
Abstract
The prehydrolysis liquor from the prehydrolysis Kraft process is rich in sugars and could thus serve as a sustainable feedstock for the production of various chemicals. However, its industrial utilization is impeded by the presence of fermentation inhibitors and extensive lignin precipitation, the latter receiving only little attention in the literature.
In order to provide a feedstock suitable for biotechnological or chemical conversion, the prehydrolysis liquor from eucalyptus wood must be detoxified whilst preventing the precipitation of lignin. To increase the yield of monomeric sugars, acid posthydrolysis should be investigated.
Various solvents and solvent mixtures were screened for the high temperature liquid–liquid extraction of isothermally separated prehydrolysis liquor. Their capability to prevent lignin precipitation and to extract fermentation inhibitors was assessed using mass balances and size-exclusion chromatography. Based on the solvent screening, a process for simultaneous posthydrolysis and liquid–liquid extraction of eucalyptus prehydrolysis liquor was proposed and investigated using statistic experimental design.
Liquid–liquid extraction using aliphatic alcohols effectively prevents lignin precipitation, and the addition of 25% (w/w) tri-n-octylamine was found to increase the overall inhibitor extraction efficiency. The conditions for the simultaneous posthydrolysis were investigated using a Box-Behnken experimental design, allowing for a maximum monomeric sugar yield of 83.0% at a sugar purity of 91.6%.
The simultaneous posthydrolysis and liquid-liquid extraction (SIMPLLE) process thus avoids industrial-level problems associated with lignin precipitation. It provides a carbohydrate-rich stream with low levels of fermentation inhibitors, enabling further conversion to value added products.
Similar content being viewed by others


1 Introduction
Prehydrolysis Kraft (PHK) pulping has progressively become the major technology for producing dissolving pulp from various wood species [1]. In terms of resource efficiency and biorefining, PHK has the potential to utilize the hemicellulose fraction extracted during prehydrolysis [2].
Prehydrolysis is typically carried out at temperatures of 150–200 °C using steam or liquid hot water [3, 4]. At these conditions, the acetyl groups are cleaved from the hemicellulose polymers, yielding free acetic acid (HAc) that decreases the pH of the prehydrolysis liquor (PHL) to 3–4. The released protons further catalyze the hydrolysis of hemicellulose and cellulose. However, the cellulose degradation rate constant is low compared to the hemicellulose degradation rate, due to the high crystallinity of cellulose [5]. Depending on the prehydrolysis intensity, hemicellulose is degraded to soluble oligomers, monomeric sugars or further dehydrated to furfural or 5-hydroxymethylfurfural (HMF). Due to its high sugar content, PHL is regarded as an important feedstock for biorefineries in a PHK pulp mill context, potentially creating additional revenue [6,7,8].
Eucalyptus is a fast growing tree species that is widely used in the pulping industry. Eucalyptus wood is exceptionally well suited for the production of dissolving pulp, due to its morphological structure, low lignin content, low hemicellulose, and high cellulose content [9]. Additionally, eucalyptus is easy to delignify, due to a high proportion of syringyl units, a high abundance of β-O-4 linkages, and a low degree of condensation [10, 11]. Eucalyptus hemicellulose consists of glucuronoxylan, a linear polymer of 1-4 linked β-d-xylopyranosyl units, substituted with 4-O-methylglucuronic acid and acetate residues. The degree of substitution depends on the wood species. The average molar ratio of xylose to 4-O-methylglucuronic acid to acetyl groups has been reported to be 10:1:7 in hardwoods [12, 13].
During prehydrolysis, xylans are converted to mixtures of monomeric xylose and xylooligosaccharides. However, most microorganisms are not able to utilize hemicellulosic oligosaccharides directly [14]. In contrast, monomeric xylose can serve as fermentation substrate to produce various chemicals biotechnologically. Ethanol is the most studied fermentation product, but xylitol, lactic acid, fatty alcohols, and terpenes were also shown to be accessible from xylose via fermentation [15, 16]. Therefore, the hydrolysis of oligosaccharides is an essential step to utilize hemicellulose from PHL.
During prehydrolysis and oligosaccharide posthydrolysis various fermentation inhibitors are released or formed. Small organic acids, e.g., formic acid (HCOOH) and HAc, and furans, e.g., furfural and HMF, are known to inhibit biochemical processes [17,18,19]. Additionally, dissolved low molecular weight lignin fragments and phenolic compounds are toxic for many microorganisms [20, 21]. Thus, efficient detoxification strategies are of great importance to enable the industrial utilization of PHL sugars.
Lignin is also responsible for another technological hurdle hindering the utilization of PHL; upon cooling of the PHL, parts of the dissolved lignin precipitate and form sticky deposits leading to extensive fouling problems, high maintenance downtimes, and poor process economics [22].
Too prevent the above described processing issues, a number of PHL purification methods like membrane separations, adsorption processes, ion exchange, liquid–liquid extraction (LLE), enzymatic treatments, or combinations thereof were recently reported in the literature (see Table 1).
Given that the principal inhibitors are structurally divers, being either short chain carboxylic acids or aromatic compounds, some of the detoxification methods have a high specificity for one substance class, being poorly suitable to remove the other substance class. Also, in most studies dealing with detoxification of hardwood hydrolysates, the fate of lignin was not the focus or not even analyzed. The reprecipitation behavior of PHL-lignin upon cooling is almost exclusively ignored. In literature, precipitated lignin is simply filtered or centrifuged off before conducting the study. In industrial scale, these separation methods are problematic due to the sticky nature and small particle size of lignin. In this work, we tackle these existing shortcomings of the available research and sketch out a process to gain an inhibitor reduced fermentation substrate from PHL, circumventing the problem of lignin precipitation. A detailed study of solvents and reactive extractants was conducted, and the corresponding partition coefficients were determined for the principal inhibitory compounds (HAc, HCOOH, furfural, HMF, and acid soluble lignin). Our investigation revealed previously undescribed side reactions of reactive extractants and specific interactions with lignin subfractions. To reduce the need for processing equipment, the presented process comprises an optimized and integrated unit operation which we termed simultaneous posthydrolysis of oligosaccharides and liquid–liquid extraction of fermentation inhibitors (SIMPLLE).
2 Materials and methods
2.1 Materials
In this study, wood from an E. grandis × E. urophylla hybrid was used for all experiments. Wood samples were kindly supplied by Lenzing from 6 to 7 year old trees grown in Brazilian plantations. The wood chips were ground using a Retsch SM300 cutting mill (Retsch GmbH, Haan, Germany) and fractionated using an AS 200 vibratory sieve shaker (Retsch GmbH, Haan, Germany). The fraction with particle size between 2.50 and 3.55 mm was collected and used for the prehydrolysis experiments.
Unless otherwise specified, all other chemicals were of analytical grade, obtained from commercial suppliers, and used without further purification.
2.2 Prehydrolysis experiments
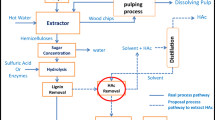
A lab-scale Parr 4875 reactor station with two 450-mL reactors was used to conduct the experiments. The first reactor used for prehydrolysis was made from T316 stainless steel and mechanically stirred at 300 rpm using two propeller mixers (four-bladed, D1 = 50 mm, D2 = 35 mm). The second reactor used for separating the PHL, LLE, and posthydrolysis was made from Hastelloy C and was stirred using two propeller mixers (four-bladed, D = 35 mm, see Scheme 1).

