
Abstract
Organic nitrates in the atmosphere are associated with photochemical pollution and are the main components of secondary organic aerosols, which are related to haze. An efficient method for determining organic nitrates in atmospheric fine particles (PM2.5) was established using synthesized standards. Four alkyl (C7–C10) nitrates and three aromatic nitrates (tolyl nitrate, phenethyl nitrate, and p-xylyl nitrate) were synthesized and characterized by 1H and 13C nuclear magnetic resonance spectroscopy. The optimal ions for quantifying and confirming the identities of the analytes were identified by analyzing the standards by gas chromatography tandem mass spectrometry. The tandem mass spectrometer was a triple quadrupole instrument. This method can obtain more accurate information of organic nitrates than on-line methods. Spiked recovery tests were performed using three spike concentrations, and the recoveries were 61.0–111.4 %, and the relative standard deviations were < 8.2% for all of the analytes. Limits of detection and quantification were determined, and the linearity of the method for each analyte was assessed. The applicability of the method was demonstrated by analyzing six PM2.5 samples. Overall, 87% of the analytes were detected in the samples. Phenethyl nitrate, heptyl nitrate, and octyl nitrate were detected in every sample. Phenethyl nitrate was found at a higher mean concentration (3.23 ng/m3) than the other analytes.

Similar content being viewed by others


Organic nitrates (ONs) are esters and derivatives containing nitrate groups. ONs are active nitrogen oxides that have important effects in the atmosphere. ONs can be divided into anthropogenic ONs and biogenic ONs. Many studies of biogenic ONs have been performed because they play important roles in the atmospheric nitrogen cycle [1, 2]. Anthropogenic ONs may, however, be the dominant ONs in the atmosphere in densely populated urban areas because of the considerable emission of anthropogenic VOCs [3]. Volatile organic compounds (VOCs) are precursors of ONs, and the VOCs present determine the types of ONs that form [4]. Long-chain alkanes, ethylbenzene, toluene, and xylene are currently the dominant VOCs in the atmosphere in Beijing [5,6,7]. Most VOCs are active [8] and can generate ONs through photochemical processes in the atmosphere. Lee et al. [9] found that ONs are generated through oxidation of anthropogenic VOCs and are the main components of secondary organic aerosols in the Uintah Basin, Utah, USA. Long-chain alkanes and aromatic compounds can give relatively high ON yields [10, 11]. The relatively high contributions (5–40%) of ONs to organic matter in atmospheric particles [12] mean that the generation and transformation of ONs affect the production and composition of secondary organic aerosols. High O3 concentrations have been found in the atmosphere in China [13, 14], and it is believed that high O3 concentrations probably cause ONs to form through photochemical processes. It is therefore necessary to monitor long chain alkyl and aromatic organic nitrate concentrations in urban areas.
Studies of aromatic nitrates have mainly been focused on organic aerosols generated from precursor aromatic hydrocarbons through photochemical reactions [18, 19], and few studies have been focused on ONs generated from aromatic hydrocarbons. Few alkyl and aromatic ON standards are available, and this has limited the development of methods for qualitatively and quantitatively determining ONs. There is still a lack of methods for qualitatively and quantitatively determining long-chain alkyl and aromatic nitrates in the atmosphere. Currently, ONs are mainly determined using on-line monitoring instruments [15]. Online monitoring systems give fast responses and small errors, but cannot determine specific ONs and therefore can only determine total ON concentrations. Long-chain alkyl nitrates in the gas phase are currently determined by gas chromatography (GC) with electron capture detection and by GC mass spectrometry (MS) [16, 17]. Analytes can often be unambiguously identified, and structural information for unknown compounds can be acquired by MS but not using an electron capture detector. Tandem MS (MS/MS) offers advantages over standard MS in terms of qualitative and quantitative analyses: more accurate information of ONs can be obtained by GC-MS/MS and less interference for GC-MS/MS to detect the trace compounds in the environment. These advantages make it easier to determine the accurate concentration of ONs. Hence, it is necessary to develop a GC-MS/MS-based analytical method. In this study, ON standards were synthesized, and a GC-MS/MS method for determining alkyl and aromatic ONs in the atmosphere was established.
