
Abstract
Nanoflow liquid chromatography coupled with a captive spray ionization time-of-flight tandem mass spectrometer (nLC/CSI-QTOF-MS/MS) was used in the structural determination of polar steroid compounds of starfish Lethasterias fusca. A total of 207 compounds including 106 asterosaponins, 81 glycosides of polyhydroxysteroids, and 14 polyhydroxylated steroids were detected and characterized by MS and MS/MS. Twenty compounds among them were unambiguously identified using authentic standard compounds, isolated earlier from this and other starfish species. The other compounds were tentatively characterized by accurate mass measurement and comparing retention times and characteristic MS/MS fragmentation patterns with reference standards. Moreover, fragmentation behaviors of a series of pure standards of starfish polar steroids and polyhydroxysteroid compounds detected in L. fusca have been extensively investigated and characteristic fragmentation pathways were described and used for the characterization of unknown compounds.

Similar content being viewed by others


Introduction
A distinguishing feature of the starfish metabolome is the high content of steroid compounds of a great structural diversity including steroid hormones, free sterols, and polar steroid compounds such as polyhydroxysteroids, related mono-, bi-, and rare triosides, glycosides with cyclic carbohydrate chains, and oligoglycosides named asterosaponins. Currently, more than 800 polar steroid compounds have been isolated from different species of starfish, and the variety of structures of these compounds seems inexhaustible [1,2,3,4,5].
Presently, about 150 asterosaponins are known, which are the most polar steroid compounds of starfish. While a 3-O-sulfated 3β,6α-dihydroxysteroid tetracyclic nucleus with a 9(11)-double bond is a general structural characteristic of asterosaponins, side chains of aglycones as well as oligosaccharide chains show a significant natural structural variety. Most aglycones of asterosaponins have cholestane-type side chains, but ergostane (24-methyl-cholestane), stigmastane (24-ethyl-cholestane), and aglycones with shortened side chains also were described [1,2,3,4,5]. The side chains of aglycones may contain hydroxy and/or ketone groups and double bonds in different locations. Carbohydrate chains are located at the C-6 of aglycone and have, as a rule, of four to six monosaccharides with branching at the second unit. Some hexaosides contain oligosaccharide chains with two branches at the second and third monosaccharide units. Usually carbohydrate chains of asterosaponins include hexoses (glucose, galactose), pentoses (arabinose, xylose), and deoxyhexoses (quinovose, fucose) linked, as a rule, by β-glycosidic bonds.
Polyhydroxysteroids contain four to nine hydroxy groups in the tetracyclic nucleus and side chains. Polyhydroxysteroids typically have hydroxy groups at 3β, 6 (α or β), 8, 15 (α or β), and 16β positions, though some polyhydroxysteroids contain hydroxy groups at 4β, 5α, 7 (α or β), and rarely at the 14α position. Structures of side chains of polyhydroxysteroids are widely different. Most polyhydroxysteroids have cholestane side chains with a hydroxy group at C-26 or C-24 (C-28 or C-29 in ergostane or stigmastane types of side chains, respectively). Glycosides of polyhydroxysteroids have a polyhydroxylated steroid nucleus and, as a rule, one or two sugar units located at steroid moiety, either to side chains or to a steroid nucleus and side chain simultaneously. Glycosides of polyhydroxysteroids usually have pentose (arabinose, xylose, or their methylated derivates) or hexose (glucose, galactose) as monosaccharide units and can have sulfate group. Typically, a sulfate group is located in the steroid nucleus at C-15 or in the side chain, but polyhydroxysteroids with sulfate groups at C-3, C-6, or C-16 were also described [1,2,3,4,5]. Additionally, a large number of glycosides with sulfated monosaccharides were isolated from different starfish species.
Starfish receive a lot of attention because of their steroid metabolites with unique chemical structures and also due to their broad spectrum of biological effects such as antiviral, cytotoxic, antifungal, antibacterial, anti-inflammatory, anticancer, analgesic, and neuritogenic actions [1,2,3,4,5].
Study of the physiological activities and biological functions of natural compounds is not possible without determination of the exact chemical structure of these metabolites. However, identification and full de novo structure elucidation of starfish steroid metabolites still remains a challenging task due to the great diversity of these compounds and complexity of steroid fractions consisting of hundreds of compounds. The methodologies that have been developed from classical natural product chemistry include isolation of individual compounds and establishing structures by different approaches. The structure elucidation of a new compound is always performed with a set of independent methods, such as one- (1D) and two-dimensional (2D) nuclear magnetic resonance spectroscopy (NMR), mass spectrometry (MS), or other spectroscopic methods and chemical derivatization. Although NMR methods have been successfully used, the utility of NMR is limited due to the amount of material required for analysis.
Mass spectrometry, especially liquid chromatography/mass spectrometry (LC/MS), has become the primary approach for investigating the structures of metabolites in complex mixtures of natural product extracts [6, 7]. The characteristic fragmentations in MS/MS spectra allow determining main structural features for new steroid glycosides such as the presence of sulfate groups or a specific substituent, sequencing oligosaccharide chains, and establishing aglycone structures. It is known that MS approaches usually have great advantages for the structural analysis of individual starfish steroid metabolites [8,9,10,11,12,13]. Recently, LC-ESI MS/MS methods were used for analysis of polar steroid metabolomes of the Far Eastern starfishes Aphelasterias japonica [6] and Patiria pectinifera [7]. Totally, 68 and 72 polar steroid metabolites were found and characterized, respectively, and reasonable proposals for all glycoside structures were given. As a result, a large number of new minor compounds not previously found in these species of starfish were discovered. Also we have applied LC/MS-based targeted metabolomics to evaluate changes in the steroid metabolome of the starfish P. pectinifera under the influence of such environmental factors and stresses [8]. It was shown that LC-ESI MS/MS is a quite applicable approach for the profiling of starfish extracts and the data obtained can be useful for comparing steroid metabolomic profiles of different starfish species and populations for ecological, dietary, and biosynthetic studies.
In this paper, we describe the application of a nanoflow liquid chromatography/tandem mass spectrometer with captive spray ionization (nLC/CSI-QTOF-MS/MS) for the profiling and characterization of the polar steroid constituents of the starfish Lethasterias fusca. The starfish L. fusca (order Forcipulatida, family Asteriidae) is a common species in the Northwestern Pacific area. The previous investigations of this starfish have led to the isolation of 14 steroid compounds, comprising 3 polyhydroxysteroids, 6 glycosides of polyhydroxysteroids [9], and 5 asterosaponins [10]. It has been shown that asterosaponin lethasterioside A considerably inhibits (up to 90%) the formation of cancer cell colonies [10]. The approach we used allows obtaining a large pool of MS data for starfish polar steroids and offers tentatively structures for detected compounds without isolating individual metabolites, which may be of practical significance in the further use of MS methods for studying steroids of different structure classes.
Materials and Methods
Chemicals
Acetonitrile (UHPLC grade) and water (LC/MS grade) were obtained from Panreac (Barcelona, Spain), and methanol (HPLC grade) was obtained from J.T. Baker (Deventer, Netherlands). All other chemicals were of analytical grade or equivalent.
Animal Material
Specimens of the starfish Lethasterias fusca (order Forcipulatida, family Asteriidae) were collected at Posyet Gulf, the Sea of Japan, in August 2017. Species identification was carried out by B.B. Grebnev (G.B. Elyakov Pacific Institute of Bioorganic Chemistry of the Far Eastern Branch of Russian Academy of Sciences (PIBOC FEB RAS), Vladivostok, Russia). All the specimens were sexually mature and ranged in diameter from 10 to 18 cm; the identification of sex of the animals was not performed. The voucher specimen No. PIBOC-2017-08-LF is preserved in the collection of G.B. Elyakov Pacific Institute of Bioorganic Chemistry of the Far Eastern Branch of Russian Academy of Sciences.
Sample Preparation and Extraction
Five freshly caught animals (wet weight of the individuals = 151 ± 97 g) were chopped and subjected to the triple extraction with ethanol (totally 1.5 L, for 10 h). The ethanol extracts were filtered, combined, and evaporated in vacuo. A lipid-containing sample of dried extract was subjected to the liquid-liquid extraction with a solvent combination of chloroform:methanol:water (CHCl3/MeOH/H2O 8:4:3, v/v/v) to a final dilution 30-fold in relation to the weight of the dried sample to obtain a purified fraction of polar steroid compounds. After dispersion, the whole mixture was agitated for 20 min by a shaker and centrifuged for 15 min at 1400 rpm. As a result, lower lipophilic and upper hydrophilic phases separated by a protein layer were generated. The 100 μL of the upper phase was removed and re-extracted with the solvent mixture CHCl3/MeOH/H2O 8:4:3, v/v/v to a final volume 1600 μL. After centrifugation, the upper phase was again collected (400 μL), dried in a vacuum centrifuge, and reconstituted in 150 μL 50% MeOH.
For desalting, this fraction was subjected to the solid-phase extraction (SPE). SPE process was described in detail previously [6, 7]. Briefly, SPE cartridge (BondElut C18, 100 mg/1 mL, Agilent Technologies, Santa Clara, CA, USA) was conditioned with 3 mL of acetonitrile (ACN) followed by 3 mL 0.1% formic acid (FA) in water. The 100 μL of extract was loaded into the SPE cartridge. The SPE cartridge was washed with 0.5 mL of 0.1% FA. Polar steroid compounds were eluted with 1 mL of 100% ACN. This fraction was dried and dissolved in 500 μL 80% MeOH in water (v/v) and subjected to LC/MS analyses.