Experimental setup for the prehydrolysis and subsequent isothermal PHL separation and high temperature LLE
Fifty grams of wood were placed inside the reactor, and deionized water was added to achieve a liquid to wood ratio (L/W ratio) of 3:1. This ratio was chosen because it was the lowest possible ratio still allowing stirring of the mixture. A low L/W ratio is favorable for industrial processes due to lower energy demand for heating, cooling, and concentrating the PHL [39]. Accordingly, a constant temperature of 170 °C and a prehydrolysis factor (P-factor) of 500 were chosen as representative conditions. The P-factor concept was introduced by Sixta [39] and describes the prehydrolysis intensity, allowing to compare prehydrolysis at different temperatures. The reactor was heated to the desired temperature within approximately 20 min, after which the temperature was held constant. After the P-factor of 500 was reached, the PHL was separated from the wood residue by displacement with nitrogen gas to a preheated second reactor. The P-factor describes the prehydrolysis intensity and allows to compare prehydrolysis at different temperatures. It was calculated according to Sixta et al. from the recorded temperature and time data according to Eq. (1):
where t is the prehydrolysis time, krel is the relative rate of acid-catalyzed glycoside hydrolysis, and T is the temperature. An activation energy for eucalyptus xylan hydrolysis EA,H of 125.6 kJ mol−1 was used in the calculation.
The connection tube between the reactors was heated to 170 °C, and the second reactor was pressurized with equal pressure (7.8 bar) as in the prehydrolysis reactor using nitrogen gas. A wire mesh at the bottom of the prehydrolysis reactor and a metal sieve (74-μm pore size) in the connection tube were used to avoid particle transfer to the second reactor. The PHL was introduced directly to the bottom of the second reactor via an immersion tube. This isothermal displacement was done to avoid precipitation of any PHL constituents in the connection tube upon cooling due to temperature or pressure differences. After completing the PHL transfer, the prehydrolysis reactor was immediately cooled to room temperature by immersion in an ice bath. Only 40% of the PHL were transferred to the second reactor, because 55% were trapped in the pores of the hydrolyzed wood chips and 5% were lost, remaining in the connection tube and filtration unit. Lignin precipitation was observed on the wood chips and in the connection tube upon cooling.
The second reactor contained saturated steam for experiments with no subsequent treatment of the PHL. For conducting high temperature LLE, an organic solvent with or without added acid catalyst was present.
Six organic solvents, comprising various functional groups, like alcohols, ethers, aromatics, and ketones were selected: 1-butanol, 1-hexanol, 1-octanol, cyclopentyl methyl ether (CPME), MIBK, and toluene. Additionally, a hydrophobic deep eutectic solvent (DES) was investigated. The DES, a 2:1 mixture of decanoic acid and tri-n-octylphosphine oxide (TOPO), was selected based on a review of current literature and subsequent preliminary experiments using seven selected DES (see section S1 in Online Resource 1).
The second reactor was stirred at 700 rpm to ensure complete dispersion of biphasic mixtures and the temperature was varied between 100 and 150 °C. Samples were withdrawn after distinct time intervals from the bottom drain valve. The valve was purged with twice the dead volume prior to withdrawing a few milliliters of sample. After the desired time, the second reactor was also cooled to room temperature by submerging it into an ice bath.
2.3 PHL workup
The extracted, biphasic PHL was centrifuged at 4000 rpm for 10 min to ensure complete phase separation, and the phases were separated into an organic and aqueous fraction. If insoluble parts were present, they were separated via centrifugation, washed with deionized water, and dried overnight under reduced pressure at 40 °C.
The wood residue was washed with 2 L of deionized water to remove residual PHL and subsequently dried over night at 40 °C and 200 hPa. All the obtained fractions and the wash filtrate were weighed and analyzed in order to establish mass balances. Relative experimental error of the replicates was below 3%, except for HMF and HCOOH, where the error was below 5%. Gaps in mass balances were below 5% unless otherwise stated.
2.4 Analysis
2.4.1 Sugar analysis
Monomeric sugars were analyzed by high performance anion exchange chromatography with pulsed amperometric detection according to Schlackl et al. [40]. More precisely, a Dionex CarboPac SA10 4*250 mm column was used as the separation column in a Dionex ICS 5000 + HPLC system with pulsed amperometric detection. Deionized water (HPLC grade) and a 0.35 M sodium hydroxide solution were used as eluents at a flow rate of 1 mL min−1. The injection volume was 10 μL. Aqueous samples were filtered (0.2 μm PTFE syringe filter) and diluted to the desired concentration, whereas organic samples were extracted with the appropriate amount of deionized water and filtered prior to analysis.
To determine the total carbohydrate composition of the organic and aqueous fractions, samples were hydrolyzed at 110 °C for 50 min using a 9-fold excess of sulfuric acid (0.67 M) prior to sugar analysis. Sugar composition of solid wood and wood residue samples was determined employing a two stage sulfuric acid hydrolysis; 0.5 mL of sulfuric acid (12.1 M) were added to 40 mg of finely ground sample and stirred for 3.5 h. Then the mixture was diluted with 8.5 mL of deionized water and heated to 110 °C for 90 min. Furfural and HMF that are formed during total hydrolysis due to extensive carbohydrate degradation were quantified and added to the amount of xylose and glucose respectively. The amount of soluble oligomeric sugars in organic and aqueous fractions was calculated as the difference between the amount of sugars before and after total hydrolysis.
2.4.2 Inhibitor analysis
Klason lignin and acid soluble lignin (ASL) in wood, wood residue, and liquid samples were quantified according to Leschinsky et al. [41] and following TAPPI T222 om 98 and TAPPI UM 250 (Klason lignin) and TAPPI 1991 (ASL). Klason lignin determination is basically a gravimetric analysis of the residue after complete acid hydrolysis of a biomass, pulp or wood sample, and acid soluble lignin or lignin in liquid samples is photometrically determined at 205 nm.
Phenolic monomers were identified and quantified using the GC-MS method reported by Süss et al. [42] by means of a Shimadzu GC-MS-QP 2020 with an HP-SM5 capillary column (60 m × 0.25 mm × 0.25 μm). The oven temperature was increased from 50 to 300 °C at a 10°C min−1 heating rate. Holding times of 5 and 8 min were set for oven temperatures of 120 °C and 280 °C, respectively. A split ratio of 10 was used with helium as the carrier gas. The injection temperature was set to 250 °C. Five milligrams of the samples were dissolved in 1-mL ethyl acetate, mixed with 100-μL internal standard (toluene) and injected directly. Forty-one possible monomer substances that can be formed during lignin depolymerization were used as standards for external calibration. The concentrations ranged from 300 to 0.3 mg L−1. Aqueous samples were acidified using sulfuric acid, then 40 mL were extracted three times with ethyl acetate (20 mL, 10 mL, 10 mL), dried with Na2SO4 and analyzed. For the analysis of organic samples, 50 mL of the sample were extracted three times with 10 mL NaOH solution (w = 3.33 %) and subsequently two times with deionized water. The combined aqueous phases were afterwards treated similarly to the aqueous samples.
