
Abstract
Extraction of lupeol from lupin hulls has been carried out using supercritical CO2 extraction technology under different operating conditions in order to obtain value-added extracts from the raw material of industrial lupin. The operational parameters used include CO2 pressure and flow and sequential depressurization fractionation. The highest lupeol recovery (96.8%) has been obtained using 320 bar and 50 g/min of CO2. For sequential depressurization, the best results were obtained with a CO2 density close to 728 kg/m3 providing up to 92% of lupeol in the extract and an enrichment factor of 1.2. Despite this high enrichment, lupeol recovery decreases to 50% after fractionation. Better extraction recoveries would have been expected and thus further studies are necessary to improve the extraction recovery of extracts with a high lupeol composition.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Food agro-industry generates around 12 to 40% of food waste at different steps in the supply chain according to the European Commission [1]. These raw materials have a strong negative impact on both environment and economy, and consequently, in the consumer population. Biomass such as husk, seeds, hulls and pulp remnants are considered by-products of food processing industry [2]. However, several studies have shown that many of these by-products have relevant contents of bioactive molecules with potential health benefits that could be extracted and used as ingredients in functional foods or nutraceutical supplements [3]. Besides, current strategies for food waste management, such as incineration, fabrication of compost or animal feeding, are not based on environmentally friendly concepts and optimum valorization. For this reason, promotion of new strategies directed to reduce the waste material and to create value-added products have become necessary for environmental sustainability and circular economy [4, 5].
Lupinus, commonly known as lupin, is a genus of plants of the legume family Fabaceae. Lupin seeds represent a promising nutrient source for livestock and humans [6]. The use of lupin seed flour rich in protein and fiber is spreading rapidly, and it has been used recently in bakery and meat products [7,8,9]. However, lupin seed hulls, which accounts for about 20% of the total weight of the seed, have been considered as by-products with increasing interest [10]. This material shows low value for animal feeding because it provides low metabolizable energy due to its high content of indigestible fiber. In our opinion, this concept of metabolizable energy should be reviewed for the optimum animal welfare.
Interestingly, some studies have shown the enormous potential of lupin hulls as a novel source of health promoting food ingredients and, in particular, as a source of dietary fiber and phytochemicals [10, 11]. The fat content of lupin hulls is very low, around 1.5 to 2.4% of the total fat content, and for this reason there is a lack of published data related to oil extraction from them. Despite this, several studies have demonstrated significant abundance of lupeol in fat isolated from lupin hulls [12, 13].
Lupeol is a triterpene alcohol associated to several bioactive properties. A large number of in vitro and in vivo studies have shown its extensive range of pharmacological effects and benefits for human health, such as anticancer, anti-inflammatory, antioxidant, antimicrobial, and also cardiovascular, neurodegenerative and hepatic protection effects [14, 15]. Different plants and fruits, such as olive, mango, aloe vera, oak and elm leaves, show a moderately low concentration of lupeol (3–880 µg/g) [15]. In any case, the majority of the previous studies, which have focused on the extraction of lupeol from lupin seed hulls, have shown not to be environmentally friendly due to the use of conventional organic solvents, such as the chloroform:methanol mixture or hexane [15]. In some cases, sonication and microwaves have been employed to obtain lupeol from raw materials [16].
Due to the low concentration in which bioactive compounds are found in plant tissues, an effective and selective extraction technology becomes necessary to arise an efficient isolation. In this line, special attention has been paid to the development of processes based on the application of green technologies and the use of environmentally-friendly solvents, to rule out the use of conventional and well-known toxic solvents, such as hexane [17, 18]. In recent years, supercritical fluid extraction (SFE) and, in particular, the use of carbon dioxide (CO2) have become an interesting extraction technique, not only because CO2 is a substance generally recognized as safe (GRAS), but also because it is used in a large number of industrial applications [19]. This technology shows interesting advantages, such as high selectivity and reproducibility, and thereby, faster mass transfer and limited extraction time, and it involves low and mild extraction temperatures [18]. In addition, supercritical CO2 can change to the gaseous state and thus it can be eliminated from the solid extract, providing solvent-free extracts. CO2 can still be recycled through recirculation in the supercritical system in order to avoid environmental issues as well as to decrease energy and other operational costs [20]. The selection of operational parameters, mainly temperature and pressure, has a critical impact on the final composition of the extracts, which translates into the selectivity of the extraction process [21]. The use of supercritical CO2 is manly focused in the extraction of non-polar compounds due to its non-polar nature, and thus lupeol would be a target molecule for this type of solvent [22].