Materials and Methods
Chemicals
Pesticide-grade acetonitrile and dichloromethane were obtained from J.T. Baker (Phillipsburg, NJ, USA). Ultra-pure water was produced using a Milli-Q system (EMD Millipore, Billerica, MA, USA). Liquid chromatography grade n-nonane and analytical grade brominated alkanes and aromatic compounds were obtained from Macklin (Shanghai, China). Silver nitrate was purchased from Jinke (Tianjin, China). Tetramethylsilane was obtained from Innochem (Beijing, China).
Instrumental Method
The ONs were determined using a Trace 1310 gas chromatograph coupled to a TSQ 8000 Evo tandem mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Separation was achieved using a J&W Scientific DB-5MS GC column (30 m long, 0.25 mm i.d., 0.1 μm film thickness; Agilent Technologies, Santa Clara, CA, USA). The oven temperature program started at 40 °C, which was held for 10 min, then increased at 4 °C∙min−1 to 260 °C, which was held for 3 min. The mass spectrometer ion source and transfer line were kept at 280 and 290 °C, respectively. The MS/MS instrument was operated in electron impact ionization mode and selected reaction monitoring mode. The collision voltages and scanning time windows for the analytes are shown in Table S3 in the Supplementary Material. The injection volume was 1.0 μL, and splitless injection mode was used. The carrier gas was helium, and the flow rate was 1.0 mL min−1.
The analytes were analyzed by nuclear magnetic resonance spectroscopy (NMR) using an Avance-600 MH system (Bruker, Billerica, MA, USA). Tetramethylsilane was used as an internal standard and deuterochloroform (CDCl3) was used as the solvent for the analytes.
Sample Collection and Preparation
Fine particulate (PM2.5) samples were collected at Minzu University of China in Beijing (116.19° E, 39.57° N) using a TH-16A small-volume sampler (Tianhong, Wuhan, China). The PM2.5 samples were collected on 47-mm diameter Whatman glass fiber filters (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA). For each sample, 10 m3 was passed through a filter at a flow rate of 16.7 L min−1. The samples were collected on the roof of a building. Samples were collected in different seasons, and then each sample was wrapped in aluminum foil and sealed in a plastic bag, then stored at − 20 °C until they were analyzed.
When a sample was analyzed, the filter was cut into pieces and sonicated with 15.0 mL of CH2Cl2 for 15.0 min. The sample was extracted in the same way with fresh CH2Cl2 three more times, then the four extracts were combined. The extract was then stored at 4 °C for 12 h, then evaporated almost to dryness using a rotary evaporator. The residue was dissolved in 5.0 mL of CH2Cl2, then the extract was centrifuged at 3000 rpm for 5.0 min. A small amount of n-nonane was added to the supernatant, then the mixture was evaporated under a gentle stream of high purity nitrogen without any heat applied until 100.0 μL of n-nonane remained.
Method validation
The recovery, calibration curve linearity, precision, and detection limit were determined for each analyte to assess the performance of the analytical method. Six samples spiked with each analyte were analyzed to determine the analyte recoveries achieved using the method. This was performed at each of three spike concentrations. The instrument detection limit (IDL) and method detection limit (MDL) were defined as the concentrations giving signal-to-noise ratios of 3 and 10, respectively. The precision was defined as the relative standard deviation (%) of the recoveries found in replicate samples. Standard solutions at different concentrations were analyzed, a calibration curve for each analyte was produced, and the linearity of a calibration curve was assessed using the correlation coefficient (R2).
Results and discussion
Synthesis of standards
The appropriate brominated alkanes and aromatic compounds were treated with silver nitrate to produce C7–C10 nitrates, p-xylyl nitrate, tolyl nitrate, and phenethyl nitrate standards [17]. The structures and molecular weights of the synthesized ONs are shown in Table 1. Excess AgNO3 was dissolved in an appropriate amount of acetonitrile, then the selected brominated alkane or aromatic compound was added, and the mixture was stirred with a magnetic stirrer at room temperature for 2–3 days. The mixture was then filtered, then 80.0 mL of ice-cold ultra-pure water and 60.0 mL of CH2Cl2 were added. The mixture was then shaken well to achieve an effective liquid–liquid extraction. The CH2Cl2 layer was evaporated to give the pure synthetic standard. The reaction between a brominated organic compound and silver nitrate is shown below.
The synthesized ONs were characterized by GC-MS/MS, 1H NMR, and 13C NMR. The full-scan mass spectrum of each ON was obtained to obtain the relative abundances of the different fragment ions and identify the most appropriate fragment ions to monitor. Some example mass spectra are shown in Figures 1(a) and 3, S3, and S5.