Furfural, HMF, HAc, and HCOOH were analyzed using liquid chromatography according to our previous study [43]. More precisely, furfural and HMF were separated using a Thermo BDS Hypersil C8 (4.6 mm 250 mm, 5 μm) analytical column at room temperature and a Dionex P680 isocratic pump operating at a flow rate of 1 ml min−1. Both compounds were detected using a Dionex UVD 340 UV detector operating at a wavelength of 277 nm and quantified by external calibration. An aqueous solution of acetonitrile (w = 20%) was used as an eluent, the sample volume was 20 μl, and the run time was set to 7.5 min per sample. HAc and HCOOH were separated using a Dionex GS50 equipped with a Kontron 465 autosampler, a Dionex IonPac AS11-HC (4 mm 50 mm) guard column, and a Dionex IonPac AS11-HC (4 mm 250 mm) separation column. A Dionex ED 50 conductivity detector with an ASRS Ultra II (4 mm) suppressor was used to quantify both acids by external calibration. Samples were eluted using a 15 min gradient from 1 mmol L−1 of sodium hydroxide to 31 mmol L−1 during 15 min and a at a flow rate of 1.5 ml min−1. This gradient was preceded by 8 min of isocratic elution at 1 mmol L−1 sodium hydroxide and followed by a one minute gradient back to 1 mmol L−1 and a 6-min isocratic hold at 1 mmol L−1.
Distribution coefficients were calculated from the mass concentrations of inhibitors in the organic and aqueous phase according to Eq. (2):
where Dx is the distribution coefficient for inhibitor x, βx,o, and βx,a are the mass concentrations of inhibitor x in the organic and aqueous phase respectively.
Extraction efficiencies were calculated from the mass concentrations in the aqueous phase according to Eq. (3):
where EEx is the distribution coefficient for inhibitor x, βx,0 is the average mass concentration of inhibitor x in untreated PHL, and βx,a is the mass concentrations of inhibitor x in the aqueous phase.
Sugar and inhibitor analysis results for all experiments can be found in Online Resource 2.
2.4.3 Molar mass distribution
Molar mass distribution was determined by size-exclusion chromatography (SEC) as described by Schlackl et al. [26, 40]. Size exclusion chromatography (SEC) measurement was performed using four columns connected in a series. The first was a PSS MCX 1000 (10 μ, 8 × 50 mm) column, the second and third were PSS MCX 1000 (10 μ, 8 × 300 mm) columns, and the fourth was a PSS MCX 10,000 (10 μ, 8 × 300 mm) column. The injection volume was 80 μL. 0.5 M sodium hydroxide with a flowrate of 1 mL min−1 used as eluent. The system was equipped with two detectors. The refractive index detector ERC RI-101 was used for quantification, and the viscosity detector WGE ETA2010 was used for the measurement of the molecular weight by means of universal calibration. To analyze organic samples, sample preparation similar to the phenolic monomer analysis was used, as described in Section 2.4.2.
2.4.4 Optimization of simultaneous posthydrolysis and LLE
A Box-Behnken design was used to evaluate the influence of the three parameters (sulfuric acid concentration wH2SO4, TOA concentration wTOA and posthydrolysis temperature T) on the simultaneous hydrolysis and extractive detoxification of PHL, and to reveal the optimum process conditions. The center point was replicated 3 times and the investigated ranges of wH2SO4, wTOA, and T were 1.0% (w/w)–5.0% (w/w), 0–50% (w/w), and 100–150 °C, respectively. Sugar monomer yield (Ysugar), sugar purity (wsugar), extraction efficiencies of lignin (EEASL), and total inhibitors (EEinh) were chosen as responses. Additionally, the response for carboxylic acids (EECA) and furans (EEF) extraction efficiency can be found in section S5 in Online Resource 1. Design Expert statistical software (version 13.0.4.0, Stat-Ease Inc., Minneapolis, MN, USA) was used to design the experiment, for analysis of variance (ANOVA) and model evaluation.
3 Results and discussion
3.1 Composition of native wood and untreated PHL
The composition of the wood sample used in this study is given in Table 2 and is in good agreement with other results for the composition of eucalyptus wood [41, 44]. During water prehydrolysis, the wood constituents are partially hydrolyzed and dissolved in the PHL. The composition of the resulting wood residue is also given in Table 2. As expected only a small amount of glucose is extracted during prehydrolysis, as cellulose is hardly affected by the applied conditions. In contrast, hemicellulose-sugars are readily extracted, whereby the individual sugars are hydrolyzed at different rates. This is in accordance to the work of Sixta [39] dealing with the kinetics of eucalyptus prehydrolysis in more detail. In total, 77.2% of hemicellulose sugars and HAc, as well as 23.0% of Klason lignin and 72.4% of ASL were extracted during prehydrolysis at 170 °C, a L/W ratio of 3:1 and a P-factor of 500, which is in good agreement with values reported previously [41, 44]. The PHL composition given in Table 3 shows that hemicellulose oligomers are the major constituents of the PHL accounting for 42.9% of the dry substance (DS) and only 34% of the total sugar content are monomers.
The PHL is obtained as a brown turbid suspension that separates into a clear brown solution and a brown precipitate upon centrifugation. Solids were not only contained in the PHL, but also adhered on the reactor walls. The brown precipitate was identified as lignin by ATR-IR and accounts for w = 9.5% of the DS present in the prehydrolysate. Soluble lignin was quantified by UV spectrometry and represent w = 12.5% of the DS, giving a total lignin content of w = 22.0% of the DS in the PHL. During the industrial scale operation, high amounts of insoluble lignin species tend to deposit on equipment parts, leading to processing problems and high cleaning effort using large amounts of white liquor [39]. Additionally, the soluble lignin species are inhibitors for fermentative processes and need to be removed to enable the production of sugar-based byproducts from PHL.
3.2 Prevention of lignin precipitation and high temperature extractive detoxification
To avoid the problems associated with lignin precipitation, the isothermally separated PHL was introduced trough an immersion tube directly into various organic solvents in the second reactor. The organic solvents were preheated to a lower temperature of 120 °C to avoid high vapor pressures.
Lignin precipitation was only assessed visually because quantification of precipitated lignin did not lead to reproducible results, due to difficult recovery of all solids. In some cases, lignin was not found to precipitate from the PHL, nor was it found to adhere to the reactor walls. These experiments are marked with a check mark in Fig. 1. Only the investigated alcohols solubilized all lignin species efficiently. All the other solvents could not prevent lignin precipitation. Sugar losses were low for all the investigated solvents. Significant losses were only observed using 1-butanol, where 9.4% of monomeric sugars and 5.9% of total sugars were dissolved in the organic phase.