The main aim of the present work is the extraction of lupeol from lupin hulls by-products by using green technologies, in particular supercritical CO2, to obtain value-added ingredients. In the present study, different operational parameters such as CO2 pressure and flow have been explored. A fractionation process by sequential depressurization has been carried out for the obtention of lupeol enriched extract. The results obtained by using supercritical CO2 extraction have been compared with those obtained by using solid–liquid conventional extraction with hexane and concepts such as composition and recovery for the different extraction processes have been discussed in depth. Supercritical CO2 shows to be a promising and clean extraction method to obtain lupeol with high purity and good recoveries, even better than those obtained when using hexane. Moreover, this technology has the potential to be scaled-up for food industry applications.
2 Materials and methods
2.1 Materials
White lupin hulls and white lupin seeds (Lupinus albus) were supplied by Inveja SAS-Lup'ingredients (Haute-Goulaine, France). The material was stored under vacuum and refrigeration conditions (4 °C). For the SFE, CO2 with a purity of 99.98% was obtained from Carburos Metálicos (Madrid, Spain). Standards of stigmasterol ≥ 95%, lupeol ≥ 98% and 1,3-diolein ≥ 99% were purchased from Merck (Darmstadt, Germany). The lupin seed oil, used as a standard for triacylglycerols, was obtained from white lupin seeds by pressing with a domestic oil expeller (Wartmann®, brand model WM-OP-1402A, Tilburg, The Netherlands). Hexane (HEX), methyl tert-butyl ether (MTBE), methanol (MeOH) and chloroform (CLF), were supplied by Macron (Avantor Performance Material, Center Valley, USA).
2.2 Methods
2.2.1 Conditioning
The grinding of the lupin hulls was carried out using an impact miller IKA-M20 (Werke Staufen, Germany). Lupine seed hulls were subjected to grinding at 20.000 rpm for 90 s, followed by 60 s at rest, and another 90 s at 20,000 rpm. The resulting flour was vacuum packaged into a bag and stored at 4 °C.
2.2.2 Granulometry
To determine the particle size distribution of the lupin hulls flour after milling, 50 g of flour were introduced into a TZA 1491 electric sieve shaker (Bunsen SA., Madrid, Spain) with four sieves with different pore sizes (> 1 mm, 1–0.5 mm, 0.5–0.25 mm, 0.25–0.125 mm) and a container, in which particles less than 0.125 mm were collected. The samples were placed for 6 min in the sieve shaker with ultrasonic vibration, with an amplitude of 1.8 mm and cycles of 9 s on and 1 s off. Finally, each fraction was collected and weighed. Granulometry is shown in Table 1.
2.2.3 Moisture
The moisture content of lupin hulls flour was determined by gravimetry using a precision analytical balance (± 0.1 mg) after drying 10 g of samples in an oven at 105 °C until constant weight. These determinations were carried out in triplicate.
2.2.4 Determination of total fat content at analytical scale
The total fat content of lupin hulls flour was determined at analytical scale using the Folch method [23], with some modifications. In brief, 200 mL of chloroform:methanol (2:1 v/v) were added to 10 g of hulls and mixed with an Ultraturrax T18 basic IKA (Staufen, Germany) at speed between 11,800 and 16,200 rpm for 2 min, until an homogeneous mixture was obtained, that was maintained by magnetic stirring at 800 rpm during 60 min. The sample was then vacuum filtered and distilled water was added to the organic phase in a 1:5 ratio. The mixture was mixed in a vortex for 1 min and centrifuged for 5 min at 10,000 rpm. Then, the lower phase was collected and evaporated in a Büchi B-480 rotary evaporator (Uster, Switzerland) at 40 °C, under 10 mbar until constant weight. This procedure was carried out in triplicate.
2.2.5 Solid–liquid hexane extraction
Solid–liquid extraction was carried out using the method described by Piccirilli et al. [13]. Hence, 150 g of flour from lupin hulls with particle size between 250 and 500 µm were extracted 4 times consecutively with 250 mL of hexane by magnetic stirring at 900 rpm. The extraction conditions were 45 °C and 15 min each time. The solid and liquid phases were separated by vacuum filtration in a kitasato flask during 15 min and the hexane was evaporated up to an approximate a volume of 10 mL in a Büchi B-480 rotary evaporator (Uster, Switzerland) at 40 °C and 10 mbar. The volume was transferred to a 50 mL centrifuge tube, and it was left during 15 h at 25 °C for crystallization and, consequently, precipitation of lupeol. For the separation of the rest of hexane from the crystallized lupeol, a centrifuge process at 10.000 rpm during 30 min was carried out. After this, the hexane phase was recovered in a evaporate flask and the solid phase of lupeol (product 1) was washed with another 10 mL of hexane, remained at 25 °C for 15 h, and then centrifugated at 10.000 rpm during 30 min. The hexane phase of the washings (product 2) was recovered and evaporated at 40 °C, under 10 mbar until constant weight. The solid phase, rich in lupeol, was dried under nitrogen until constant weight. This procedure was carried out in triplicate.