(a) Mass spectrum and (b) possible cleavage modes for octyl nitrate
The structural characteristics of the ONs were investigated by 1H NMR and 13C NMR. The 1H NMR and 13C NMR data for the synthetic ONs are shown in Tables S1 and S2 of the supplementary, respectively. The signals for protons in the R–CH2–O–NO2 group in the alkyl nitrates had chemical shifts of 4.37–4.45 ppm, and the signals for protons in the R–CH2–O–NO2 group in the aromatic nitrates had chemical shifts of 4.64–5.42 ppm, indicating that each compound contained the –ONO2 group. The downfield shifts indicated that strong electron-withdrawing atoms or groups were present. Muthuramu et al. [20] found that the presence of a strong electron-withdrawing group (e.g., –ONO2) will cause the 1H NMR signals for protons in an R–CH2–O–NO2 group to be shifted downfield more than the signals for protons in the presence of other types of C atoms. Lopes et al. [21] found that the signal for a proton bound to an oxygenated saturated aliphatic C atom (i.e., H–C–O) in an alcohol, polyol, ether, ester, or ON (R–CH2–O–NO2) will have a chemical shift of between 3.3 and 4.1 ppm. Rindelaub et al. [22] found that the signal for the proton attached to the C atom in the R–CH–OH group of a synthesized “α-pinene-derived organic nitrate” had a downfield chemical shift of 4.1 ppm. The signal for a proton attached to a C–O group will be more strongly downfield shifted than the signal for a proton attached to another type of C atom because of the strong electronegativity of O atom.
Qualitative and Quantitative Analysis of Alkyl Nitrates by GC-MS/MS
The mass spectrum of octyl nitrate is shown in Figure 1a. The spectrum contains peaks at m/z ratios characteristic of alkanes, e.g., m/z 43.15 ([CH3(CH2)2]+) and m/z 57.13 ([CH3(CH2)3]+), at relatively high abundances. Peaks for [NO2]+ (at m/z 46.07) and [CH2ONO2]+ (at m/z 76.07) were also abundant. [NO2]+ is a characteristic ion for alkyl nitrates [16, 17, 23]. Saturated aliphatic hydrocarbon ONs can also produce the [CH2ONO2]+ ion, so the [CH2ONO2]+ and [NO2]+ ions could both be used to indicate the presence of an alkyl nitrate. The [CH2ONO2]+ and [NO2]+ ions were therefore used to identify the alkyl nitrates in our method.
Possible cleavage modes for octyl nitrate are shown in Figure 1b. We believe that [NO2]+ ions would have been produced not only through the cleavage of the molecular ion M+ (m/z 175) but also from [CH2ONO2]+. The [CH2ONO2]+ ion (m/z 76.07) can be used to distinguish alkyl nitrates from alkanes and was relatively abundant. The [CH2ONO2]+ ion was therefore used as the primary ion. [CH2ONO2]+ could fragment and produce [NO2]+ (m/z 46.07), so [CH2ONO2]+ and [NO2]+ were used to quantify the alkyl nitrates. The optimized collision voltage was 15 eV. The C7–C10 alkyl nitrate standard total ion chromatogram is shown in Figure 2. The identification ions and quantification ion pairs for the C7–C10 alkyl nitrates are shown in Table 1.

Total ion chromatogram for the C7–C10 alkyl nitrates
Qualitative and Quantitative Analysis of Aromatic Nitrates by GC-MS/MS
Aromatic nitrates have more complex structures than alkyl nitrates. A pair of ions was selected for each aromatic nitrate for use in selective reaction mode. The characteristic ion for each analyte was used as the primary ion, and a suitable product ion was selected after investigating the cleavage modes.
The mass spectrum for tolyl nitrate is shown in Figure 3. The spectrum contains an M+ ion at m/z 153.08, an ion indicative of a nitrate [NO2]+ at m/z 46.07, a [C7H7O]+ ion at m/z 107, and ions characteristic of aromatic hydrocarbons (e.g., [C6H5]+ at m/z 77.10 and [C7H7]+ at m/z 91.11) at relatively high abundances. Possible cleavage modes for tolyl nitrate in electron impact ionization mode are shown in Figure 4. After the impact of an electron, tolyl nitrate will form the molecular ion M+ through the loss of one electron from the O atom in the C–O group. [C6H5]+ will be produced through cleavage of the C–C bond at the connection between the methyl and phenyl groups. M+ could also be produced through the loss of an electron from a C atom in the phenyl group. In this cleavage mode, the O atom will receive an electron after heterolysis of the C–O bond, then the O atom will combine with a H atom released by the phenyl group to form a hydroxy moiety. This could produce [C7H7O]+. The ion at m/z 105.08 was probably produced though the dehydrogenation of [C7H7O]+. The ions at m/z 65.11, 51.07, and 39.12 would have been produced through the cleavage of the phenyl group. In summary, the cleavage pattern and fragment ions allowed the tolyl nitrate structure to be deduced from the mass spectrum. The M+ ion at m/z 153.08 was used as the primary ion, and the [NO2]+ ion at m/z 46.07 was used as the product ion for characterizing tolyl nitrate.