Chromatography and Mass Spectrometry
Nanoflow liquid chromatography was performed using an UltiMate 3000 RSLCnano System (Dionex, Sunnyvale, CA, USA), equipped with a pump module, a column compartment (NCS-3500 RS), and an autosampler (WPS-3000TPL RS). Chromatographic separation was performed on an Acclaim PepMap RSLC column (75 μm × 150 mm, nanoViper, C18, 2 μm, 100 A; Thermo Scientific) with a cartridge-based trap column μ-Precolumn (300 μm × 5 mm, C18, 5 μm, 100 A; Thermo Scientific) at a column temperature of 40 °C. The mobile phases consisted of water containing 0.1% FA as solvent A and acetonitrile containing 0.1% FA as solvent B. The gradient profile began with 34% B at 400 nL/min flow rate for 5 min, increased to 58% B from 6 to 20 min, and then to 80% B from 20 to 70 min, from 80 to 99% B from 70 to 71 min, isocratic at 99% of eluent B to 80 min, and finally decreased to 34% B to 81 min for equilibration of the column during 15 min. The injection volume was 0.2 μL.
The Bruker Impact II Q-TOF mass spectrometer (Bruker Daltonics, Bremen, Germany) equipped with a CaptiveSpray ionization source (Bruker Daltonics, Bremen, Germany) was used to record the MS spectra within m/z range of 100–2000 and tandem spectra within m/z range 50–1500. The capillary voltage was set to 1300 V, and the drying gas was heated to 150 °C at the flow rate 3 L/min. Collision-induced dissociation (CID) product ion mass spectra were recorded in auto-MS/MS mode, the threshold for precursor ion isolation was set to 1000, and an active exclusion after five spectra was used. The collision energy was set automatically from 30 to 140 eV according to the molecular masses of precursor ions chosen for fragmentation with an isolation window width of 3 Da. Nitrogen was used as the collision gas.
The mass spectrometer was calibrated using the ESI-L Low Concentration Tuning Mix (Agilent Technologies, Santa Clara, CA, USA) under conditions recommended by the manufacturer. Additionally, a lock-mass calibration with hexakis(1H,1H,3H-tetrafluoropropoxy)phosphazine (922.0098 m/z in positive mode; 966.0007 m/z in negative mode; Agilent Technologies, Santa Clara, CA, USA) was performed; a calibrant was applied to the inner wall of the air filter of CaptiveSpray source. The instrument was operated using the otofControl (ver. 4.0, Bruker Daltonics, Bremen, Germany) and the data were analyzed using the DataAnalysis Software (ver. 4.3, Bruker Daltonics, Bremen, Germany).
Standards of Polar Steroid Metabolites
Forty-three polar steroid metabolites previously isolated by our group from the starfish L. fusca [9, 10], Patiria (=Asterina) pectinifera [11,12,13], Aphelasterias japonica [14,15,16], Asterias rathbuni [17], Diplasterias brucei [18], Linckia laevigata [19], Hippasterias kurilensis [20], Ogmaster capella [21], Leptasterias ochotensis [22], Pentaceraster regulus [23], and Choriaster granulatus [24] were used as standards of polar steroids. Structures of these compounds were established using different methods including 1D and 2D NMR spectra. Compounds were dissolved in 80% MeOH (1 μg/mL) and 0.2 μL of solution was subjected to nLC/CSI-QTOF-MS/MS analysis in the same condition as the L. fusca sample. The full list of the standards of polar steroids and the obtained qualitative information is given in Supplementary Table S1 (Supplementary Material).
Results and Discussion
Fragmentation Study of Standards of Starfish Polar Steroids
For the identification of fragmentation patterns and retention behavior of various polar steroid metabolites from starfish, 43 authentic compounds, including 5 asterosaponins, 28 glycosides of polyhydroxysteroids, and 10 polyhydroxysteroids, isolated earlier from L. fusca and other species were analyzed by nLC/CSI-QTOF-MS/MS. Pure standards of polar steroids were analyzed in the same condition as L. fusca sample. A detailed fragmentation analysis with accurate mass measurement provided by QTOF-MS discovered correlations of fragmentation patterns of compounds with their structural characteristics. Since majority of polar steroid metabolites of starfish contain sulfate groups, the negative ion mode LC/MS experiments showed much higher sensitivity than that in the positive ion mode. In addition, the negative ion spectra have revealed characteristic fragmentations of aglycone moieties and oligosaccharide chains of these compounds. The list of the analytes as well as the obtained data, including the retention times, accurate mass measurements of precursor and product ions, are listed in Supplementary Table S1.
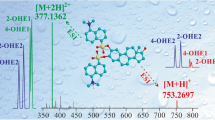
The fragmentation analyses of asterosaponins lethasteriosides A and B, thornasteroside A, anasteroside A, and luidiaquinoside, isolated earlier from L. fusca [10], were performed by nLC/CSI-QTOF-MS/MS. The negative product ion spectra of asterosaponins yielded several characteristic mass losses provided information about side chain structures in aglycones, presence of sulfate groups, and sequences of monosaccharide units in carbohydrate chains. In the negative product ion spectra, an intense Y-type product ion series (nomenclature according to Domon and Costello [25], example is demonstrated in Figure 1a) associated with the cleavages of glycosidic bonds and corresponding sequential losses of sugar units was observed. For example, a negative product ion spectrum of lethasterioside A displayed the [M–Na]− precursor ion at m/z 1227.55 and showed fragment peak arising from the cleavages of glycosidic bonds at m/z 1081.49 [M–Na–Fuc (or Qui)]−, 935.43 [M–Na–Fuc–Qui]−, 919.44 [M–Na–Fuc–Gal]−, 773.38 [M–Na–Fuc–Qui–Gal]−, 641.34 [M–Na–Fuc–Qui–Gal–Xyl]−, 495.28 [M–Na–Fuc–2×Qui–Gal–Xyl]−, 477.27 [M–Na–Fuc–2×Qui–Gal–Xyl–H2O]−, and fragment peak at m/z 96.9 [HSO4]− (Figure 2a). Spectra of thornasteroside A and luidiaquinoside displayed a mass loss of fragment of 100 Da as well as the Y–100 product ion series (Figure 2b). This fragmentation corresponds to the loss of C6H12O molecule associated with the C-20–C-22 bond cleavage and 1H transfer, that is characteristic of asterosaponins containing an aglycone with a 20-hydroxy-cholestan-23-one side chain [1].

Fragmentation nomenclature used for asterosaponins (a) and glycosides of polyhydroxysteroids (b)

The MS/MS spectra of [M–Na]− precursor ion of authentic standards of lethasterioside A (a) and luidiaquinoside (b) isolated from L. fusca [10]
Structural characterization of polyhydroxysteroids and glycosides of polyhydroxysteroids is challenging due to a great diversity of compounds of this class. However, fragmentation under CID condition revealed specific patterns according to structural features of polyhydroxysteroids and glycosides of polyhydroxysteroids. Spectra of polyhydroxysteroid compounds and related glycosides contain fragmentation peculiarities enabling the determination of a number of hydroxy groups and structures of the side chains and steroid nuclei. In general, polyhydroxysteroid compounds under CID conditions tend to lose H2O molecules [26]. A recent study on the fragmentation of highly hydroxylated brassinosteroids showed that water loss occurs readily from vicinal OH groups at the initial fragmentation step, forming an epoxide structure [27]. Recently, we demonstrated that it is possible to distinguish between starfish polyhydroxysteroids with different orientations of the hydroxy group at C-15 in the steroid nucleus via tandem MS. Tandem mass spectra of polyhydroxysteroids showed intensive peaks of product ion series [M–H–n×H2O]− and [M–H–n×H2O–2H]−. In the spectra of 15α-derivatives, the intensities of product ions [M–H–n×H2O–2H]− (n = 1, 2, 3) were higher than the intensities of the corresponding product ions [M–H–n×H2O]−, whereas 16β-derivatives exhibited more intensive product ion series [M–H–n×H2O]− [28].
Moreover, several studies showed [6, 7] that there is a correlation between chromatographic behavior and structural features of polyhydroxysteroid compounds. Specifically, configurations of hydroxy groups influenced strongly the retention time. For instance, in RP-LC the retention times of 15α-derivatives were shorter than those of 15β-derivatives. Elongation of a side chain increases the retention times while the introduction of an additional hydroxy group or particularly sugar unit decreases retention times [6, 7].
Below we discuss fragmentation patterns of some standards used for characterization of compounds detected in the analyzed L. fusca sample. The nomenclature systems denoting the steroid nuclear and side chain fragment ions proposed by Griffiths [26] and denoting glycoconjugate fragment ions proposed by Domon and Costello [25] were used at fragmentation analyses throughout this work; some examples are demonstrated in Figure 1b.