In the extraction processes, two main responses were evaluated:
-
a. Composition (wt%), defined as (weight of x-compound in a fraction/weight of entire fraction) × 100
-
b. Recovery (%), defined as (weight of x-compound in a fraction/weight of x-compound in Biomass) × 100
2.2.6 Supercritical CO2 extraction and fractionation though sequential depressurization
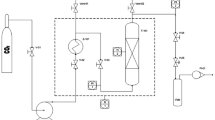
A basic extraction scheme for supercritical fluid extraction (SFE) with CO2 is shown in Fig. 1. In brief, 150 g of flour from lupin hulls with particle size between 250 and 500 µm were used to perform a supercritical CO2 extraction in a homemade pilot-plant device comprising a 350 cm3 cylindrical extraction vessel (EV), and two different separators (S1 and S2), 270 cm3 capacity each, with independent control of temperature and pressure. In general, the ratio between height and diameter of the cylindrical EV is recommended to be 5–7. The equipment design implies a batch procedure to incorporate the lupin seed hulls flour in the EV. CO2 is first heated (HE2) up to the desired temperature and then pumped (P1) from the bottom of the EV, up to the desired extraction pressure. For adequate pumping and to prevent cavitation of the pump, precooling of the solvent is required (HE3). In this study, the use of a co-solvent was not considered, due the relative low polarity of lupeol and the advantages associated to the use of pure CO2.

Scheme of supercritical CO2 extraction of lupin seed hulls flour. P1: CO2 pump; P2: cosolvent pump; HE1, HE2, HE3: heat exchangers; EV: extraction vessel; S1, S2: separator cells; V, V1, V2: back pressure regulator valves; ST: CO2 storage tank; F: filter
At the exit of the extractor, the supercritical solvent with the extracted solute flows through a depressurization valve (V) to a separator 1 (S1), in which due to the lower pressure the extracts are separated from the solvent and collected. The remaining extract still soluble in CO2 can be collected in a separator (S2), that is around 20 bar. This way permits the fractionation of the extract in two fractions (on-line) by setting suitable temperatures and pressures in the fractionation units. In this study, the SFE conditions were 180, 250, 320 bar of pressure at 40 °C during 90 min, at 50 or 100 g/min CO2 flows. For the sequential fractionation of the extracts, the extraction was carried out at 320 bar and 50 g/min of CO2 flow for the column, and three different CO2 densities, 137, 642, 728, and 808 kg/m3, were set in S1. S2 was maintained under constant conditions of 20 bar and 20 °C.
In all the CO2 extraction and fractionation processes the composition (wt%) and recovery (%) were evaluated as explained in Sect. 2.2.5. In addition, the enrichment factor defined as the ratio between the lupeol composition in S1 and the lupeol composition in the original extract obtained at selected conditions, were calculated.
2.2.7 GC–MS method
All samples and extracts obtained were analyzed by gas chromatography (GC) coupled to mass spectrometry (MS). The GC–MS system consisted of an Agilent GC Series 6890N (Santa Clara, CA, USA) with on column injection and flame ionization detector (FID), and an Agilent single quadrupole MS detector model 5975C (Santa Clara, CA, USA), with electron energy set at 70 eV and the mass range at 50–700 m/z. The chromatographic separation was based on the method developed by Torres et al. [24]. A HP-5MS capillary column (7 m × 0.25 mm internal diameter × 0.25 μm) was used. The injection volume was 0.2 µL. Temperatures of injector and detector were 50 and 340 °C, respectively. The program of temperatures started at 60 °C, increasing at 42 °C min−1 until 250 °C. This temperature was maintained for 20 min and then increased up to 340 °C at 25 °C min−1, which was maintained for 20 min. The flow rate of carrier gas (helium) was maintained at 1 mL/min. The GC–MS control and data processing was performed using Chem-Station (Agilent Technologies) software. For the compound identification, manual spectral matching was ascertained by using the mass spectral library of National Institute Standard and Technology (NIST) version 2.0.
Quantitative analysis of lipid compounds (sterols, lupeol, diacylglycerols and triacyclglycerols) using the external standard method with pure compounds. For that matter, calibration curves with each standard from 0.2 to 6 mg mL−1 were prepared and analyzed.
2.2.8 Statistical analysis
All experiments were carried out in triplicate and the data were expressed as mean ± standard deviation. The statistical significance of the differences between the groups was measured by one-way analysis of variance (ANOVA) and post-hoc Tukey HSD test and parametric Student´s t-test for independent samples. Statistical significance was defined at the level of p < 0.05. All statistical evaluations were performed using Origin (version 9.0 for Windows; OriginLab Corporation, Northampton Northampton, USA).