Full scan mass spectrum for tolyl nitrate, acquired using a m/z ratio scanning range of 30–300

Possible cleavage paths for tolyl nitrate
The phenethyl nitrate mass spectrum is shown in Figure S3, and possible phenethyl nitrate cleavage modes are shown in Figure S4. The M+ (m/z 167.11), [C7H7]+ (m/z 91), [C5H5]+ (m/z 65.09), [C8H8]+ (m/z 104.13), and [NO2]+ (m/z 46.05; the characteristic ion) ions had the highest relative abundances. [C8H9NO3]+ (m/z 167.11) and [NO2]+ (m/z 46.05) were used to identify phenethyl nitrate. M+ (m/z 167.11) was used as the primary ion, and [NO2]+ (m/z 46.05) was used as the product ion.
The p-xylyl nitrate mass spectrum is shown in Figure S5, and possible p-xylyl nitrate cleavage modes are shown in Figure S6. A clear molecular ion peak (m/z 167.10) was found, and [C8H9O]+ (m/z 121.12), [C8H9]+ (m/z 105.12), [C7H7]+ (m/z 91.11), [C6H5]+ (m/z 77.09), and [NO2]+ (m/z 46.05) peaks were also present. [C8H9NO3]+ (m/z 167.10) was used as the primary ion, and [NO2]+ (m/z 46.05) was used as the product ion.
Characteristic ions of aromatic hydrocarbons, e.g., [C6H5]+ (m/z 77.09) and [C7H7]+ (m/z 91.11), were relatively abundant in the spectra but the responses were too low for them to be good primary ions. These ions are also prone to interferences because of the large numbers of complex aromatic compounds found in environmental media. The relatively low abundance of [CH2ONO2]+ in an aromatic nitrate spectrum acquired in selective reaction mode means [CH2ONO2]+ and [NO2]+ make a poor quantitative ion pair because of unsatisfactory responses. We therefore used [NO2]+ as the primary ion and used a collision voltage of 10 eV. [NO2]+ will be cleaved to form other product ions very little at a low collision voltage such as 10 eV, so we used [NO2]+ as the product ion. The aromatic nitrate total ion chromatogram is shown in Figure 5. The qualitative and quantitative ion pairs for the aromatic nitrates are shown in Table 1.

Total ion chromatogram for tolyl nitrate, phenethyl nitrate, and p-xylyl nitrate
Optimization of the Instrumental Method
The gas chromatography parameters were optimized to maximize the responses. Tests were performed at different injector temperatures to identify the temperature giving the strongest response for each analyte. The peak areas found using the different injector temperatures are shown in Figure 6. Increasing the injector temperature did not improve the response. The optimal injector temperature was between 180 and 200 °C. The responses for most of the analytes were better at 180 °C than at higher temperatures, but the heptyl nitrate and octyl nitrate responses were best at 200 °C. However, the heptyl nitrate and octyl nitrate responses at 200 and 180 °C were not very different, so an injector temperature of 180 °C was used subsequently.

Responses (as peak areas) for the organic nitrate analytes at different injector temperatures
Linearity, Limits of Detection, and Limits of Quantification
Calibration curves of 0.01 to 20.0 ng μL−1 were used. These covered the entire ON concentration ranges found in real PM2.5 samples. The correlation coefficients (R2) were all > 0.9994. The IDL was defined as the concentration giving a signal-to-noise ratio of 3, and the MDL was defined as the concentration giving a signal-to-noise ratio of 10. The IDLs were 1.0–10.0 pg, and the MDLs were 0.1–1.0 pg m−3. The aromatic nitrate limits of detection were higher than the alkyl nitrate limits of detection. The ON IDLs and MDLs are shown in Table 2.