Pycnopodioside C has 5α-cholestane-3β,6α,8,15β,24-pentaol aglycone and sulfated glucose in the side chain at C-24 [14]. Fragmentation of this compound showed diagnostic pattern specific for all compounds with similar structures. The MS/MS spectrum revealed the most prominent ion B0 at m/z 241.00 [C6H9O8S]− which is characteristic of sulfated hexose unit [29, 30]. In addition, this spectrum contains a weak characteristic fragment peak at m/z 369.12 (‘E) [C14H25O9S]− arising from cleavage of the C-17–C-20 bond via a 1,4-H2 elimination reaction [26]; a peak at m/z 397.16 (D2) [C16H29O9S]− that arises from cleavage of the C-13–C-17 and C-15–C-16 bonds; and a peak at m/z 467.19 (C3) [C20H35O10S]− arising from cleavage of the C-8–C-14 and C-12–C-13 bonds (Figure 3a). Thus, the presence of weak fragment ions ‘E, D2, and C3 in the spectrum along with the intense ion B0 indicates that the glycoside has an aglycone with a cholestane-type side chain and sulfated monosaccharide at C-24.

The MS/MS spectra of [M–Na]− precursor ion of authentic standards of pycnopodioside C (a) and asteriidoside H (b)
The spectrum of aphelasteroside D having 5α-cholestane-3β,5,6β,8,15α,29-hexaol aglycone and sulfated glucose in the side chain at C-29 [14] contains fragment peaks at m/z 341.09 (‘H) [C11H19O9S]− and 355.11 (‘G) [C12H21O9S]− arising from cleavage of the C-23–C-24 and C-22–C-23 bonds, respectively, and very weak peaks of fragment ions at m/z 425.19 (D2) [C18H33O9S]−, 397.15 (‘E) [C16H29O9S]−, and 241.00 (B0) [C5H7O7S]− associated with 2’-O-sulfated glucose.
Analysis of the MS/MS data of Δ22 glycosides of polyhydroxysteroids also showed the presence of characteristic fragmentation. For example, the MS/MS spectrum of asteriidoside H having Δ22 side chain and a sulfated pentose at C-24 [18] contains an intensive fragment peak at m/z 295.05 [C10H15O8S]−, arising from C-22–C-23 double bond cleavage and 2H transfer (ion “G). Weak fragment peaks at m/z 307.05 [C11H15O8S]− (ion ‘F-2H) arising from cleavage of the C-20–C-22 bond, 339.11 [C13H23O8S]− corresponding to the loss of the side chain (ion ‘E), 351.11 [C14H23O8S]−, and 365.13 [C15H25O8S]− formed by D-ring bond cleavage were also observed (ions D1 and D2, respectively, Figure 3b). Thus, low abundant fragment ions ‘F-2H, ‘E, D1, and D2 and the intense ion “G are formed as a result of specific fragmentation characteristics for glycosides with Δ22 and sulfated monosaccharide moiety at C-24.
The spectrum of glycoside asteriidoside L with 28-O-sulfo-ergost-22-ene side chain [18] also shows the formation of an intense product ion “G with m/z 177.02 [C6H9O4S]− arising from C-22–C-23 double bond cleavage. It should be noted that double bond cleavage in CID condition is rare, while cleavages of allyl and vinyl bonds are common in tandem spectra of unsaturated fatty acids, lipid, and other compounds [26, 31]. The authors of the recent study proposed that cleavage across the double bond involved a double bond rearrangement, homolytic cleavage across the previous double bond followed by resonance stabilization and β-hydrogen abstraction [32]. A similar mechanism can explain the presence of the product ion “G in spectra of Δ22 glycosides of polyhydroxysteroids.
In contrast to the compounds with the sulfate group in the side chain or at C-3, glycosides with sulfate at C-15 or C-16 exhibited limited fragmentation. The spectrum of coscinasteroside B with 5α-cholestane-3β,6β,8,15α,24-pentaol 15-O-sultated as aglycone and xylose in the side chain at C-24 [21] showed a weak fragment peak at m/z 513.29 associated to the loss of pentose, along with ion “D3 at m/z 387.15 and the most prominent peak at m/z 96.96 [HSO4]−.
In the product ion spectra of glycosides with sulfated monosaccharide unit, the A- and X-type product ions formed by cross-ring cleavages of sulfated monosaccharide were detected. It was reported that a negative product ion spectrum provides diagnostic product ions from cross-ring cleavages allowing distinction between isomeric monosaccharides with different positions of the sulfate group [29, 30]. The obtained fragmentation patterns of glycosides of polyhydroxysteroids are in good agreement with these data. For instance, the presence of fragment peak at m/z 166.96 [C3H3O6S]− in the product ion spectra of glycosides with sulfated hexose (product ion B0 at m/z 241.00 [C6H9O8S]−) corresponded to the sulfate group at 2’ or 3’. The presence of fragment ions at m/z 164.99 [C4H5O5S]− and 168.98 [C3H5O6S]− corresponded to the hexose unit with the sulfate group at 4’ or 6’. However, we were not able to distinguish hexoses sulfated at 2’-O- or 3’-O- and hexoses sulfated at 4’-O- or 6’-O- positions.
Thus, characteristic fragmentation pathways discussed herein as well as accurate mass measurements and retention behavior can reveal the number of hydroxy groups, structure of side chains, number and position of double bonds and sugar moieties. Unfortunately, CID was inefficient for rings and side chain fragmentation and so the specific functional group locations were determined on the basis of literature data and biosynthetic considerations.
The Workflow for Characterization of the Polar Steroid Compounds from L. fusca
Ethanolic extracts of starfish are complicated mixtures containing hundreds of compounds. Beside polar steroids, starfish extracts contain significant amounts of various other compounds, including lipids. To obtain a purified fraction of polar steroid compounds, liquid-liquid extraction with a solvent combination of chloroform:methanol:water was used followed by desalting by SPE. Preliminary LC/MS experiments showed the absence of polar steroids in the chloroform layer and lipid contamination in the water-methanol layer.
For characterization of the polar steroid compounds from starfish extract, the next workflow was used. First, the in-house database including names, structures, and the information on the retention times, accurate mass measurements of precursor and product ions of the polar steroid compounds from L. fusca and other starfish was compiled. Some characteristic diagnostic fragmentations and retention behavior were established and used for identification of unknown compounds. The purified fraction of polar steroids was subjected to nLC/CSI-QTOF-MS/MS. Nanoflow liquid chromatography coupled with captive spray ionization increased the sensitivity and reduced the noise level compared to previous studies in which the conventional LC/ESI-MS was used [6, 7]. Profiling of steroid compounds from the starfish L. fusca allowed a multitude of different steroid compounds to be detected. The base-peak and extracted ion chromatograms of the L. fusca are shown in Figure 4. The accurate mass measurement provided by QTOF-MS allowed proposing molecular formulas for 207 detected compounds, and MS/MS experiment provided extensive characteristic fragmentation. Due to the wide variability of chemical structures of starfish steroid compounds and the inherent limitation of the MS technique, only the most probable structures could be approximately constructed. The stereochemistry and some details of exact structures could not be unambiguously differentiated by the LC/MS without comparing with the authentic standards or application of additional approaches such as NMR.

The base peak ion chromatogram of the polar steroid fraction of L. fusca analyzed by nLC/CSI-QTOF-MS in negative ion mode (a) and extracted ion chromatograms of detected compounds (b)
Majority of product ion spectra included characteristic neutral losses of sugar moieties (162 Da for hexoses, 146 Da for deoxyhexoses, and 132 Da for pentoses), indicating glycosidic nature of detected compounds. Glycosides exhibited precursor ions within m/z range of 1100–1400 are asterosaponins. A strategy for identification of unknown polyhydroxysteroid glycosides was as follows. First, we check the molecular formulae and fragment peaks at m/z 96.96 [HSO4]− that clearly indicate the presence of sulfate group. Next, we analyze product ion peak arising by cleavage of glycoside bond. The presence of B0 ions with m/z 241.00 [C6H9O8S]−, 225.01 [C6H9O7S]−, and 210.99 [C5H7O7S]− associated with sulfated monosaccharide unit whereas the presence of intense Y-type ions associated with non-sulfated compounds or sulfated aglycone. The elemental composition of aglycone provided information on double bonds and amount of hydroxyl group. Localization of hydroxyl group was based on fragmentation, literature data, and biosynthetic considerations. Next, characteristic fragment peaks are helpful for localization of sugar moiety and establishment of structure details. The presence of fragment ions ‘E, D2, C3, and B0 indicates that the compound has a sulfated monosaccharide in the side chain while fragment ions b1, d1, d2, and e–15 associated with the sulfate group at C-3 of steroid aglycones.
A strategy for identification of unknown asterosaponins was as follows. We first analyze intense Y-type product series associated with the sequential losses of monosaccharide units in the negative product ion spectra. The Y-type product ions provided information on amount of units, branchings, and sequence of monosaccharides in the oligosaccharide chain. Next, the product ion Y0 gives information about the elemental composition of aglycone, amount of hydroxyl groups, and double bonds besides 9(11)-double bond. The presence of characteristic mass difference between precursor ion and first intense product ion and the presence product ion series [Yn–X] are helpful for establishment of structure of side chain of aglycone. Finally, after comparison with known compounds and compounds in the database, most possible structures are proposed based obtain data.
As a result of our studies, 207 components were detected and structurally characterized, including 106 asterosaponins, 6 native aglycones of asterosaponins, 81 glycosides of polyhydroxysteroids, and 14 polyhydroxylated steroids (Figure 5; Table 1; Supplementary Tables S2 and S3).