3 Results and discussions
3.1 Total fat content of lupin hulls
The lupin hulls had 0.76 ± 0.05% of total fat content as determined by the Folch method [23]. These values are, in general, lower than those found by Zhong et al. [7], where the total fat content ranged between 1.6 and 2.4 wt%. This difference in the total fat content can be attributed to the variety of the seeds, which in the present case were sweet lupin hulls, and to the cultivation characteristics.
3.2 Solid–liquid hexane extraction of lupeol
A solid–liquid extraction with hexane was carried out to obtain lupeol from 150 g of lupin hulls with a moisture of 6.9 ± 0.4 wt% and particle size between 250 and 500 µm, following the method of Piccirilli et al. [13]. As shown in Table 2, the products obtained were divided into product 1 (P1) and product 2 (P2). The recovery of lupeol was higher in P1 (51.0%) than in P2 (30.3%). Sterols and triacylglycerols recoveries of P2 were higher than for P1. Comparing this data with those obtained through the official Folch method (considered as 100 wt%), it was observed that in terms of recovery, 81.3% of lupeol, 102.9% of sterols and 81.0% of triacylglycerols were obtained with the solid–liquid hexane extraction (P1 + P2). In terms of composition, P1 was the richest in lupeol (89.0 wt%) and P2 was richer in triacylglycerols (33.5 wt%) as compared to P1 (7.9 wt%). P2 had also higher composition in sterols (9.7 wt%) than P1 (3.3 wt%). These results are very similar to those found by Piccirilli et al. [13].
3.3 Supercritical CO2 extraction of lupeol
A study on the combination of pressure and temperature during supercritical CO2 extraction was conducted to obtain lupeol extracts with the highest purity and yield. Table 3 shows composition (wt%) and recovery (g/%) of the extracts obtained after supercritical CO2 extraction from lupin hulls flour with a moisture of 6.9 ± 0.44 wt% and particle size between 250 and 500 µm, using 180, 250 and 320 bar of pressure with a combination of 50 and 100 g/min of CO2 flow during 90 min.
Regarding composition, no significant differences (p > 0.05) were found in lupeol, sterols and triacylglycerol at the different conditions of CO2 pressure and flow. The composition in lupeol was found to be between 72.3 wt% and 75 wt%, the composition in sterols was from 4.9 wt% to 5.2 wt%, and the composition in triacylglycerol was from 20.4 wt% to 22.1 wt%. Identification of lupeol in the extracts was confirmed by GC–MS analysis, as shown in Fig. 2.

Mass spectra of lupeol obtained by GC–MS. (A) Lupeol standard at 5 mg/mL. (B) Extract obtained with supercritical CO2 at 250 bar and 100 g/min. The probability for identification according to the GC–MS library is indicated
As shown in Table 3, significant differences (p < 0.05) were observed in lupeol recovery when comparing different pressures at the CO2 flows investigated. Thus, at 50 g/min, lupeol recovery was significantly enhanced as the pressure increased, leading values of 73.7%, 82.9% and 96.9%, at 180, 250 and 320 bar, respectively. Same pattern was observed at 100 g/min CO2 flow between 180 and 250 bar, obtaining lupeol recoveries of 85.8% and 91.8%, respectively. However, at 100 g/min CO2 flow no statistical differences (p > 0.05) were observed between 250 (91.8%) and 320 bar (87.2%).
In general, recovery of lupeol increases with increasing CO2 flow, from 50 to 100 g/min at 180 and 250 bar, with significant increase of 17% and 10.7%, respectively. However, in the case of 320 bar no statistical differences (p > 0.05) were observed between both CO2 flows. It is possible that in this particular case, CO2 could take preferential pathways which would not happen at other pressure and flow combinations. The dead space and the axial dispersion inside the EV could create a non-ideal pattern due to the presence of preferential pathways, and this could influence negatively in the fluid phase along the extractor, resulting in a decrease in the motion forces which given the mass transfer rate causes a drop in the overall extraction efficiency [25]. The extraction efficiency is also related with the enhancement of the surface area of contact with the solvent. Although the particle size of the flour is the same for all the extraction conditions, the influence of size should be considered to avoid preferential paths [26].
Regarding the recovery of sterols, even if no significant differences (p > 0.05) were found for the different combinations studied, the combination 320 bar and 100 g/min represent the best for sterols recovery (94.6%). In the case of triacylglycerols, no significant differences were observed between the different treatments.
When the results obtained with supercritical CO2 extraction are compared to those obtained with the Folch method, it can be observed that the conditions 320 bar and 50 g/min of CO2 permit to obtain the largest amount of lupeol (96.9%). Again, no significant differences were found for sterols and triacylglycerols recoveries comparing supercritical CO2 extraction and the Folch method.