Recoveries and Precision of the Method
The ON recoveries were defined as the ratios between the amounts of ONs found in the spiked filter sample extracts and the amounts of ONs in the standards spiked into the samples. The performance of the method was tested using three spike levels (10, 100, and 500 ng of each ON). The recoveries are shown in Table 3. Six replicate spiked samples were analyzed at each spike level. Three blank samples were analyzed, and ONs were not detected in any blank sample. The relative standard deviations (%) were 2.3–8.2%, indicating that the method had a good degree of repeatability. ONs are poorly photostable, so light must be excluded throughout the sample preparation process. In a previous experiment, we found not excluding light gave unsatisfactory results (recoveries 61.2–156.3%, relative standard deviations > 10.2%).
ON Concentrations in Real PM2.5
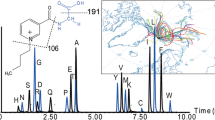
The method was used to analyze six real PM2.5 samples and two blank samples. No ONs were detected in the blank samples. Overall, 87% of the seven analytes were detected in the real samples. Phenethyl nitrate, heptyl nitrate, and octyl nitrate were detected in all of the samples, and the concentrations are shown in Table S4. Phenethyl nitrate was found at the highest concentration (3.23 ng m−3). The mean decyl nitrate concentration was 2.30 ng m−3. Total ion chromatograms of some real samples are shown in Figure 7. The peaks at 20.28, 23.87, 27.28, and 30.55 min were assigned to C7, C8, C9, and C10 alkyl nitrates, respectively, and the peaks at 21.56 and 25.11 min were assigned to tolyl nitrate and phenethyl nitrate.

Total ion chromatograms for organic nitrates in real PM2.5 samples
Conclusions
A GC-MS/MS method for determining alkyl and aromatic nitrates was established, and reliable and effective qualitative and quantitative ions were identified by analyzing synthesized ONs standards by GC-MS/MS. The method was used to determine ON concentrations in real PM2.5 samples collected in Beijing. Phenethyl nitrate, heptyl nitrate, and octyl nitrate were detected in all of the samples. The mean phenethyl nitrate concentration was higher than the mean concentration of any other ON. Light needed to be excluded during the sample preparation process to achieve good recoveries and good degrees of precision and accuracy.
References
Ng, N.L., Brown, S.S., Archibald, A.T., Atlas, E., Cohen, R.C., Crowley, J.N., Day, D.A., Donahue, N.M., Fry, J.L., Fuchs, H.: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol. Atmos. Chem. Phys. 17, 2103–2162 (2017)
Fry, J., Kiendler-Scharr, A., Rollins, A., Wooldridge, P., Brown, S., Fuchs, H., Dubé, W., Mensah, A., Maso, M.d., Tillmann, R.: Organic nitrate and secondary organic aerosol yield from NO3 oxidation of β-pinene evaluated using a gas-phase kinetics/aerosol partitioning model. Atmos. Chem. Phys. 9, 1431–1449 (2009)
Jie, S., Wu, F., Bo, H., Tang, G., Wang, Y.: VOC characteristics, emissions and contributions to SOA formation during hazy episodes. Atmos. Environ. 141, 560–570 (2016)
Perring, A.E., Pusede, S.E., Cohen, R.C.: An observational perspective on the atmospheric impacts of alkyl and multifunctional nitrates on ozone and secondary organic aerosol. Chem. Rev. 113, 5848–5870 (2013)
Duan, H., Liu, X., Yan, M., Wu, Y., Liu, Z.: Characteristics of carbonyls and volatile organic compounds (VOCs) in residences in Beijing, China. Front. Env. Sci. Eng. 10, 73–84 (2016)
Wei, W., Li, Y., Wang, Y., Cheng, S., Wang, L.: Characteristics of VOCs during haze and non-haze days in Beijing, China: concentration, chemical degradation and regional transport impact. Atmos. Environ. 194, 134–145 (2018)
Cao, X., Yao, Z., Shen, X., Ye, Y., Jiang, X.: On-road emission characteristics of VOCs from light-duty gasoline vehicles in Beijing, China. Atmos. Environ. 124, 146–155 (2016)