Structures of glycosides of polyhydroxysteroids and polyhydroxysteroids from the starfish L. fusca characterized by nLC/CSI-QTOF-MS/MS method
Characterization of Polyhydroxysteroids and Glycosides of Polyhydroxysteroids from L. fusca
It has been reported previously that hydroxylations of polyhydroxysteroid compounds of starfish during their biosynthesis from dietary sterols or sterol sulfates [33] are generally ordered as follows: at position 6, then at positions 24 or 26 (for ergostane and stigmastane derivatives at positions 28 and 29, respectively), then at positions 15 or 8, and then at position 16 and other positions [6, 7, 34]. Structures of polyhydroxysteroid compounds earlier isolated from L. fusca confirm this hydroxylation order [9].
The analyzed metabolite profile revealed at least 95 polyhydroxysteroid compounds and related glycosides. The detected compounds were characterized by tandem MS and structures were proposed for 91 compounds (Table 1, Figure 5). MS/MS data and fragmentation details are given in Supplementary Tables S3 and S4.
In all, 14 non-sulfated polyhydroxysteroid compounds, including 5 polyhydroxysteroids and 9 glycosides of polyhydroxysteroids, were detected by nLC/CSI-QTOF-MS/MS. From non-sulfated polyhydroxysteroids, 5α-cholestane-3β,4β,6α,7α,8,15α,16β,26-octaol (4), 5α-cholestane-3β,6α,7α,8,15α,16β,26-heptaol (11), 5α-cholestane-3β,4β,6α,7α,8,15β,16β,26-octaol (15), 5α-cholestane-3β,6α,7α,8,15β,16β,26-heptaol (19), and 5α-cholestane-3β,6α,8,15β,16β,26-hexaol (24) were unambiguously identified using authentic standards earlier isolated from this and other starfish species [9, 13]. Non-sulfated glycosides of polyhydroxysteroids fuscaside B (5), distolasteroside D1 (10), distolasteroside D2 (14), pycnopodioside A (18), and desulfated minutoside A (21) [9] were identified by co-chromatographic and MS analysis with the authentic standards. The diagnostic ions of glycosides of polyhydroxysteroids were hardly identifiable due to their structural diversity. Their fragmentation mainly involves cleavage of glycoside bonds and subsequent dehydroxylation of the aglycone. Fragmentation of the steroid nucleus is most often weak and nonspecific. For example, the spectrum of fuscaside B (5) contains a weak product ion B2 at m/z 291.23 [C19H31O2]−, arising from the loss of xylose and cleavage of the C-6–C-7 and C-9–C-10 bonds of aglycone. Biosides distolasteroside D1 (10) and distolasteroside D2 (14) (Δ22-derivate of 10) produced ion B3; at the same time, the spectra of related monosides pycnopodioside A (18) and desulfated minutoside A (21) contain ion B2.
The MS spectrum of glycoside 1 displayed a [M–H]− ion at m/z 599.38 and the MS/MS spectrum showed the loss of the pentose unit (peak at m/z 467.34 [C27H47O6]−). The characteristic product ion d2 at m/z 293.18 [C17H25O4]− and intense ions [M–H–Pent–n×H2O–2H]− led to the glycoside identification as 24-O-pentosyl-5α-cholestane-3β,6,8,15α,16β,24-hexaol (configurations of C-3 and C-16 are β since these configurations are typical for polyhydroxysteroids and related glycosides [1]). Glycoside 2 has a hexose residue and aglycone with seven hydroxy groups. Its MS/MS spectrum showed D2 and d2–H2O product ions at m/z 333.19 and 293.18, respectively. Presence of d2–H2O ion instead of d2 is probably associated with vicinal hydroxy groups at C-6 and C-7. Thus, compound 2 was assigned as 24-O-hexosyl-5α-cholestane-3β,6,7,8,15α,16β,24-heptaol. Compound 7 produced the only Y0 ion at m/z 481.35 [C28H49O6]− and no further structural information and so was tentatively characterized as 28-O-pentosyl-5α-ergostane-3β,6,8,15,16β,28-hexaol. Glycoside 12 displayed a [M–H]− ion peak at m/z 613.40 and its analysis by tandem MS yielded the aglycone ion at m/z 451.34 [C27H47O5]− and B2 ion, similarly to the spectrum of pycnopodioside А (18). Thus, compound 12 was tentatively identified as 24-O-hexosyl-5α-cholestane-3β,6,8,15β,24-pentaol.
Glycoside 107 was identified as pycnopodioside C with 5α-cholestane-3β,6α,8,15β,24-pentaol aglycone and sulfated glucose in the side chain at C-24 [14] by comparing its retention time, elemental composition, and MS/MS spectrum with those of authentic standard. The MS/MS spectrum produced the most prominent ion B0 at m/z 241.00 [C6H9O8S]− and weak characteristic product ions ‘E at m/z 369.12 [C14H25O9S]−, D2 at m/z 397.15 [C16H29O9S]−, and C3 at m/z 467.19 [C20H35O10S]−. Characteristic product ions at m/z 241.00, 369.12, 397.15, and 467.19 were observed in MS/MS of a [M–Na]− ion with m/z 825.39 of compound 46. Also, this spectrum showed Y0 ion at m/z 693.35 associated with the loss of pentose. These data along with shortened retention time indicated the presence of the pentose unit at C-3. Therefore, 46 was tentatively identified as 3-O-pentosyl-24-O-sulfohexosyl-5α-cholestane-3β,6,8,15,24-pentaol.
The MS spectra of compounds 155 and 169 showed [M–Na]− ions with m/z 663.34 and their MS/MS spectra contained B0 ion peak at m/z 210.99 [C5H7O7S]−, which are characteristic of the sulfated pentose unit, and low abundant ions ‘E at m/z 339.11 and D2 at m/z 367.14. Glycoside 155 was identified as 24-O-sulfopentosyl-5α-cholestane-3β,6,8,15,24-pentaol and 169 probably was its isomer at C-15 according to different retention behaviors. According to the obtained data, compounds 92 and 100 ([M–Na]− ions at m/z 795.38) have similar structures as 155 and 169 with the additional pentose unit at C-3, which is confirmed by shortened retention times and Y0 product ions (m/z 663.34) in the MS/MS spectra. Glycosides 172 and 202 displayed [M–Na]− ions at m/z 677.36 and B0 at m/z 225.01 [C6H9O7S]− which are characteristic of the methylated sulfated pentose unit. Compound 172 was assigned as 24-O-methylsulfopentosyl-5α-cholestane-3β,6,8,15,24-pentaol and 202 as its isomer at C-15. According to the MS data, glycosides 108 and 145 most probably are analogues of 172 and 202 with pentose units at C-3. Glycoside 53 has a [M–Na]− ion at m/z 709.35 and produced ion B0 at m/z 241.00 and ‘E ion at m/z 369.13 exhibiting a fragmentation pattern of pycnopodioside C (107). However, the characteristic product ion D2 at m/z 413.15 differed by 16 Da from those in spectra 107. Thus, 53 was characterized as 16-hydroxy derivative of pycnopodioside C, 24-O-sulfohexosyl-5α-cholestane-3β,6,8,15,16β,24-hexaol. It must be noted that patterns of fragmentation of sulfated hexoses of 46 and 53 were similar to pycnopodioside C (107) indicating the presence of 6’-O-sulfated hexoses in 46 and 53.
The MS spectrum of compound 124 exhibited a [M–Na]− ion peak at m/z 679.34. The MS/MS spectrum of this ion showed a fragment peak at m/z 210.99 corresponding to sulfated pentose. Fragment peaks at m/z 339.11 and 383.14 were also observed in the spectrum indicating the presence of additional hydroxy group at C-16 compared with 172. Thus, compound 124 was assigned as 24-O-sulfopentosyl-5α-cholestane-3β,6,8,15,16β,24-hexaol. Polyhydroxysteroid 136 ([M–Na]− ion at m/z 531.30) was shown in the MS/MS spectrum product ions ‘E at m/z 207.07 and D2 at m/z 235.10 exhibiting the fragmentation pattern characteristic of 5α-cholestane-3β,6,8,15,24-pentaol 24-O-sulfate. Despite the fact that polyhydroxysteroid compounds usually have the sulfate group at C-26, 136 most probably has 24-O-sulfate because it is known that 26-O-sulfate derivatives appear at shorter retention times on RP-LC [6].
The product ion spectra of glycosides 57, 83, 120, and 126 contained peaks of ions at m/z 327.08 (‘H) [C11H19O9S]− and 341.09 (‘G) [C12H21O9S]− arising from cleavage of the C-23–C-24 and C-22–C-23 bonds, respectively, besides ‘E and D2 ions. This fragmentation pattern most probably corresponds to the ergostane side chain with sulfated hexose at C-28. Glycoside 57 showed [M–Na]− ion at m/z 839.41 and produced prominent product ions at m/z 707.37 (Y0), 689.36 (Z0), and 241.00 (B0) by the loss of pentose and sulfated hexose units. Weak product ions with m/z 481.21 (C3), 465.36 [C28H49O5]−, 411.17 (D2), 383.14 (‘E), 341.09 (‘G), and 327.08 (‘H) indicated the presence of sulfated hexose in the side chain and ergostane aglycone with five hydroxy groups. Therefore, 57 was assigned as 3-O-pentosyl-28-O-sulfohexose-5α-ergostane-3β,6,8,15,28-pentaol. The spectra of compounds 83 and 120 contained peaks of [M–Na]− ions at m/z 723.36 and produced same B0, ‘E, ‘G, and ‘H ions as in the spectrum of 57. However, D2 ions at m/z 427.17 were 16 Da heavier than those of glycoside 57 indicating the presence of the hydroxy group at C-16. Therefore, 83 and 120 were characterized as isomers of 28-O-sulfohexosyl-5α-ergostane-3β,6,8,15,16β,28-hexaol. Compound 126 showed in the MS/MS spectrum the same fragmentation as 57, except Y0 and Z0 ions, and was characterized as 28-O-sulfohexosyl-5α-ergostane-3β,6,8,15,28-pentaol. The spectra of glycosides 57, 83, 120, and 126 exhibited the same patterns of fragmentation of sulfated hexoses that were similar to fragmentation of 6’-O-sulfated glucose in pycnopodioside C (107). All spectra had fragment peaks at m/z 198.99 [C4H7O7S]−, 180.98 [C4H5O6S]−, 164.99 [C4H5O5S]−, and 138.97 [C2H3O5S]− indicating the presence of 6’-O-sulfated hexoses.