To understand the efficiency of the extraction procedure, it is important to consider the solvent-to-feed ratio, i.e., the proportion of CO2 used in relation to the amount of material being processed to obtain the target compound [20]. In the case of 50 g/min CO2 flow, the ratio is 30:1, and in the case of 100 g/min, a ratio of 60:1 is obtained. It must be noticed that the increase in the recovery of lupeol is not substantially larger (10–11%) when the largest CO2 flow is employed. Thus, we can conclude that the most efficient flow rate to carry out the recovery of lupeol from lupin hulls using supercritical CO2 would be 50 g/min, since the solvent-to-feed ratio is lower, and the extraction recovery is very similar to that achieved with 100 g/min CO2 flow.
From the comparison of the recovery and composition of the extracts obtained by supercritical CO2 extraction and by solid–liquid extraction with hexane, it can be observed that the results are much more favorable when employing supercritical CO2, since extracts with a similar composition are obtained in less time in comparison with the solid–liquid hexane extraction (90 min vs. 33 h, respectively). Besides, the recovery with the use of supercritical CO2 extraction represents an increase of lupeol extraction yield of 19% compared to that with the hexane solid–liquid extraction.
Table 4 summarizes previous studies on supercritical CO2 extraction of lupeol from different plant matrices with similar conditions to those used in the present study. Felföldi-Gáva et al. studied extracts from Alnus Glutinosa bark, where lupeol represents 14.3 g/100 g of the extract. These results were obtained at 450 bar, 60 °C, 116.7 g/min CO2 flow and 60 min [27]. It should be noted that, even with a higher pressure, these authors attained 5.4 times lower lupeol content than that of the present study. De Souza et al. obtained 35.3 g/100 g of lupeol acetate in the extracts from Archium Lappa leaves through supercritical CO2 at 200 bar, 62 °C, 50 min, 1.4 g CO2/min and ethanol (EtOH) (2:1; EtOH w:w of solids). In this case, the use of EtOH as modifier can enhance the extraction yield, also leading to co-extraction of more polar compounds [28]. Accordingly, Casas et al. investigated lupeol extraction from Acacia Dealbata flowers using CO2 at 300 bar, 45 °C, 180 min, 25 g CO2/min and 10% EtOH (w:w; EtOH:CO2), which provided 18 g/100 g lupeol content in the extracts [29]. In the study of Nakuerte et al., conditions of 250 bar, 55 °C, 2 h, 25 g CO2/min were used to obtain up to 23.2 g/100 g of lupeol in supercritical extracts from Matricaria recutita chamomile florets [30]. In general, lupeol composition achieved of the aforementioned investigations are remarkably lower than those attained in the present work, where lupeol concentration from lupin hulls was around 75 g/100 g in the extract. This could be explained by the differences in parameters of the supercritical extraction processes (e. g. density of the CO2) as well as by the diverse raw materials used.
One of the most relevant advantages of the present work is that the use of solvents as modifiers was not necessary. Thus, an efficient extraction of lupeol from lupin hulls flour, in terms of composition and recovery, was attained by using pure supercritical CO2. These results would inform us about the chemical form of the lupeol to be extracted from a given matrix. Several plants produce abundant glycosylated triterpenoids (with a sugar part in their structure), which are called saponins. When this occurs, ethanol is needed as a modifier during the supercritical CO2 extraction because it increases the molecular weight and polarity, aiding to the extraction of saponins [31]. However, as in the present study the use of modifiers was not necessary, this would indicate that lupeol from lupin hulls is present in its aglycone form.
Thus, the use of supercritical CO2 to obtain lupeol from lupin hulls not only entails advantages in terms of composition and recovery, but also provides extracts free of solvent that have not been subjected to high temperatures. The sustainable revaluation of lupin hulls using supercritical CO2 is evident, since not only it is possible to isolate an ingredient with high bioactive potential, but also the starting material remains intact and useful for further use for conventional purposes.
Besides, the composition of the extracts obtained by supercritical CO2 and by the Folch method are very similar to each other. This could indicate that pure supercritical CO2 can be as efficient as the chloroform: methanol (2:1; v:v) mixture in order to extract lupeol from this raw material, even though being solvents with different polarity. The different extraction conditions employed with supercritical CO2 led to different amounts of total lipid content in the extracts, but without any selectivity for different lipid compounds. For a subsequent enrichment in lupeol of the extracts, strategies such as fractionation of the extract in different fractionation units in a supercritical plant could be employed.
3.4 Fractionation of lupeol extract by sequential depressurization
Once the optimal conditions for lupeol extraction from lupin hulls were achieved, the extract was fractionated in two separators S1 and S2. The aim of this fractionation was to separate lupeol and triacyclglycerols to increase the purity of the lupeol extracts. For this purpose, different supercritical CO2 densities were used in S1 by changing the pressure and temperature. S2 was always around 20 bar to quantitatively recover the extract. At this pressure (20 bar), CO2 has a negligible solubilization capacity and all substances dissolved in CO2 precipitate and can be recovered in this separator.
