TYK2) inhibitor, compared with placebo and apremilast in moderate to severe plaque psoriasis: results from the POETYK PSO-1 study annual meeting of the American Academy of Dermatology. Virtual meeting, 2021.
Acknowledgements
Funding
Funding for writing, editorial support, and Rapid Service Fee was provided by Bristol Myers Squibb, Princeton, NJ, USA.
Medical Writing, Editorial, and Other Assistance
Professional medical writing and editorial assistance was provided by Julianne Hatfield, PhD, of Peloton Advantage, LLC, an OPEN Health company, Parsippany, NJ, USA, and was funded by Bristol Myers Squibb, Princeton, NJ, USA.
Author Contributions
George Martin contributed to the conceptualization, drafting, and revisions of this manuscript, and read and approved the final manuscript.
Disclosures
George Martin has served as a paid consultant to AbbVie, Almirall, Bausch Health, Bristol Myers Squibb, Dermavent, Galderma, Leo Pharma, Lilly, Pfizer, and UCB; has served on the scientific advisory board for AbbVie, Almirall, Bausch Health, Bristol Myers Squibb, Dermavent, DUSA/SUN, Horizon, Incyte, Janssen, Leo Pharma, Lilly, and UCB; has received speaking fees from Almirall, Dermavent, Incyte, Leo Pharma, and UCB; and is a stockholder of Doc Martin’s of Maui (Sunscreen Co.).
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Martin, G. Novel Therapies in Plaque Psoriasis: A Review of Tyrosine Kinase 2 Inhibitors. Dermatol Ther (Heidelb) 13, 417–435 (2023). https://doi.org/10.1007/s13555-022-00878-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-022-00878-9