Distribution coefficients D for the extraction of the main inhibitors from eucalyptus PHL with organic solvents. The y-axis is interrupted from 4–7 to display the high D values of furfural with MIBK. A corresponding table can be found in Section S8 in Online Resource 1. Green checkmarks indicate that a solvent prevented lignin precipitation, a red x shows that lignin precipitation was not prevented
The partition coefficients for lignin calculated from the experimental results are shown in Fig. 1 and decrease in the order 1-butanol > 1-hexanol > CPME > MIBK > 1-octanol > DES > toluene. The extraction efficiency of a solvent for lignin is governed by its molar mass and chemical structure (see Section 3.3). Depending on the structural features of the lignin, different solvent characteristics (donor-, acceptor-, pi-bonding) may be more or less important. We found a significant linear correlation between the lignin extraction efficiency (EEASL) and the Hansen solubility parameter for hydrogen bonding δH (detailed information can be found in section S2 in Online Resource 1). Other research groups have also found a correlation between the ability of a solvent to form hydrogen bonds and lignin solubility [45]. In the literature δH values in the range of 11.3 for hardwood Kraft-lignin to 16.9 for milled wood lignin are reported [46]. Solvents with a δH value matching the one of lignin show the highest solubility. This observation can be explained by the polymeric structure of lignin, bearing high quantities of aliphatic and aromatic hydroxyl groups that are susceptible to hydrogen bond formation with the solvent. However, some groups reported contradictory results. Ribeiro et al. [47] reported a significant correlation of Kraft lignin solubility with δP, whereas the δH parameter was regarded as the most important by other research groups [45, 48]. In conclusion the influence of solvent parameters on lignin solubility is complex and Hansen’s solubility model may be a good starting point for solvent selection but is not capable of fully predicting the lignin solubility. This is due to additional factors not incorporated in the model (e.g., steric factors) and the complex, heterogeneous structure of lignin. Furthermore, lignin structure is dependent on various factors like wood species (hardwood vs. softwood) and processing conditions (e.g., Kraft lignin, MWL, organosolv lignin, prehydrolysis lignin) making it hard to compare results and predict solubilities.
It should be mentioned that the partition coefficient of lignin in various solvents is different from EEASL. This is explained by the fact that not only ASL is extracted from the aqueous PHL into the organic phase, but also other species that would have precipitated without any solvent.
Besides lignin, also small organic acids and furanic compounds are known inhibitors for many sugar-based fermentation processes. Therefore, the extraction of HCOOH, HAc, furfural, and 5-hydroxymethylfurfural (HMF) to the organic phase was investigated. The distribution coefficients D for the inhibitors and for their total sum can be also found in Fig. 1. Carboxylic acids show the lowest D value for all the investigated solvents, with HAc values ranging from 0.00 for toluene to 0.35 for 1-butanol. The extraction efficiency decreases in the order 1-butanol > 1-hexanol > 1-octanol > DES > MIBK > CPME > toluene and is thought to be correlated with the polarity and the ability of the solvent to form hydrogen bonds. D values for HCOOH are in a similar range (0.03–0.51) to those of HAc, with 1-butanol again showing the highest value. However, the second highest D was observed in MIBK, followed by DES, 1-hexanol, 1-octanol, CPME, and toluene. D for HCOOH is in line with the dielectric constant ε of the solvent, with D values increasing with increasing ε value.
The extraction efficiencies of the furanic compounds HMF and furfural are generally higher than for carboxylic acids. HMF is more hydrophilic than furfural, due to its hydroxymethyl group and is therefore harder to extract to an organic solvent. The D values of HMF are in the range of 0.03–1.66. Butanol was the only solvent capable to extract HMF at a D > 1. The higher extraction efficiency of DES for HCOOH can be explained by the superior ability of TOPO to form hydrogen bonds with acidic protons, compared to hydroxy groups. Due to its hydrophobic nature, furfural is readily extracted to all organic solvents tested, with D values of 1.86–7.50, which corresponds to a furfural reduction of 75–93% in the aqueous phase.
The total inhibitor distribution coefficient Dtot decreases in the order 1-butanol > 1-hexanol > CPME > MIBK > 1-octanol > DES > toluene. For 1-butanol the Dtot value is 1.02 and the total inhibitor amount in the aqueous phase is reduced by 63%.
3.3 Molar mass distribution of extracted lignins
The size-exclusion chromatograms of native eucalyptus PHL after prehydrolysis (P = 500, T = 170 °C, L/W ratio = 3) was measured using a RI and UV detector. The RI-chromatogram in Fig. S3 in Online Resource 1 shows a bimodal distribution. The first peak shows a maximum at 176 Da and the second peak a maximum abundance at 562 Da. The low dispersity first peak can be ascribed to the soluble sugar fraction, consisting mainly of monomeric sugars and short oligomers (see Section 3.1). The second peak that is ascribed to soluble lignin and higher sugar oligomers has a broader molar mass distribution, ranging from around 350 Da to 10 kDa, with a maximum abundance at 562 Da and a shoulder at around 1000 Da.
The UV signal in Fig. 2 reveals the presence of multiple lignin species with maxima at 172 Da, 374 Da, and 810 Da. The various phenolic monomers responsible for the first maximum were identified and quantified by GC-MS.

Size-exclusion chromatograms with UV detection for the aqueous (a) and organic (b) phases of PHL after LLE with various solvents
In total, 1.13 g L−1 of phenolic monomers were found in untreated PHL with 4-vinylguaiacol, syringaldehyde, vanillin, and syringic acid being the most abundant species (detailed tables can be found in section S4 in Online Resource 1). The extraction efficiency of lignin monomers reached values between 48 and 100% of the monomers transferred to the organic phase. The observed trend is similar to the trend in the total ASL reduction, except for MIBK showing the second highest extraction efficiency for lignin monomers after 1-butanol.
The second maximum in Fig. 2 is found at 374 Da, which is in the region of phenolic dimers, with β-O-4, β-β resinols, and stilbene structures previously found in eucalyptus PHL. These structures are formed via radical coupling mechanisms via intermediates originating from the homolytic cleavage of β-O-4 linkages [22].
The maximum of the largest peak is at 800 Da and has a broad distribution up to 4 kDa, with a shoulder at 1.5 kDa. This peak is ascribed to water-soluble oligomeric lignin species.
The SEC UV-chromatograms of the aqueous phases in Fig. 2a show an increasing ratio of the dimer peak to the 800 Da peak with increasing overall extraction efficiency of the solvent. This indicates that lignin oligomers are more easily extracted with an organic solvent than dimeric species. In literature, a decrease of the hydroxyl group content with increasing molecular weight in technical lignins is described [22, 49]. Given the highly comparable quality of the lignin in this study, it is not speculative to postulate that the corresponding higher lipophilicity, compared to lignin dimers explains the higher extraction rate using organic solvents.
The SEC UV-chromatograms of the organic samples in Fig. 2b show a bimodal molar mass distribution with maxima at 800 Da and 1400–1600 Da. Lower molecular species are only observed as small shoulders. With increasing extraction efficiency, the area ratios shift towards the higher molecular species. Further, the molar mass maxima of the large lignin species shift to higher molar masses. Therefore, we speculate that these high molar mass species are responsible for the precipitation of lignin in the conventional prehydrolysis process.
3.4 Influence of reactive extractants on the inhibitor extraction efficiency
As explained in Section 3.2, small carboxylic acids are most difficult to extract with organic solvents due to their polar, protic character. To separate carboxylic acids from aqueous solutions, reactive extraction can be used. Therefore, multicomponent solvents are usually employed, in which the extractant reacts with the solute to alter its distribution coefficient. Diluents are used to improve phase separation and reduce extractant losses. 1-Hexanol was chosen as diluent, as it prevented the precipitation of lignin and exhibited a good inhibitor extraction efficiency (see Section 3.2). Although 1-butanol showed a better extraction performance, it has a high mutual solubility with water and its application led to the highest sugar losses. Expected high solvent and sugar losses and a more complex downstream processing eliminate 1-butanol as a diluent in reactive PHL-extraction. It must be pointed out that the solubility of 1-hexanol (5.9 g L−1) in a range that is toxic to most microorganisms [50]. However, several strategies can be employed to reduce or eliminate solvent toxicity. Residual solvent can be removed by steam stripping, salting out, adsorption, or washing the aqueous phase with a non-toxic solvent [51,52,53]. Alternatively, sugars can be isolated by precipitation with an antisolvent, subsequent washing and drying [54].