Table 5 shows the results of the different fractionations performed. The values indicate the separator (S1 or S2) where lupeol was mostly recovered. Recovery was calculated as the amount of the identified compound out of the total of each compound extracted (S1 + S2).
Regarding the different densities used, it was observed that when the CO2 density in S1 is close to 800 kg/m3, the extract is soluble and almost completely recovered in S2. On the contrary, when CO2 density is around 137 kg/m3, the extract precipitates in S1 and only negligible amounts of extracts are recovered in S2. As can be seen, as the density of CO2 decreases, its solubilization capacity decreases, as all the extract precipitates in S1. However, in terms of extract composition, there is no significant difference (p > 0.05) in lupeol composition between these densities (~ 85 wt%).
Intermediate densities (642 and 728 kg/m3) were tested to fractionate the extracts into two different portions. Thus, at densities of about 728 kg/m3 in S1, the total extract was fractionated into two different products. The product on S1 contained 92.5 wt% of lupeol. However, the lupeol recovery represents almost 50% of the total lupeol in the extract. This result indicates that to obtain an enrichment factor of about 1.2, 50% of total lupeol cannot be recovered in S1 and will precipitate in S2 mixed with triglycerides. Furthermore, for a density of about 642 kg/m3 the lupeol composition was 87 wt% and 91.5 wt% of the total lupeol in the extract was recovered in this separator.
From the results shown above, a compromise must be found between purity or composition and recovery. To increase the purity of lupeol above 90 wt%, about 50% of the total lupeol remains soluble at this CO2 density (728 kg/m3), and precipitates in S2. It is therefore concluded that lupeol is less soluble in supercritical CO2 than the triacylglycerols. This is explained by the fact that the purity of lupeol increases in S1. As the density is slightly reduced, the CO2 solubilization capability decreases and vice versa.
Figure 3 shows a comparison of chromatograms of extracts obtained after solid–liquid extraction with hexane and the different supercritical fractionations mentioned above.

Chromatograms obtained after extractions showing the identification of lupeol, sterols and triacylglycerols. A) Solid–liquid extraction with hexane. B) Fractionation 4, extract in S2. C) Fractionation 3, extract in S1
Fractionation of the extract represents a strategy that slightly increases the purity of lupeol, but with the drawback that a significant fraction of the total lupeol cannot be recovered in S1. It is an interesting strategy to assess the relative solubility of the two main ingredients of the extract (lupeol and triacylglycerols) and to partially eliminate triacylglycerols from the extracts, which could interfere in subsequent analyses of lupeol biological activity tests.
4 Conclusions
For the first time, lupeol has been extracted from lupin seed hulls by means of supercritical CO2 fluid with the consequent revalorization of a waste product. These results were compared with those obtained with conventional solid–liquid extraction with hexane showing that the use of supercritical CO2 is more advantageous. The highest lupeol recovery by supercritical CO2 extraction was 96.8%, which was achieved using 320 bar and 50 g/min of CO2 flow rate. In terms of lupeol enrichment, sequential depressurization was also efficient in separating lupeol and triacylglycerols, thereby increasing the purity of lupeol in the extracts. A compromise between composition and recovery had to be found and it could be concluded that lupeol is less soluble in supercritical CO2 than the triacylglycerols. Furthermore, fractionation of the extract increases lupeol composition somewhat to 92.5 wt% when the density is about 728 kg/m3. However, a significant fraction of the total lupeol cannot be recovered. In any case, fractionation represents an interesting strategy for the evaluation of the relative solubility of the main extract ingredients (lupeol and triacylglycerols in this case) and thus partially removes triacylglycerols from the extracts. The next step would be to test the biological activity of supercritical lupeol extracts obtained from a by-product such as lupin hulls.
Data availability
Not applicable.