Glycosides 162, 163, 181, 185, 197, and 204 showed fragmentation patterns similar to compounds 57, 83, 120, and 126, but product ions ‘G and ‘H were 14 Da heavier, corresponding to stigmastene side chains of their aglycones. In general, the fragmentations of compounds 162, 163, 181, 185, 197, and 204 were very similar to the fragmentation pattern of the aphelasteroside D which has 5α-stigmastane-3β,5,6β,8,15α,29-hexaol aglycone with 2’-O-sulfo-β-D-glucopyranose at C-29 [14]. However, the retention time of the authentic standard of aphelasteroside D did not match with any of the detected compounds in L. fusca. MS spectra of glycosides 163, 181, and 197 displayed [M–Na]− ions with m/z 737.38. Their MS/MS spectra all had B0 at m/z 241.00 [C6H9O8S]− which are characteristic of sulfated hexose unit and very weak peaks of product ions at m/z 441.18 (D2) [C18H33O10S]−, 397.15 (‘E) [C16H29O9S]−, 355.11 (‘G) [C13H23O9S]−, 341.09 (‘H) [C12H21O9S]− and aglycone ion at m/z 495.37 [C29H51O6]−. According to these data, 163, 181, and 197 were tentatively assigned as isomers of 29-O-sulfohexosyl-5α-stigmastane-3β,6,8,15,16β,29-hexaol. The product ion spectrum of compound 204 displayed the same ion series, but all peaks were 30 Da (CH2O) lighter than those of 163, 181, and 197 indicating sulfated pentose instead of sulfated hexose unit. Thus, 204 was assigned as 29-O-sulfopentosyl-5α-stigmastane-3β,6,8,15,16β,29-hexaol. Glycoside 162 also showed a similar fragmentation pattern, but its [M–Na]− ion at m/z 753.37 was 16 Da heavier than in 163. It is feasible that 162 is 29-O-sulfohexosyl-5α-stigmastane-3β,6,7,8,15,16β,29-heptaol with an additional hydroxy group at C-7. Contrariwise, glycoside 185 contains one oxygen less and is suggested to be 16-dehydroxy derivative of 163, as indicated by the product ion D2 at m/z 425.18, which is of 16 Da lighter than the same ion in 163. About the structure of the monosaccharide residue, we can note that the product ion spectra of glycosides 162, 163, 181, 185, and 197 contained peaks of fragment ions at m/z 166.97 [C3H3O6S]− indicating the presence of 2’-O- or 3’-O-sulfated hexose units in these glycosides [29].
Glycosides 55 and 59 both exhibited [M–Na]− ions at m/z 663.34. MS/MS spectra of 55 and 59 showed Y0 and Z0 ions at m/z 531.30 and 513.29 indicating the loss of the pentose unit. These spectra also contained weak fragment peaks at m/z 207.07 (‘E) [C8H15O4S]−, 235.10 (D2) [C10H19O4S]−, 305.14 (C3) [C14H25O5S]−, and 375.19 (B3) [C18H31O6S]− arising from cleavage of the C-7–C-8 and C-9–C-10 bonds, and 459.24 (A4) [C23H39O7S]− arising from cleavage of the C-4–C-5 and C-1–C-10 bonds. The corresponding product ions as well as the shortened retention time indicated the presence of the sulfate group at C-26 and 5α-cholestane-3β,6,8,15,26-pentaol aglycone. Therefore, 55 and 59 were tentatively identified as isomers of 3-O-pentosyl-5α-cholestane-3β,6,8,15,26-pentaol 26-O-sulfate. Glycoside 75 has [M–Na]− ion peak at m/z 677.36 in its spectrum and the similar product ion series, but all the peaks were 14 Da (CH2) heavier. According to these data and the retention behavior, 75 was identified as 3-O-pentosyl-5α-ergostane-3β,6,8,15,26-pentaol 26-O-sulfate. The elemental composition of 23 (C32H55O14SNa), according to its accurate mass of 695.3320, indicated two additional oxygen atoms compared with 55. The difference between ions ‘E at m/z 207.07 and D2 at m/z 251.10 and the difference between ions B3 at m/z 391.18 and A4 at m/z 491.23 showed additional 7- and 16-hydroxy groups. Thus, 23 was preliminary identified as 3-O-pentosyl-5α-cholestane-3β,6,7,8,15,16β,26-heptaol 26-O-sulfate.
Glycosides 45, 48, 68, 81, 96, and 109 have the sulfate group at C-3 of steroid aglycones as follows from the MS analysis. The MS/MS spectra contained ring fragment ions b1 at m/z 191.04 [C7H11O4S]−; d1 ions at m/z 345.14 [C16H25O6S]−; d2 ions at m/z 375.15 [C17H27O7S]−; and side-chain cleavage e–15 ions formed by concerted cycloelimination reactions that involve six-membered ring transition states followed by elimination of 18β-methyl [26] and ‘f ions. According to literature data, this fragmentation pattern is a classical fragmentation pattern for a saturated steroid with 3-O-sulfate [26]. The difference between d2 and e–15 ions determines the presence of the 16-hydroxy group. Glycoside 45 yielded product ion e–15 at m/z 403.14 [C18H27O8S]−, while in the spectrum of 48, a product ion e–15 at m/z 387.15 [C18H27O7S]− was observed. Both compounds gave Y0 and Z0 ions indicating the presence of the hexose unit and weak unusual Y0–C3H8 ions probably associated with C-24–C-25 bond cleavage. On this basis, 45 and 48 were assigned as 24-O-hexosyl-5α-cholestane-3β,6,8,15,16β,24-hexaol 3-O-sulfate and 24-O-hexosyl-5α-cholestane-3β,6,8,15,24-pentaol 3-O-sulfate, respectively. Glycosides 68 and 81 showed similar fragmentation under CID condition, except there were no Y0 and Z0 ions corresponding to the loss of pentose moiety. Compounds 68 and 81 probably are analogues of 45 and 48, respectively, with the pentose unit instead of hexose. Glycosides 96 and 109 have an additional double bond compared with 68 and 81, as indicated by their molecular formula (C32H53O13SNa) and retention behavior. The product ion spectra of 96 and 109 showed the fragmentation pattern analogous of 68 and 81 and according to these data, a double bond is localized in the side chain, probably at 22(23). Thus, glycosides 96 and 109 were characterized as Δ22-derivatives of 68 and 81, respectively. Collisional activation of compound 141 produced characteristic ring and side-chain fragmentations but did not produce Y0 ion and so was assigned as 5α-cholestane-3β,6,8,15,16β,24-hexaol 3-O-sulfate. The product ion spectrum of glycoside 64 revealed Y0 and Z0 ions corresponding to the loss of pentose unit and showed characteristic fragmentation of 3-O-sulfated compounds, but all the diagnostic ions were 132 Da (C5H8O4) heavier. In addition, this spectrum contained a product ion B0 at m/z 210.99 [C5H7O7S]−, which is characteristic of the sulfated pentose unit. Based on these data, 64 was tentatively identified as 3-O-sulfopentosyl-24-O-pentosyl-5α-cholestane-3β,6,8,15,24-pentaol. The spectrum of glycoside 84 showed the fragmentation patterns similar to those of 96 and 109, but 84 has ergostene aglycone and probably was 28-O-pentosyl-5α-ergost-22-ene-3β,6,8,15,16β,2-hexaol 3-O-sulfate.
The series of polyhydroxysteroids 63, 101, 117, 133, 146, and 153 have sulfate groups identified by molecular formulae and fragment peaks at m/z 96.96 [HSO4]−. However, their MS/MS spectra did not contain fragment peaks characteristic of 3-O-sulfated steroids or steroids with sulfate in the side chain. According to literature data, in most cases, the sulfate group in polyhydroxysteroids is located at C-6, but in glycosides of polyhydroxysteroids at C-15. The product ion spectra of these polyhydroxysteroids exhibit extensive fragmentation, including the loss of the sulfate group and sequential neutral losses of H2O molecules and cleavages in the tetracyclic nucleus. The relative intensity of peaks of fragment ions belonging to the series [M–H–n×H2O]− and [M–H–n×H2O–2H]− enabled us to distinguish between stereoisomers with the different orientations of the hydroxy group at C-15 [28]. According to the obtained data, we assume that all the compounds are 6-O-sulfated derivatives and 63 was assigned as 5α-cholestane-3β,4,6,7,8,15α,16β,26-octaol 6-O-sulfate, 101 as 5α-cholestane-3β,6,7,8,15α,16β,26-heptaol 6-O-sulfate, 117 and 133 as 15α- and 15β-isomers of 5α-cholestane-3β,6,8,15,16β,26-hexaol 6-O-sulfate, respectively, 146 as 5α-ergost-22-ene-3β,6,7,8,15α,16β,26-heptaol 6-O-sulfate, and 153 as 5α-ergost-22-ene-3β,6,8,15,16β,26-hexaol 6-O-sulfate.