A structurally diverse set of nitrogen and phosphorous containing reactive extractants (see Table 4) with Lewis base character were screened to potentially improve the LLE of carboxylic acids. Concentrations of 25 % (w/w) in 1-hexanol were employed, representing a commonly used solvent composition [33, 38]. Higher concentrations are sometimes used to further improve the extraction efficiency; however, an increased concentration usually increases the viscosity, leading to slower or incomplete phase separation. Additionally, higher extractant losses to the aqueous phase are reported, potentially leading to toxic effects on microorganisms [55, 56].
As can be seen in Fig. 3, the addition of every tested reactive extractant increased D of carboxylic acids compared to 1-hexanol. TBP and TOPO only led to a slight improvement of the HAc extraction, whereas the N-based extractants increased DHAc substantially. The addition of tertiary amines resulted in the highest DHAc with values of 0.81 and 0.83 for TOA and THA respectively. This can be explained by the higher basicity of tertiary amines due to the inductive effects of the alkyl groups.

Influence of the addition of 25 % (w/w) reactive extractants on the distribution coefficients D for the LLE of the main inhibitors from eucalyptus PHL with 1-hexanol. The y-axis is interrupted from 3.5 to 5 to display the high D values of HCOOH. A corresponding table can be found in Section S8 in Online Resource 1
Addition of TBP and TOPO resulted in a slight increase of the HCOOH distribution, similar to that observed for the HAc distribution. In contrast, with amine extractants the D values were increased extensively. However, it was observed that the total amount of HCOOH was significantly higher in samples extracted with amines. Using tertiary amines approximately 20% more HCOOH was formed, whereas 2.0 and 2.6 times more HCOOH was formed in the presence of DOA and DHA respectively. At the high temperatures of the PHL extraction, the basicity of the amines and the reactivity of secondary amines cause a partial degradation of sugars and furans. Thereby, HCOOH is split off, leading to increased concentrations.
HMF and furfural show similar trends upon LLE with various reactive extractants. In accordance with the increased HCOOH values, diamines lead to the degradation of furanic compounds. Only 7.4% and 17% of the initial furfural, and 40% and 55% of the initial HMF were found after using DOA and DHA as extractants. Small losses of furans were observed for TBP. All amine extractants only slightly reduce the D of furans, except diamines and TBP, leading to extensive furan degradation.
Except for the diamines, reactive extractants decreased DASL, and this effect was strongest for P-based compounds. On the other hand, DHA and DOA showed the highest DASL and SEC confirmed that the diamines could selectively reduce lignin fragments of ca. 400 kDa (see Fig. S3 and Fig. S4 in Online Resource 1), which are particularly toxic [17].
In Fig. 3, the D values of the total inhibitor amount are also shown for the investigated extractants. Using TOPO, the total inhibitor extraction was lower compared to neat 1-hexanol. The addition of 25 % (w/w) TBP did not lead to significant improvements either.
All amine extractants were able to improve the total inhibitor extraction efficiency. Dtot was increased from 0.75 in pure 1-hexanol to 0.92 and 1.02 with added TOA and THA respectively. This is mainly due to the higher D for carboxylic acids, whereas other inhibitors were not significantly affected. The better extraction of ASL by diamines leads to the highest Dtot for DHA and DOA with values of 1.11 and 1.28 respectively. Despite these values, the loss of virtually all furanic compounds must be considered. Furthermore, diamines bear a reactive N-H bond which may lead to other side-reactions, extractant losses, difficult regeneration, and the formation of carcinogenic nitrosamines [57]. The high water solubility of THA compared to TOA is a disadvantage and makes downstream processing more complex; thus, we decided to use a system composed of 25 wt% TOA in 1-hexanol for further study.
3.5 Simultaneous posthydrolysis and liquid–liquid extraction
As described in Section 3.1, only 39% (w/w) of the sugars in the PHL are present as monomers. To make the total sugar content accessible for biotechnological or chemocatalytic conversion, the oligomeric sugars must be hydrolyzed to the respective monomers. Sugar oligomers may be hydrolyzed by enzymes, or acids at elevated temperatures. Enzymatic hydrolysis has several disadvantages such as low space-time yield and high enzyme costs [58]. Acidic posthydrolysis is a cost-effective and energy saving method, especially if it is performed directly after prehydrolysis without intermediate cooling. However, it may to lead to severe fouling problems due to condensation reactions of lignin and furans (furfural and HMF) [43]. These problems may be avoided by the simultaneous extraction of lignin, furans, and eventually, their condensation products into an organic phase during acidic hydrolysis. We therefore investigated the possibility of simultaneous posthydrolysis and extraction of lignin and other inhibitors, using sulfuric acid and the TOA-1-hexanol system described in Section 3.4. To show the influence of sulfuric acid concentration wH2SO4, TOA concentration wTOA and posthydrolysis temperature T on sugar hydrolysis and the extraction of inhibitors, a response surface study was performed. A Box-Behnken design was used to evaluate the influence and optimal conditions of the three parameters. Sugar monomer yield (Ysugar), sugar purity (wsugar), extraction efficiencies for lignin (EEASL), and total inhibitors (EEinh) were chosen as responses. Additionally, the response for carboxylic acids (EECA) and furans (EEF) can be found in section S5 in Online Resource 1. The 16 randomized experimental runs with parameter ranges and the corresponding response values are given in Table 5.
Y sugar in the aqueous phase varied between 26.9 and 83.4%, with wsugar varying between 62.3 and 91.6%, EEASL varied between 51.2 and 85.4%, and EEinh varied between 64.1 and 83.2%.
By applying the ANOVA method to the experimental data, given in Table 5, the reduced quadratic equations for predicting the responses were obtained and are given in section S7 in Online Resource 1. The statistical significance of the mathematical models was evaluated by checking the F values, lack-of-fit values, and model probability values given in section S6 in Online Resource 1. No significant lack-of-fit was observed for any of the responses, thus showing that the model equations are suitable to describe the measured data. The coefficients of determination R2 for Ysugar, wsugar, EEASL, and EEinh were 0.834, 0.944, 0.966, and 0.998 respectively, indicating a consistent model fit. All adjusted R2 and predicted R2 values are in reasonable agreement with each other and the parity plots show that the predicted values match the experimental values well (for details see section S6 in Online Resource 1).
Response surface plots in Fig. 4 and Fig. 5 show the influences of wH2SO4, wTOA, and T on Ysugar (Fig. 4a, Fig. 4b), wsugar (Fig. 4c, Fig. 4d), EEASL (Fig. 5a, Fig. 5b), and EEinh (Fig. 5c, Fig. 5d). Ysugar and wsugar increase with increasing H2SO4 concentration because the hydrolysis of sugar oligomers is catalyzed by protons. The results show that the sugar hydrolysis rate is higher than the formation rate of inhibitors like carboxylic acids and furans, as it was also observed by Borrega et al. [59]. On the contrary, higher concentrations of TOA decrease the sugar yield and purity due to a decreasing pH value. High temperatures have a negative effect on Ysugar and wsugar due to considerable sugar degradation. Interestingly, the influence of the reaction parameters is less pronounced at higher temperatures which is believed to be a consequence of faster oligomer hydrolysis at higher pH values (low wH2SO4 and high wTOA) and higher inhibitor formation balancing each other out. Additionally, at 150 °C, the second dissociation constant for sulfuric acid is nearly one order of magnitude lower than at 100 °C, thus decreasing the influence of wH2SO4 at higher temperatures [60].