References
Patinha Caldeira C, Barco Cobalea H, De Laurentiis V, Sala S (2019) Review of studies on food waste accounting at Member State level. Publ Off Eur Union. https://doi.org/10.2760/340637
Mallek-Ayadi S, Bahloul N, Kechaou N (2018) Chemical composition and bioactive compounds of Cucumis melo L. seeds: Potential source for new trends of plant oils. Process Saf Environ Prot 113:68–77. https://doi.org/10.1016/j.psep.2017.09.016
Rao M, Bast A, de Boer A (2021) Valorized food processing by-products in the EU: Finding the balance between safety, nutrition, and sustainability. Sustainability 13:4428. https://doi.org/10.3390/su13084428
Gómez-García R, Campos DA, Aguilar CN, Madureira AR et al (2021) Valorization of food agro-industrial by-products: From the past to the present and perspectives. J Environ Manage 299:113571. https://doi.org/10.1016/j.jenvman.2021.113571
Brunetti L, Leuci R, Colonna MA, Carrieri R et al (2022) Food industry byproducts as starting material for innovative, green feed formulation: A sustainable alternative for poultry feeding. Mol 27:4735. https://doi.org/10.3390/molecules27154735
Johnson SK, Clements J, Villarino CBJ, Coorey R (2017) Lupins: Their unique nutritional and health-promoting attributes. In: Taylor JRN and Awika JM (eds) Gluten-Free ancient grains. Cereals, pseudocereals, and legumes: Sustainable, nutritious, and health-promoting foods for the 21st century. Elsevier, Amsterdam, pp 179–221. https://doi.org/10.1016/B978-0-08-100866-9.00008-X
Zhong L, Ali H, Fang Z et al (2020) Lupin seed coat as a promising food ingredient: Physicochemical, nutritional, antioxidant properties, and effect of genotype and environment. Int J Food Sci Technol 55:1816–1824. https://doi.org/10.1111/ijfs.14460
Leonard W, Hutchings SC, Warner RD, Fang Z (2019) Effects of incorporating roasted lupin (Lupinus angustifolius) flour on the physicochemical and sensory attributes of beef sausage. Int J Food Sci Technol 54:1849–1857. https://doi.org/10.1111/ijfs.14088
Malekipoor R, Johnson SK, Bhattarai RR (2022) Lupin kernel fibre: Nutritional composition, processing methods, physicochemical properties, consumer acceptability and health effects of its enriched products. Nutrients 14:2845. https://doi.org/10.3390/nu14142845
Zhong L, Fang Z, Wahlqvist ML et al (2018) Seed coats of pulses as a food ingredient: characterization, processing, and applications. Trends Food Sci Technol 80:35–42. https://doi.org/10.1016/j.tifs.2018.07.021
Clements J, Dracup M, Galwey N (2002) Effect of genotype and environment on proportion of seed hull and pod wall in lupin. Aust J Agric Res 53(10):1147–1154. https://doi.org/10.1071/AR01156
Pilkington M (2013) Characterisation of lupin-derived lupeol with a metabolomics study of the impact and potential neuroprotection of lupeol. PhD Thesis, Murdoch University. Perth, Australia
Piccirilli A, Legrand J, Broutin N (2004) Extract from the pods of lupin seeds containing lupeol. WO2002085827A1 Patent
Liu K, Zhang X, Xie L et al (2021) Lupeol and its derivatives as anticancer and anti-inflammatory agents: Molecular mechanisms and therapeutic efficacy. Pharmacol Res 164:105373. https://doi.org/10.1016/j.phrs.2020.105373
Sohag AAM, Hossain MT, Rahaman MA et al (2022) Molecular pharmacology and therapeutic advances of the pentacyclic triterpene lupeol. Phytomedicine 99:154012. https://doi.org/10.1016/j.phymed.2022.154012
Lerma-Torres JM, Navarro-Ocaña A, Calderón-Santoyo M et al (2019) Preparative scale extraction of mangiferin and lupeol from mango (Mangifera indica L.) leaves and bark by different extraction methods. J Food Sci Technol 56:4625–4631. https://doi.org/10.1007/s13197-019-03909-0
Krakowska-Sieprawska A, Rafińska K, Walczak-Skierska J et al (2021) Promising green technology in obtaining functional plant preparations: Combined enzyme-assisted supercritical fluid extraction of flavonoids isolation from Medicago sativa leaves. Materials (Basel) 14:2724. https://doi.org/10.3390/ma14112724
Buszewski B, Rafińska K, Cvetanovic A et al (2019) Phytochemical analysis and biological activity of Lupinus luteus seeds extracts obtained by supercritical fluid extraction. Phytochem Lett 30:338–348. https://doi.org/10.1016/j.phytol.2019.02.014
Santos SA, Villaverde JJ, Silva CP et al (2012) Supercritical fluid extraction of phenolic compounds from Eucalyptus globulus Labill bark. J Supercrit Fluids 71:71–79. https://doi.org/10.1016/j.supflu.2012.07.004
Knez Z, Markocic E, Leitgeb M, Primozic M, KnezHrncic M, Skerget M (2014) Industrial applications of supercritical fluids: A review. Energy 77:235–243. https://doi.org/10.1016/j.energy.2014.07.044
Buszewski B, Szultka-Mlynska M (2012) Past, present, and future of solid phase extraction: A review. Crit Rev Anal Chem 42:198–213. https://doi.org/10.1080/07373937.2011.645413