Compound 139 was identified as coscinasteroside B [21] by comparing its product ion spectrum and retention time with authentic standard. Compound 139 was the only glycoside that has 15-O-sulfate group. Earlier isolated from L. fusca fuscaside A was not found in the analyzed sample, although fuscaside B (5), the desulfated derivative of fuscaside A, was identified.
All other detected glycosides of polyhydroxysteroids have a double bond as indicated by increased retention times compared with saturated analogue, molecular formulae, and ring and double bond equivalents. It is known that glycosides of polyhydroxysteroids usually have the double bond in the side chain at C-22. Analysis of the MS/MS data of asteriidoside H, containing cholest-22-ene side chain in aglycone moiety, showed the characteristic fragmentation. Fragment ions “G, ‘F-2H, ‘E, D1, and D2 are characteristic of glycosides with Δ22 and sulfated monosaccharide moiety at C-24. Similar fragmentation pattern was observed in spectra of glycosides 76, 85, 148, 154, 165, 171, 183, 189, and 196. Glycosides 76 and 85 displayed a [M–Na]− ions at m/z 837.39 and their MS/MS spectra showed the loss of the pentose unit, B0 at m/z 241.00 [C6H9O8S]−, which is characteristic of the sulfated hexose unit, prominent ion “G at m/z 339.08 and weak fragment peaks at m/z 381.12 (‘E), 395.14 (D1), and 409.15 (D2). In addition, the MS analysis showed the presence of isomeric monosaccharide residues—76 has 2’-O- or 3’-O-sulfated hexose and 85 has 4’-O- or 6’-O-sulfated hexose. Therefore, 76 and 85 were assigned as isomers of 3-O-pentosyl-28-O-sulfohexosyl-5α-ergost-22-ene-3β,6,8,15,28-pentaol. Compounds 154 and 171 showed similar fragmentations except for Y0 ions and probably are analogues of 76 and 85, having no pentose units at C-3. MS/MS of both 148 and 165 revealed D2 ions at m/z 423.16 that is 16 Da heavier than those ions of 154 and 171 indicating the presence of 16-hydroxy group. According to that, 148 and 165 were characterized as isomers of 28-O-sulfohexosyl-5α-ergost-22-ene-3β,6,8,15,16β,28-hexaol. Both glycosides 183 and 189 exhibited fragmentation pattern of 148, but all characteristic ring and side-chain fragmentation ions were 14 Da heavier, corresponding to stigmastene side chains of their aglycones. Also, the spectra of 183 and 189 contained fragment peaks at m/z 166.97 [C3H3O6S]− indicating the presence 2’-O- or 3’-O-sulfated hexoses [29]. Accordingly, 183 and 189 were tentative characterized as isomers of 29-O-sulfohexosyl-5α-stigmast-22-ene-3β,6,8,15,16β,29-hexaol. The MS/MS spectrum of 196 was very similar to the spectrum of asteriidoside H, except for the absence of Y0 ions. It is probable that 196 is 24-O-sulfopentosyl-5α-cholest-22-ene-3β,6,8,15,24-pentaol.
Fragmentation of series of glycosides 72, 82, 99, 102, 103, 114, 147, 151, 167, 184, and 188 also led to G-, F-, E-, and D2-types of ions, but G and F ions were 2 Da heavier than those in the spectra of Δ22 derivatives. Analysis of fragmentation indicated that in the case of double bond cleavage, the formed ion was deficient in two hydrogens as compared to a fragment ion formed by homolytic fragmentation at the same bond. Therefore, it can be assumed that these glycosides are Δ20(22)-derivatives, although this location of double bond is not typical of glycosides of polyhydroxysteroids. Glycosides 72 and 99 displayed [M–Na]− ions at m/z 721.35. Their product ion spectra gave B0 ions at m/z 241.00 [C5H7O7S]−, which are characteristic of the sulfated hexose unit; fragment ions at m/z 341.09 [C12H21O9S]−, arising from C-22–C-23 bond cleavage (ion ‘G) and 353.09 [C13H21O9S]− arising from C-20–C-22 double bond cleavage (ion ‘F), 381.12 [C15H25O9S]− correspond to the loss of the side chain (ion ‘E) and 425.15 [C17H29O10S]− formed by D-ring bond cleavage (D2). The MS analysis showed also the presence of epimeric monosaccharide residues—72 has 4’-O- or 6’-O-sulfated hexose and 99 has 2’-O- or 3’-O-sulfated hexose. According to these data, 72 and 99 were tentatively assigned as isomers of 28-O-sulfohexosyl-5α-ergost-20(22)-ene-3β,6,8,15,16β,28-hexaol. Glycoside 103 was characterized as 16-dehydroxy derivative of 72. Glycosides 102, 147, and 167 exhibited fragmentation patterns resembling those of 72 and 99, but all the characteristic ring and side-chain fragmentation ions were 14 Da heavier, corresponding to stigmastene side chains of aglycones. In addition, fragmentation of sulfated hexose units observed in all the spectra indicated the presence of different epimers of sulfated hexoses. Accordingly, 102, 147, and 167 were assigned as isomers of 29-O-sulfohexosyl-5α-stigmast-20(22)-ene-3β,6,8,15,16β,29-hexaol. The MS analysis of 151 showed the presence of the sulfated pentose unit and allowed it to be identified as 28-O-sulfopentosyl-5α-ergost-20(22)-ene-3β,6,8,15,16β,28-hexaol. Compounds 184 and 188 exhibited similar fragmentation and were characterized as isomers of 28-O-sulfopentosyl-5α-ergost-20(22)-ene-3β,6,8,15,28-pentaol. Glycoside 114 produced Y0 and Z0 ions and fragmentation pattern of 184 and so was characterized as 3-O-pentosyl-28-O-sulfopentosyl-5α-ergost-20(22)-ene-3β,6,8,15,28-pentaol. The spectrum of glycoside 82 showed Y0 and Z0 ions corresponding to the loss of pentose units. The side chain of 82 does not have monosaccharide units, but its spectra exhibit fragmentation patterns of Δ20(22)-derivatives; therefore, the structure of 82 probably is 3-O-pentosyl-5α-ergost-20(22)-ene-3β,6,8,15,28-pentaol 28-O-sulfate. The spectrum of glycoside 115 showed the fragmentation pattern characteristic of Δ20(22)-derivatives, but it has the cholestene side chain and thus might be characterized as 24-O-sulfohexosyl-5α-cholest-20(22)-ene-3β,6,8,15,24-pentaol.
Collisionally induced fragmentation of 69, 71, 91, 94, 113, 127, 134, 164, and 179 was similar to those of a series of Δ22 glycosides. However, additional ion peaks ‘I and ‘H were observed, indicating other positions of sulfated monosaccharide units. The spectra of 94, 113, 127, 164, and 179 contain peaks of unusual fragment ions ‘I and ‘H at m/z 299.04 [C9H15O9S]− and 327.08 [C11H19O9S]− corresponding to the loss of sulfated hexose with fragments C3H6 and C5H10, respectively. These fragments are probably related to the presence of an ergostene side chain with 26-O-sulfohexose unit. The spectra also contain “G ions at m/z 339 indicating Δ22 double bond and an unusual prominent ion E–15 at m/z 367.11 that has never been observed anywhere before. Other fragments were typical of glycosides with the sulfated monosaccharide unit in the side chain and corresponded to the loss of the side chain (‘E) and D-ring bond cleavage (D1 and D2). Therefore, glycosides 113 and 127 were suggested to be isomers of 26-O-sulfohexosyl-5α-ergost-22-ene-3β,6,8,15,16β,26-hexaol, 164 and 179 isomers of 26-O-sulfohexosyl-5α-ergost-22-ene-3β,6,8,15,26-pentaol, and 94 3-O-pentosyl-26-O-sulfohexose-5α-ergost-22-ene-3β,6,8,15,26-pentaol. The spectra of 134 showed peaks of Y0 and Z0 ions corresponding to the loss of pentose units and fragmentation pattern that corresponded to 3-O-pentosyl-5α-ergost-22-ene-3β,6,8,15,26-pentaol 26-O-sulfate. The spectra of 69, 71, and 91 contained fragment ion ‘I at m/z 285.03 [C8H13O9S]−, corresponding to the loss of sulfated hexose with fragment C3H6, intensive fragment ion E–15 at m/z 353.09 [C13H21O9S]−, and ‘E ion at m/z 367.11 [C14H23O9S]−. These data indicate that glycosides 69, 71, and 91 have 27-nor-24-methyl-cholestane aglycones with sulfated hexose at C-26. Thus, 71 and 91 were characterized as isomers of 26-O-sulfohexosyl-27-nor-5α-ergost-22-ene-3β,6,8,15,16β,26-hexaol and 69 as 3-O-pentosyl-26-O-sulfohexosyl-27-nor-5α-ergost-22-ene-3β,6,8,15,26-pentaol. Glycoside 56 shows fragment ions ‘I at m/z 299.04 [C9H15O9S]−, ‘E ion at m/z 367.11 [C14H23O9S]−, and D2 ion at m/z 409.12 [C16H25O10S]−. According to these data, 56 was assigned as 26-O-sulfohexosyl-5α-cholest-22-ene-3β,6,8,15,16β,26-hexaol. It is necessary to point out that all glycosides of this type (except 134) have sulfated hexoses. Compounds 56, 69, 71, 94, 113, and 164 showed fragmentation of sulfated monosaccharide characteristic of 4’-O- or 6’-O-sulfated hexose, and 91, 127, and 179 showed fragmentation characteristic of 2’-O- or 3’-O-sulfated hexose.