Response surface plots showing the influences of sulfuric acid concentration wH2SO4 and TOA concentration wTOA on the sugar yield Ysugar at 100 °C (a) and 150 °C (b). The lower graphs show the influences of sulfuric acid concentration wH2SO4 and TOA concentration wTOA on the sugar purity wsugar at 100 °C (c) and 150 °C (d)

Response surface plots showing the influences of sulfuric acid concentration wH2SO4 and TOA concentration wTOA on the extraction efficiency of lignin EEASL at 100 °C (a) and 150 °C (b). The lower graphs show the influences of sulfuric acid concentration wH2SO4 and TOA concentration wTOA on the overall inhibitor extraction efficiency EEinh at 100 °C (c) and 150 °C (d)
Lignin extraction is favored at low T, low wTOA, and high wH2SO4. This is in accordance with the fact that lignin is less soluble in water at low pH and can be precipitated with sulfuric acid. At higher temperatures, EEASL is lower and less dependent on the extraction conditions. This may be explained by changes in the chemical structure of lignin due to condensation reactions. The extraction of total inhibitors decreases with increasing wTOA, especially at low T. At higher T, the extraction efficiency decreases and is independent on the TOA concentration. The influence of the pH changes with the extraction temperature; at low temperatures, increasing wH2SO4 increases the EEinh, which is due to the better extraction of lignin and carboxylic acids. It is known that the extraction efficiency of carboxylic acids is higher at low pH values, because of their less ionic character.
However, the reaction conditions depend largely on the further downstream processing and utilization of the PHL. If a high extraction efficiency for carboxylic acids is required, the addition of TOA may be beneficial. On the other hand, no TOA, low T and high wH2SO4 are the best conditions to maximize the yield and purity of the resulting sugar solution. When low temperatures are used, the optimum reaction time is easy to control, and only little side-reactions occur. However, hydrolysis rates are low and reaction times up to 6 h may be required. Higher wH2SO4 helps to increase the reaction rates but also raises chemical costs and material costs due to corrosion problems.
4 Conclusions
Liquid–liquid extraction of isothermally separated eucalyptus PHL was studied using a carefully selected set of solvents. Aliphatic alcohols were able to prevent the precipitation of lignin fragments that would be detrimental in industrial operation. We assume that hydrogen bonding between the solvent and hydroxyl groups of high molar mass lignin species is essential for their dissolution. It was shown that furfural and HMF were easily extracted into the organic phase, whereas the distribution coefficients for ASL and carboxylic acids were low. The application of a mixture of 25% (w/w) TOA in 1-hexanol as an extracting agent led to low sugar losses and increased the distribution coefficients for HAc and HCOOH, without a significant change in the removal of the other inhibitors. To maximize the yield and purity of monomeric sugars from PHL a biorefinery process for the simultaneous posthydrolysis with H2SO4 and liquid–liquid extraction (SIMPLLE) was tested and optimized using a Box-Behnken experimental design. Under optimal conditions a monomeric sugar yield of 83.0% at a sugar purity of 91.6% was achieved. It was shown that the process conditions can be easily adapted to achieve desired product properties (e.g., purity, content of certain inhibitors, monomer/oligomer yield).
Thus, the SIMPLLE process offers an efficient and versatile method for the hydrolysis and detoxification of eucalyptus PHL. Fouling due to precipitation is prevented by lignin species removal, enabling industrial implementation. For industrial realization, the development of an efficient solvent regeneration is crucial, because even low amounts of impurities remaining in the extraction solvent were reported to deteriorate the extraction efficiency in long term operation [61]. Additionally, the toxicity of the SIMPLLE treated PHL for specific microorganisms needs to be properly addressed and further purification steps (e.g., solvent stripping) may be required.
Abbreviations
- PHK :
-
prehydrolysis Kraft
- PHL :
-
prehydrolysis liquor
- HCOOH :
-
formic acid
- HAc :
-
acetic acid
- FU :
-
furfural
- HMF :
-
5-(hydroxymethyl)furfural
- DES :
-
2:1 mixture of decanoic acid and tri-n-octylphosphine oxide
- MIBK :
-
methyl isobutyl ketone
- CPME :
-
cyclopentyl methyl ether
- LLE :
-
liquid–liquid extraction
- TOPO :
-
tri-n-octylphosphine oxide
- TOA :
-
tri-n-octylamine
- TBP :
-
tri-n-butylphosphate
- L/W ratio :
-
liquid to wood ratio
- P-factor :
-
prehydrolysis factor
- WR :
-
wood residue
- PTFE :
-
polytetrafluoroethylene
- ASL :
-
acid soluble lignin
- SEC :
-
size-exclusion chromatography
- w H2SO4 :
-
sulfuric acid mass fraction
- w TOA :
-
tri-n-octylamine mass fraction
- Y sugar :
-
yield of monomeric sugars
- w sugar :
-
monomeric sugar purity
- EE ASL :
-
lignin extraction efficiency
- EE inh :
-
total inhibitor extraction efficiency
- EE CA :
-
carboxylic acid extraction efficiency
- EE F :
-
extraction efficiency of furans
- DS :
-
dry substance
- ButOH :
-
1-butanol
- HexOH :
-
1-hexanol
- OctOH :
-
1-octanol
- ANOVA :
-
analysis of variance
- THA :
-
tri-n-hexylamine
- DHA :
-
di-n-hexylamine
- DOA :
-
di-n-octylamine
References
Li J, Zhang H, Duan C et al (2015) Enhancing hemicelluloses removal from a softwood sulfite pulp. Bioresour Technol 192:11–16. https://doi.org/10.1016/j.biortech.2015.04.107
Kumar H, Christopher LP (2017) Recent trends and developments in dissolving pulp production and application. Cellulose 24:2347–2365. https://doi.org/10.1007/s10570-017-1285-y
Mateos-Espejel E, Radiotis T, Jemaa N (2013) Implications of converting a kraft pulp mill to a dissolving pulp operation with a hemicellulose extraction stage. TAPPI J 12:29–38. https://doi.org/10.32964/TJ12.2.29
Lehto JT, Alén RJ (2015) Chemical pretreatments of wood chips prior to alkaline pulping - a review of pretreatment alternatives, chemical aspects of the resulting liquors, and pulping outcomes. BioResources 10. https://doi.org/10.15376/biores.10.4.Lehto
Deguchi S, Tsujii K, Horikoshi K (2008) Crystalline-to-amorphous transformation of cellulose in hot and compressed water and its implications for hydrothermal conversion. Green Chem 10:191–196. https://doi.org/10.1039/B713655B