Lee, C.-L.: Tracing atmospheric oxidation through organic nitrates. UC Berkeley. (2014)
Lee, L., Wooldridge, P.J., Degouw, J., Brown, S.S., Bates, T.S., Quinn, P.K., Cohen, R.C.: Particulate organic nitrates observed in an oil and natural gas production region during wintertime. Atmos. Chem. Phys. 15, 10677–10708 (2015)
Aiko, M., Ziemann, P.J.: Yields of beta-hydroxynitrates and dihydroxynitrates in aerosol formed from OH radical-initiated reactions of linear alkenes in the presence of NOx. J. Phys. Chem. A. 113, 599 (2009)
Lim, Y.B., Ziemann, P.J.: Products and mechanism of secondary organic aerosol formation from reactions of n-alkanes with OH radicals in the presence of NOx. Environ. Sci. Technol. 39, 9229–9236 (2005)
Rollins, A.W., Browne, E.C., Min, K.-E., Pusede, S.E., Wooldridge, P.J., Gentner, D.R., Goldstein, A.H., Liu, S., Day, D.A., Russell, L.M.: Evidence for NOx control over nighttime SOA formation. Science. 337, 1210–1212 (2012)
Wang, M., Shao, M., Chen, W., Lu, S., Wang, C., Huang, D., Yuan, B., Zeng, L., Zhao, Y.: Measurements of C1–C4 alkyl nitrates and their relationships with carbonyl compounds and O3 in Chinese cities. Atmos. Environ. 81, 389–398 (2013)
Wang, T., Xue, L., Brimblecombe, P., Lam, Y.F., Li, L., Zhang, L.: Ozone pollution in China: a review of concentrations, meteorological influences, chemical precursors, and effects. Sci. Total Environ. 575, 1582–1596 (2017)
Day, D., Wooldridge, P., Dillon, M., Thornton, J., Cohen, R.: A thermal dissociation laser-induced fluorescence instrument for in situ detection of NO2, peroxy nitrates, alkyl nitrates, and HNO3. J. Geophys. Res.-Atmos. 107, ACH 4-1–ACH 4-14 (2002)
Dambach, M.: Semivolatile long chain C6–C17 alkyl nitrates as trace compounds in air. Chemosphere. 33, 393–404 (1996)
Luxenhofer, O., Schneider, E., Ballschmiter, K.: Separation, detection and occurrence of (C2–C8)-alkyl- and phenyl-alkyl nitrates as trace compounds in clean and polluted air. Fresenius J. Anal. Chem. 350, 384–394 (1994)
Sato, K., Takami, A., Isozaki, T., Hikida, T., Shimono, A., Imamura, T.: Mass spectrometric study of secondary organic aerosol formed from the photo-oxidation of aromatic hydrocarbons. Atmos. Environ. 44, 1080–1087 (2010)
Forstner, H.J., Flagan, R.C., Seinfeld, J.H.: Secondary organic aerosol from the photooxidation of aromatic hydrocarbons: Molecular composition. Environ. Sci. Technol. 31, 1345–1358 (1997)
Muthuramu, K., Shepson, P.B., O'Brien, J.M.: Preparation, analysis, and atmospheric production of multifunctional organic nitrates. Environ. Sci. Technol. 27, 374–383 (1993)
Lopes, S.P., Matos, J.T.V., Silva, A.M.S., Duarte, A.C., Duarte, R.M.B.O.: 1H NMR studies of water- and alkaline-soluble organic matter from fine urban atmospheric aerosols. Atmos. Environ. 119, 374–380 (2015)
Rindelaub, J.D., Borca, C.H., Hostetler, M.A., Slade, J.H., Lipton, M.A., Slipchenko, L.V., Shepson, P.B.: The acid-catalyzed hydrolysis of an α-pinene-derived organic nitrate: kinetics, products, reaction mechanisms, and atmospheric impact. Atmos. Chem. Phys. 16, 15425–15432 (2016)
Luxenhofer, O., Ballschmiter, K.: C4-C14-alkyl nitrates as organic trace compounds in air. Fresenius J. Anal. Chem. 350, 395–402 (1994)
Acknowledgements
This work was supported by the National Natural Science Foundation of China [grant no. 91744206] and the Beijing Science and Technology Planning Project [grant no. Z181100005418016]. We also thank Dr. Gareth Thomas for his help in grammatical editing of this paper.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(PDF 457 kb)
Rights and permissions
About this article

Cite this article
Yang, X., Luo, F., Li, J. et al. Alkyl and aromatic nitrates in atmospheric particles determined by gas chromatography tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 30, 2762–2770 (2019). https://doi.org/10.1007/s13361-019-02347-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-019-02347-8