Glycosides 131 and 138 also produced fragment ions ‘I at m/z 285.03 [C8H13O9S]− and ‘H at m/z 313.06 [C10H17O9S]−, corresponding to 27-nor-24-methyl-cholestane aglycones with sulfated hexose at C-26, but ions ‘G and ‘F were characteristic of Δ20(22)-derivatives. The MS analysis also showed that 131 has 4’-O- or 6’-O-sulfated hexose and 138 has 2’-O- or 3’-O-sulfated hexose. Thus, compounds 131 and 138 were assigned as isomers of 26-O-sulfohexosyl-27-nor-5α-ergost-20(22)-ene-3β,6,8,15,26-pentaol.
Compound 119 was the only glycoside which had a disaccharide fragment as indicated by the fragment peaks at m/z 419.05 [C12H19O14S]−, 403.06 [C12H19O13S]−, 387.06 [C12H19O12S]−, 361.05 [C10H17O12S]−, 339.08 [C12H19O9S]−, and 315.04 [C9H15O10S]−. Product ion “G with m/z 501.13 [C18H29O14S]− indicates the presence of Δ22 double bond. Thus, 119 was suggested to be 28-O-[sulfohexosyl-hexose]-5α-ergost-22-ene-3β,6,8,15,28-pentaol.
The structures of glycosides 58, 125, 142, and polyhydroxysteroid 30 were not assigned due to poor quality of the mass spectra.
Characterization of Asterosaponins from L. fusca
The analyzed L. fusca sample showed the presence at least 112 asterosaponins and native aglycones of asterosaponins, including 28 hexaosides, 66 pentaosides, 7 triosides, and 5 “shortened” asterosaponins with the one-monosaccharide units at C-6 as well as 6 native aglycones of asterosaponins. The detected asterosaponins were characterized by tandem MS (Table 1, Supplementary Table S2). Among detected asterosaponins, lethasterioside A (198), lethasterioside B (159), thornasteroside A (118), anasteroside A (166), and luidiaquinoside (122) were unambiguously identified using authentic standards earlier isolated from L. fusca [15].
Epimeric monosaccharides as well as types of glycosidic bonds cannot be exactly established only by the MS technique, but feasible structural assignments can be made for the detected compounds. The carbohydrate chains of majority of known asterosaponins have similar structures. The second monosaccharide (quinovose, xylose, or glucose) is attached by β-1,3 glycosidic bond to the first monosaccharide unit (most often quinovose). The branching unit (always quinovose) is connected to the second monosaccharide by β-1,2-bond, while third and fourth sugars in the main chain are connected by β-1,4 and β-1,2 glycosidic bonds. Thus, based on obtained data and common structural patterns, tentatively structures of oligosaccharide chains of detected asterosaponins may be proposed (Table 1).
The analyzed sample contained 28 hexaosides and an absolute majority of them have terminal deoxyhexose and hexose units. Oligosaccharide chain of hexaosides can have two branches at the second and third monosaccharide units or one branching at the second monosaccharide. Structure of oligosaccharide chain and number of branches can be determined by complimentary use of positive and negative product ion spectra as shown earlier [7]. In the case of hexaosides from L. fusca, the spectra do not contain the characteristic fragment ions [M–Na–2×dHex]− indicating that all the hexaosides from L. fusca have a main oligosaccharide chain with five monosaccharides with only one branching.
Majority of the detected hexaosides from L. fusca have oligosaccharide chains that closely relate to each other. All the hexaosides bear deoxyhexose as the fourth sugar and as a branching monosaccharide unit, and the most of them contain hexose as the fifth, pentose (xylose) as the second, and deoxyhexose (quinovose) as the first sugars. Nineteen hexaosides contain Hex–dHex–(3rd unit)–Xyl (-Qui)–Qui–carbohydrate chains, 11 of them have the same Hex–dHex–Pent–Xyl (-Qui)–Qui–carbohydrate chains and various aglycones.
From 66 detected pentaosides, 38 compounds have deoxyhexose as the second monosaccharide and 42 compounds contain deoxyhexose as the third monosaccharide whereas the most of known asterosaponins have a pentose as the second and hexose as the third units. Frequently, the first sugar was deoxyhexose (quinovose) or hexose, but some asterosaponins with rare and untypical monosaccharide units were also found.
Ten asterosaponins (32, 33, 35, 74, 77, 135, 190, 191, 195, and 203) have hydrated 6-deoxy-xylo-4-hexulose (DXU) as first sugar unit that confirmed by prominent product ion series [M–Na–H2O]− (M = molecular ions and Y-type ions (except Y0)), along with neutral losses of monosaccharides and fragment C6H10O5 in the negative product ion spectra [35]. Fragmentations of oligosaccharide chains of compounds 180 and 187 display neutral losses of four monosaccharide units and fragment C6H6O3. Previously, we proposed that it could be unsaturated sugar with 4’-keto group and 2’(3’)-double bond due to loss of H2O [7]. The MS/MS spectra of compounds 61, 89, 156, 186, and 199 revealed an unusual product ion series [M–Na–HCN]− (M = molecular ions and Y-type ions, except Y0). Sequencing oligosaccharide chains displayed neutral losses of four monosaccharides and fragment C7H9NO4. Several similar asterosaponins containing DXU unit and unit C6H6O3 were found in the A. japonica and P. pectinifera earlier whereas unit C7H9NO4 was found in asterosaponins from A. japonica [6, 7].
Sequencing oligosaccharide chains of asterosaponin 8 displayed neutral losses of four deoxyhexoses and fragment C6H9NO3. CID spectra of 8 also provided a product ion with m/z 439.18 [C21H30O6S (aglycone) + CHO]−; 452.21 [aglycone + C2H4N]−; 466.18 [aglycone + C2H2NO]−; 480.20 [aglycone + C3H4NO]−; 536.23 [aglycone + C6H8NO2]−; and 554.25 [aglycone + C6H10NO3]−. Such fragmentation patterns could be associated with unsaturated sugar with 4’-keto and 2’-NH2 groups. As far as we know, the monosaccharide of this type was not being identified from marine sources so far.
The majority of detected asterosaponins contain deoxyhexose (quinovose) as the first sugar. We believe that glycosides without quinovose at the first position are the products of further biological oxidation of the first quinovose in their carbohydrate chains. It can be proposed that these unusual monosaccharide units are formed from quinovose. We have previously suggested that the hydrated form of 6-deoxy-xylo-4-hexulose (DXU) is formed from quinovose as a result of oxidation to 4’-keto derivative followed by hydration of carbonyl group at C-4’. It can be assumed that the elimination of the water molecule during biosynthesis and the substitution of 2’-OH to 2’-NH2 lead to the formation of the first monosaccharide unit of 8, and the elimination of two water molecules from DXU leads to the formation of the first monosaccharide units of 180 and 187.
According to MS fragmentations, compounds 6, 9, 26, 42, and 54 are “shortened” asterosaponins with the one-monosaccharide units, while asterosaponins 28, 31, 38, 39, 51, 86, and 161 are triosides. Asterosaponins with trisaccharide carbohydrate chains were rarely found in starfish. Steroid triosides, previously isolated from other starfish species, have carbohydrate chains consisted of ordinary monosaccharide residues, with the terminal monosaccharide linked by the (1 → 2) glycosidic bond. Thus, steroid triosides as well as “shortened” asterosaponins can be the biosynthetic precursor of asterosaponins with longer carbohydrate chains. This assumption is confirmed by the identification of asterosaponins found in L. fusca which has the same aglycone part and different oligosaccharide chains formed by the consequent attachments of monosaccharide units from “shortened” asterosaponins through triosides to pentaosides and hexaosides.
Fragmentation patterns of asterosaponins under CID conditions were used for the characterization of aglycones. Proposed structures of aglycones and their characteristic fragmentations are given in Table 2. According to the proposed structures of aglycones, all the detected asterosaponins of L. fusca can be divided into 23 groups (I–XXIII) according to the types of aglycones (AG I–AG XXIII).