Kaur I, Ni Y (2015) A process to produce furfural and acetic acid from pre-hydrolysis liquor of kraft based dissolving pulp process. Sep Purif Technol 146:121–126. https://doi.org/10.1016/j.seppur.2015.03.034
Huang H-J, Ramaswamy S, Al-Dajani WW et al (2010) Process modeling and analysis of pulp mill-based integrated biorefinery with hemicellulose pre-extraction for ethanol production: a comparative study. Bioresour Technol 101:624–631. https://doi.org/10.1016/j.biortech.2009.07.092
Miettinen J, Ollikainen M (2022) Economics of forest bioeconomy: new results. Can J For Res 52:426–438. https://doi.org/10.1139/cjfr-2021-0178
Kordsachia O, Patt R, Sixta H (1999) Cellulose isolation from various raw materials. Papier (Heidelberg) 53:96–108
Pascoal Neto C, Evtuguin DV, Furtado FP et al (2002) Effect of pulping conditions on the ECF bleachability of Eucalyptus globulus Kraft pulps. Ind Eng Chem Res 41:6200–6206. https://doi.org/10.1021/ie020263x
Pinto PC, Evtuguin DV, Neto CP et al (2002) Behavior of eucalyptus globulus lignin during kraft pulping. II. Analysis by NMR, ESI/MS, and GPC. J Wood Chem Technol 22:109–125. https://doi.org/10.1081/WCT-120013356
Evtuguin DV, Tomás JL, Silva AMS et al (2003) Characterization of an acetylated heteroxylan from Eucalyptus globulus Labill. Carbohydr Res 338:597–604. https://doi.org/10.1016/S0008-6215(02)00529-3
(1983) 5. Polyoses (Hemicelluloses). In: Fengel D, Wegener G (eds) Wood. DE GRUYTER, Berlin, New York
Gírio FM, Fonseca C, Carvalheiro F et al (2010) Hemicelluloses for fuel ethanol: a review. Bioresour Technol 101:4775–4800. https://doi.org/10.1016/j.biortech.2010.01.088
Lee JW, Yook S, Koh H et al (2021) Engineering xylose metabolism in yeasts to produce biofuels and chemicals. Curr Opin Biotechnol 67:15–25. https://doi.org/10.1016/j.copbio.2020.10.012
Gonçalves MCP, Romanelli JP, Cansian ABM et al (2022) A review on the production and recovery of sugars from lignocellulosics for use in the synthesis of bioproducts. Ind Crop Prod 186:115213. https://doi.org/10.1016/j.indcrop.2022.115213
Kumar V, Yadav SK, Kumar J et al (2020) A critical review on current strategies and trends employed for removal of inhibitors and toxic materials generated during biomass pretreatment. Bioresour Technol 299:122633. https://doi.org/10.1016/j.biortech.2019.122633
Zabed HM, Akter S, Yun J et al (2019) Recent advances in biological pretreatment of microalgae and lignocellulosic biomass for biofuel production. Renew Sust Energ Rev 105:105–128. https://doi.org/10.1016/j.rser.2019.01.048
Wang Q, Tian D, Hu J et al (2018) Fates of hemicellulose, lignin and cellulose in concentrated phosphoric acid with hydrogen peroxide (PHP) pretreatment. RSC Adv 8:12714–12723. https://doi.org/10.1039/C8RA00764K
Kumar V, Binod P, Sindhu R et al (2018) Bioconversion of pentose sugars to value added chemicals and fuels: recent trends, challenges and possibilities. Bioresour Technol 269:443–451. https://doi.org/10.1016/j.biortech.2018.08.042
Kang L, Lee Y, Yoon S-H et al (2012) Ethanol production from the mixture of hemicellulose prehydrolysate and paper sludge. BioResources 7:3607–3626
Leschinsky M, Zuckerstätter G, Weber HK et al (2008) Effect of autohydrolysis of Eucalyptus globulus wood on lignin structure. Part 1: comparison of different lignin fractions formed during water prehydrolysis. Holzforschung 62:645–652. https://doi.org/10.1515/HF.2008.117
Ghazali NF, Razak NDA (2021) Recovery of saccharides from lignocellulosic hydrolysates using nanofiltration membranes: a review. Food Bioprod Process 126:215–233. https://doi.org/10.1016/j.fbp.2021.01.006
Ajao O, Rahni M, Marinova M et al (2015) Retention and flux characteristics of nanofiltration membranes during hemicellulose prehydrolysate concentration. Chem Eng J 260:605–615. https://doi.org/10.1016/j.cej.2014.09.007
Pan L, He M, Wu B et al (2019) Simultaneous concentration and detoxification of lignocellulosic hydrolysates by novel membrane filtration system for bioethanol production. J Clean Prod 227:1185–1194. https://doi.org/10.1016/j.jclepro.2019.04.239
Schlackl K, Bischof RH, Fackler K et al (2020) Impact of intermolecular interactions on the nanofiltration of pulping liquor. Sep Purif Technol 250:117177. https://doi.org/10.1016/j.seppur.2020.117177
Gütsch JS, Sixta H (2012) Regeneration of spent activated charcoals used for lignin removal from prehydrolysis-Kraft prehydrolyzates. Ind Eng Chem Res 51:8624–8630. https://doi.org/10.1021/ie3006116
Wang P, Chen YM, Wang Y et al (2019) Towards comprehensive lignocellulosic biomass utilization for bioenergy production: efficient biobutanol production from acetic acid pretreated switchgrass with Clostridium saccharoperbutylacetonicum N1-4. Appl Energy 236:551–559. https://doi.org/10.1016/j.apenergy.2018.12.011
Yu Y, Christopher LP (2017) Detoxification of hemicellulose-rich poplar hydrolysate by polymeric resins for improved ethanol fermentability. Fuel 203:187–196. https://doi.org/10.1016/j.fuel.2017.04.118
Tramontina R, Brenelli LB, Sodré V et al (2020) Enzymatic removal of inhibitory compounds from lignocellulosic hydrolysates for biomass to bioproducts applications. World J Microbiol Biotechnol 36:166. https://doi.org/10.1007/s11274-020-02942-y
Stiefel S, Di Marino D, Eggert A et al (2017) Liquid/liquid extraction of biomass-derived lignin from lignocellulosic pretreatments. Green Chem 19:93–97. https://doi.org/10.1039/C6GC02270G
Ahsan L, Jahan MS, Ni Y (2013) Recovery of acetic acid from the prehydrolysis liquor of Kraft based dissolving pulp production process: sodium hydroxide back extraction from the trioctylamine/octanol system. Ind Eng Chem Res 52:9270–9275. https://doi.org/10.1021/ie401285v
Bokhary A, Leitch M, Liao BQ (2021) Liquid–liquid extraction technology for resource recovery: applications, potential, and perspectives. J Water Process Eng 40:101762. https://doi.org/10.1016/j.jwpe.2020.101762
Zautsen RRM, Maugeri-Filho F, Vaz-Rossell CE et al (2009) Liquid-liquid extraction of fermentation inhibiting compounds in lignocellulose hydrolysate. Biotechnol Bioeng 102:1354–1360. https://doi.org/10.1002/bit.22189
Cruz JM, Domı́nguez JM, Domı́nguez H et al (1999) Solvent extraction of hemicellulosic wood hydrolysates: a procedure useful for obtaining both detoxified fermentation media and polyphenols with antioxidant activity. Food Chem 67:147–153. https://doi.org/10.1016/S0308-8146(99)00106-5
Wang G, Qi S, Xia Y et al (2020) Mild one-pot lignocellulose fractionation based on acid-catalyzed biphasic water/phenol system to enhance components’ processability. ACS Sustain Chem Eng 8:2772–2782. https://doi.org/10.1021/acssuschemeng.9b06643