Fathordoobady F, Mirhosseini H, Selamat J, Manap MY (2016) Effect of solvent type and ratio on betacyanins and antioxidant activity of extracts from Hylocereus polyrhizus flesh and peel by supercritical fluid extraction and solvent extraction. Food Chem 202:70–80. https://doi.org/10.1016/j.foodchem.2016.01.121
Folch J, Lees M, Sloane Stanley GH (1957) A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem 226:497–509
Torres C, Tenllado D, Señorans F, Reglero G (2009) A Versatile GC method for the analysis of alkylglycerols and other neutral lipid classes. Chromatographia 69:729–734. https://doi.org/10.1365/s10337-008-0935-5
Zabot GL, Moraes MN, Petenate AJ, Meireles MAA (2014) Influence of the bed geometry on the kinetics of the extraction of clove bud oil with supercritical CO2. J Supercrit Fluids 93:56–66. https://doi.org/10.1016/j.supflu.2013.10.001
Sarkar S, Gayen K, Bhowmick TK (2022) Green extraction of biomolecules from algae using subcritical and supercritical fluids. Biomass Convers Biorefin 1–23. https://doi.org/10.1007/s13399-022-02309-3
Felföldi-Gáva A, Szarka S, Simandi B et al (2012) Supercritical fluid extraction of Alnus glutinosa (L.) Gaertn. J Supercrit Fluids 61:55–61. https://doi.org/10.1016/j.supflu.2011.10.003
de Souza ARC, Guedes AR, Rodriguez JMF et al (2018) Extraction of Arctium lappa leaves using supercritical CO2 + ethanol: Kinetics, chemical composition, and bioactivity assessments. J Supercrit Fluids 140:137–146. https://doi.org/10.1016/j.supflu.2018.06.011
Casas M, López-Hortas L, Díaz-Reinoso B et al (2021) Supercritical CO2 extracts from Acacia dealbata flowers. J Supercrit Fluids 173:105223. https://doi.org/10.1016/j.supflu.2021.105223
Nakurte I, Berga M, Pastare L et al (2023) Valorization of bioactive compounds from by-products of Matricaria recutita white ray florets. Plants 12:396. https://doi.org/10.3390/plants12020396
Rodrigues VH, de Melo MM, Portugal I, Silva CM (2021) Lupane-type triterpenoids from Acacia dealbata bark extracted by different methods. Ind Crops Prod 170:113734. https://doi.org/10.1016/j.indcrop.2021.113734
Beveridge TH, Girard B, Kopp T, Drover JC (2005) Yield and composition of grape seed oils extracted by supercritical carbon dioxide and petroleum ether: Varietal effects. J Agric Food Chem 53:1799–1804. https://doi.org/10.1021/jf040295q
Filip S, Djarmati Z, Lisichkov K et al (2015) Isolation and characterization of Maclura (Maclura pomifera) extracts obtained by supercritical fluid extraction. Ind Crops Prod 76:995–1000. https://doi.org/10.1016/j.indcrop.2015.07.066
Maia JD, Ávila CR, Mezzomo N, Lanza M (2018) Evaluation of bioactive extracts of mangaba (Hancornia speciosa) using low and high pressure processes. J Supercrit Fluids 135:198–210. https://doi.org/10.1016/j.supflu.2018.01.016
Martin D, Navarro Del Hierro J, Villanueva Bermejo D et al (2016) Bioaccessibility and antioxidant activity of Calendula officinalis supercritical extract as affected by in vitro codigestion with olive oil. J Agric Food Chem 64:8828–8837. https://doi.org/10.1021/acs.jafc.6b04313
De la Peña AR, Bronze MR, Matias A, Mateos-Aparicio I (2021) Triterpene-rich supercritical CO2 extracts from apple by-product protect human keratinocytes against ROS. Food Bioproc Tech 14:1–11. https://doi.org/10.1007/s11947-021-02615-0
De Melo MM, Carius B, Simoes M et al (2020) Supercritical CO2 extraction of V. vinifera leaves: Influence of cosolvents and particle size on removal kinetics and selectivity to target compounds. J Supercrit Fluids 165:104959. https://doi.org/10.1016/j.supflu.2020.104959
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. This study has been funded by Comunidad de Madrid (ALIBIRD, S2018/BAA-436 4343), Fundación Ramón Areces and Ministerio de Ciencia e Innovación (Project number PID2020–119084RB-C21). Celia Bañares thanks Ministerio de Economía y Competitividad and the European Social Fund for a pre-doctoral FPI grant (BES- 2017–080853). Assamae Chabni thanks Comunidad de Madrid for a pre-doctoral grant (PEJD-2019-PRE/BIO-14522).
Author information
Authors and Affiliations
Contributions
LV, CB and AC: Formal analysis; Investigation; Methodology; Resources; Software; Writing-original draft; Writing—review & editing. CB: Writing-original draft. CT and GR: Funding acquisition; Project administration; Resources. CT: Investigation; Methodology; Supervision; Validation.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vázquez, L., Bañares, C., Chabni, A. et al. Supercritical fluid technology for lupin hulls valorization: extraction and fractionation of lupeol. Biomass Conv. Bioref. (2024). https://doi.org/10.1007/s13399-024-05511-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13399-024-05511-7