Uslu H (2009) Reactive extraction of formic acid by using tri octyl amine (TOA). Sep Sci Technol 44:1784–1798. https://doi.org/10.1080/01496390902775893
Abdulrahman A, van Walsum GP, Um B-H (2019) Acetic acid removal from pre-pulping wood extract with recovery and recycling of extraction solvents. Appl Biochem Biotechnol 187:378–395. https://doi.org/10.1007/s12010-018-2826-z
Sixta H (2006) Handbook of pulp. Wiley-VCH; John Wiley distributor], Weinheim, Chichester
Schlackl K, Bischof RH, Samhaber W (2020) Negative retention by the nanofiltration of aqueous biomass hydrolysates derived from wood pulping. Sep Purif Technol 242:116773. https://doi.org/10.1016/j.seppur.2020.116773
Leschinsky M, Sixta H, Patt R (2009) Detailed mass balances of the autohydrolysis of Eucalyptus Globulus at 170 degrees C. BioResources 4:687–703
Süss R, Kamm B, Arnezeder D et al (2022) Homogeneously catalyzed depolymerization of lignin from organosolv medium: characterization, optimization, and minimization of coke formation. Can J Chem Eng 100. https://doi.org/10.1002/cjce.24055
Almhofer L, Bischof RH, Madera M et al (2022) Kinetic and mechanistic aspects of furfural degradation in biorefineries. Can J Chem Eng. https://doi.org/10.1002/cjce.24593
Penín L, López M, Santos V et al (2020) Technologies for Eucalyptus wood processing in the scope of biorefineries: a comprehensive review. Bioresour Technol 311:123528. https://doi.org/10.1016/j.biortech.2020.123528
Duval A, Vilaplana F, Crestini C et al (2016) Solvent screening for the fractionation of industrial Kraft lignin. Holzforschung 70:11–20. https://doi.org/10.1515/hf-2014-0346
Hansen CM (2007) Hansen solubility parameters: a user’s handbook, 2nd ed. CRC Press, Boca Raton
Ribeiro WCO, Lobosco V, Martinez PFM (2020) Solubility parameters analysis of Eucalyptus urograndis kraft lignin. BioResources 15:8577–8600. https://doi.org/10.15376/biores.15.4.8577-8600
Giummarella N, Lindgren C, Lindström ME et al (2016) Lignin prepared by ultrafiltration of black liquor: investigation of solubility, viscosity, and ash content. BioResources 11:3494–3510. https://doi.org/10.15376/biores.11.2.3494-3510
Musl O, Galler S, Wurzer G et al (2022) High-resolution profiling of the functional heterogeneity of technical lignins. Biomacromolecules 23:1413–1422. https://doi.org/10.1021/acs.biomac.1c01630
Dafoe JT, Daugulis AJ (2014) In situ product removal in fermentation systems: improved process performance and rational extractant selection. Biotechnol Lett 36:443–460. https://doi.org/10.1007/s10529-013-1380-6
Hwang YL, Olson JD, Keller GE (1992) Steam stripping for removal of organic pollutants from water. 2. Vapor-liquid equilibrium data. Ind Eng Chem Res 31:1759–1768. https://doi.org/10.1021/ie00007a022
Chang G, Bao Z, Zhang Z et al (2013) Salt-enhanced removal of 2-ethyl-1-hexanol from aqueous solutions by adsorption on activated carbon. J Colloid Interface Sci 412:7–12. https://doi.org/10.1016/j.jcis.2013.09.002
Kottenhahn P, Philipps G, Jennewein S (2021) Hexanol biosynthesis from syngas by Clostridium carboxidivorans P7 - product toxicity, temperature dependence and in situ extraction. Heliyon 7:e07732. https://doi.org/10.1016/j.heliyon.2021.e07732
Peng F, Ren J-L, Xu F et al (2009) Comparative study of hemicelluloses obtained by graded ethanol precipitation from sugarcane bagasse. J Agric Food Chem 57:6305–6317. https://doi.org/10.1021/jf900986b
Demmelmayer P, Kienberger M (2022) Reactive extraction of lactic acid from sweet sorghum silage press juice. Sep Purif Technol 282:120090. https://doi.org/10.1016/j.seppur.2021.120090
Morales AF, Albet J, Kyuchoukov G et al (2003) Influence of extractant (TBP and TOA), diluent, and modifier on extraction equilibrium of monocarboxylic acids. J Chem Eng Data 48:874–886. https://doi.org/10.1021/je020179o
Yu K, Mitch WA, Dai N (2017) Nitrosamines and nitramines in amine-based carbon dioxide capture systems: fundamentals, engineering implications, and knowledge gaps. Environ Sci Technol 51:11522–11536. https://doi.org/10.1021/acs.est.7b02597
Nakasu P, Ienczak LJ, Costa AC et al (2016) Acid post-hydrolysis of xylooligosaccharides from hydrothermal pretreatment for pentose ethanol production. Fuel 185:73–84. https://doi.org/10.1016/j.fuel.2016.07.069
Borrega M, Nieminen K, Sixta H (2011) Degradation kinetics of the main carbohydrates in birch wood during hot water extraction in a batch reactor at elevated temperatures. Bioresour Technol 102:10724–10732. https://doi.org/10.1016/j.biortech.2011.09.027
Marshall WL, Jones EV (1966) Second dissociation constant of sulfuric acid from 25 to 350° evaluated from solubilities of calcium sulfate in sulfuric acid solutions 1, 2. J Phys Chem 70:4028–4040. https://doi.org/10.1021/j100884a045
Almhofer L, Paulik C, Bammer D et al (2023) Contaminations impairing an acetic acid biorefinery: liquid-liquid extraction of lipophilic wood extractives with fully recyclable extractants. Sep Purif Technol 308:122869. https://doi.org/10.1016/j.seppur.2022.122869
Acknowledgements
We would like to acknowledge the Austrian government, the provinces of Lower Austria Upper Austria, and Carinthia, as well as Lenzing AG, for financial support. Further, we would like to express our gratitude to the Johannes Kepler University, Linz, the University of Natural Resources and Life Sciences (BOKU), Vienna, and Lenzing AG for their in-kind contributions. Furthermore, we would like to thank colleagues from Wood K + Daniela Bammer, Maria Wolfsgruber, and Markus Huemer, as well as Andreas Scherndl, Erwin Malzner, Walter Milacher, Karin Fackler, and their teams for the extraordinary support in the laboratories of Lenzing AG.
Funding
Open access funding provided by Johannes Kepler University Linz. This work was supported by the Austrian Research Promotion Agency (FFG) [grant number 844608].
Author information
Authors and Affiliations
Contributions
Lukas Almhofer: conceptualization, investigation, visualization, writing — original draft. Christian Paulik: supervision, writing — review and editing Robert H. Bischof: conceptualization, project administration, resources, writing — review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Almhofer, L., Paulik, C. & Bischof, R.H. Simultaneous posthydrolysis and liquid–liquid extraction: a SIMPLLE process to detoxify eucalyptus prehydrolysis liquor. Biomass Conv. Bioref. (2023). https://doi.org/10.1007/s13399-023-04570-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13399-023-04570-6