Groups I (3, 6, 9, 13, 20, 26, 28, 31, 37, 39, 42, 47, 51, and 86) and II (17, 22, 27, 29, 33, 35, 36, 38, 43, 50, 52, 54, 61, and 89) include asterosaponins with short-chain aglycones (AG I and AG II) containing CH3CHOH or CH3CO substituents as side chains. The (–)MS/MS spectra of these compounds displayed peaks of Y0 ions at m/z 413.20 and 411.18 Da, which are specific of asterosaponins with 3-O-sulfoasterogenol (3β,6α,20-trihydroxy-5α-pregn-9(11)-ene) and 3-O-sulfoasterone (3β,6α-dihydroxy-5α-pregn-9(11)-en-20-one) aglycones, respectively [1]. It should be noted that compounds 42, 51, 86, and 89 differ in their retention times from other asterosaponins of the corresponding groups, which can be associated with a different structure of aglycones. It is possible that the aglycones of compounds 42, 51, and 86 are stereoisomers at С-17 of 3-O-sulfoasterogenol, and aglycone of 89 is a stereoisomer at С-17 of 3-O-sulfoasterone. Previously, 3-O-sulfoisoasterone (17α-isomer of 3-O-sulfoasterone) had been isolated from Lethasterias nanimensis chelifera [36]. Compounds 34 and 73 were identified as native aglycones AG I and AG II, respectively.
Asterosaponin 8, in addition to the unique oligosaccharide chain, has a new aglycone (AG III). Fragmentation of 8 under CID conditions resulted in characteristic product ion [M–118] and product ion series [Yn–118] corresponding to the loss of C6H14O2 molecule. This fragmentation pattern most probably corresponds to aglycone with a hydroxy group at C-20 and two hydroxy groups at the cholestane side chain. Shorter retention time can be associated with a hydroxy group at C-26. Therefore, 8 most likely contains aglycone with 20,23,26-trihydroxy-cholestane side chain. Asterosaponin 16 also has aglycone (AG IV) that gives Y0 product ion at m/z 455.21 Da that has never been isolated from starfish, but this structure cannot be confirmed based on the obtained data only.
Compounds 44, 60, 70, 78, 88, 93, 106, 110, 111, 118, 122, 129, 135, 156, 161, 175, 180, 186, 187, and 199 belong to the group V. Negative product ion spectra displayed very intense characteristic product ions [M–100] and product ion series [Yn–100] associated with the loss of C6H12O molecule; Y0, Z0 product ions at m/z 511.27 and 493.26 and Y0–100, Z0–100 product ions at m/z 411.18 and 393.17. These fragmentation patterns are characteristic of aglycone with a 20-hydroxy-cholestan-23-one side chain [1]. Thus, compounds of this group have 3-O-sulfothornasterol A aglycone (AG V). Compound 205 was identified as native aglycone 3-O-sulfothornasterol A (AG V) using authentic standard.
Intense characteristic product ion [M–100] and product ion series [Yn–100] in product ion spectra of groups VI (25 and 32), VII (40, 66, and 77), and VIII (41 and 74) were also detected in spectra of asterosaponins of group V. This fragmentation pattern is specific of asterosaponins having aglycones with a 20-hydroxy-cholestan-23-one side chain, but Y0, Z0 product ions in spectra of compounds of groups VI–VIII were quite different from those of compounds with 3-O-sulfothornasterol A aglycone. Elemental composition of AG VII (C27H43O8SNa) and fragmentation patterns indicate an additional hydroxy group in steroid nucleus compared to 3-O-sulfothornasterol A that can be at С-12, as in aglycone of tenuispinoside C from Coscinasterias tenuispina [37]. Asterosaponins of group VI showed in spectrum Y0 ion at m/z 543.26 indicating the elemental composition of C27H43O9SNa for this aglycone. The additional two hydroxy group can be placed at C-8 and C-12 that was confirmed by the fragment peak at m/z 275.06 [C11H15O6S]− arising from cleavage of the C-8–C-14 and C-9–C-11 bonds in the spectrum of 25. Asterosaponins of group VIII indicated the elemental composition of aglycone AG VIII as C27H41O8SNa. Aglycones of this group most likely contain one additional hydroxy group at C-12 and a double bond compared to 3-O-sulfothornasterol A. Aglycone VIII probably has an additional double bond in the steroid nucleus, rather than a keto group as indicated by the close retention times of the asterosaponins of groups VI–VIII. Similar compounds were not reported before; however, it can be assumed that the double bond may be localized as 14(15). Compounds 174 and 178 were identified as native aglycones AG VIII and AG VII respectively.
Fragmentation of asterosaponins of the group IX (95 and 132) was similar to that of compounds in the group V. The (–)MS/MS spectra of the group IX also revealed a product ion [M–100] and product ion series [Yn–100], Y0, Z0 product ions at m/z 511.27 and 493.26 and Y0–100, Z0–100 product ions at m/z 411.18 and 393.17 Da. However, unlike the group V, the intensity of ion series [M–100] was much lower than that of the series of ions without the loss of the side chain, and intensity of Y0, Z0 product ions was much greater than that of Y0–100, Z0–100 product ions. Thus, asterosaponins of the group IX lose C6H12O fragment under CID conditions as asterosaponins of group V do but much less efficiently. These fragmentation patterns are specific to asterosaponins having aglycones (AG IX) with 20-hydroxy-cholestan-22,23-epoxy side chains [1, 38].
The spectra of asterosaponins belonging to the group X (49, 80, and 87) show Y0 ion at m/z 497.26 [C26H41O7SNa]−. In addition, low-intensity product ion series [Yn–86] corresponding to the loss of C5H10O fragment was detected in (–)MS/MS spectra of 49 and 80. This fragmentation pattern probably indicates that compounds of group X have 22,23-epoxy-24-nor-thornasterol A aglycone similar to aglycone previously found in asterosaponins from Asterias amurensis [39].
The group XI of detected asterosaponins (62 and 79) exhibits Y0 product ions with m/z 513.29 [C27H45O7S]− and low-intensity product ion series [Yn–104] corresponding to the loss of C5H12O2 fragment in the (–)MS/MS spectra. This aglycone type might be suggested to have two hydroxy groups in the side chain. In this case, asterosaponins 62 and 79 most probably have aglycones 23,24-dihydroxy-cholestane side chains.
Asterosaponin 65 (group AG XII) exhibits product ion [M–96] and product ion series [Yn–96] corresponding to the loss of C6H8O fragment as well as Y0 product ion at m/z 507.24 [C27H39O7S]− in the (–)MS/MS spectra. These fragmentation patterns could be associated with aglycone having a hydroxy group at C-20 and two double bounds and ketone in the side chain; the most probable structure of this aglycone is presented in Table 2.
The group XIII of detected asterosaponins (67, 105, 116, 130, 160, 170, and 191) in the (–)MS/MS spectra exhibits intense characteristic product ion [M–114] and product ion series [Yn–114] corresponding to the loss of C7H14O fragment, Y0, Z0 product ions with m/z 525.29 and 507.28, and Y0–114, Z0–114 product ions with m/z 411.18 and 393.17. These product ions are characteristic of asterosaponins having 3-O-sulfothornasterol B aglycone (AG XIII) with 20-hydroxy-24-methyl-cholestan-23-one side chain [1].
Fragmentation of compound 90 (group XIV) under CID conditions reveals low-intensity product ions [M–114] and [Y3–114] corresponding to the loss of C7H14O neutral fragment, Y0 product ions at m/z 557.28 [C28H45O9S]−, and fragment ion 275.06 [C11H15O6S]− formed by cleavage of the C-8–C-14 and C-9–C-11 bonds. Thus, aglycone AG XIV is similar to AG VI, but has additional 24-methyl in the side chain and probably contains 22,23-epoxy group instead 23-keto group, as indicated by the reduced intensity of [M–114] fragment ion.
The (–)MS/MS spectra of asterosaponins of group XV (97 and 98) provide Y0 product ion at m/z 527.30 [C28H47O7S]− and low-intensity product ion series [Yn–118] correspond to the loss of C6H14O2 fragment. This fragmentation pattern is similar to those of group XI, but aglycones of 97 and 98 have an additional 24-methyl in the side chain.
The group XVI (158) produces an intense product ion [M–128] and product ion series [Yn–128], and Y0–128, Z0–128 ions at m/z 411.18 and 393.17. This fragmentation patterns are characteristic of asterosaponins with 3-O-sulfo-ethylthornasterol А aglycone (AG XVI) with 20-hydroxy-24-ethyl-cholestan-23-one side chain [1].
The group XVII of detected asterosaponins (112, 166, 193, 198, 200, 201, 203, and 206) exhibits product ion peaks of Y0 ions at m/z 495.28 in their (–)MS/MS spectra, whereas the group XVIII (159, 177, and 190) and group XIX (195) show Y0 product ions at m/z 497.29 and 493.26 respectively. Fragmentation of the side chains was not observed in either group. According to literature data, the group XVII includes asterosaponins with 24,25-dihydromarthasterone aglycone (AG XVII) with cholestan-23-one side chain; group XVIII includes compounds with aglycone with 23-hydroxy-cholestan side chain and 195 has aglycone of marthasterone type with Δ24(25)-cholestan-23-one side chain [1]. Compound 207 was identified as native aglycone 3-O-sulfo-24,25-dihydromarthasterone (AG XVII) using authentic standard.
Asterosaponins of the groups XX (104, 121, 123, 128, 137, 140, 149, and 168), XXI (143 and 150), and XXII (144, 152, 157, 173, 176, 182, and 192) exhibit in their (–)MS/MS spectra product ions Y0 at m/z 497.29, 493.26, and 495.28, respectively. But unlike the groups XVII, XVIII, and XIX, a low-intensity fragment peak at m/z 411.18 was detected in the (–)MS/MS spectra of these asterosaponins. It is possible that the presence of the peak of fragment ion at m/z 411.18 was associated with C-20 hydroxy group. Thus, the group XX includes asterosaponins with aglycone having 20-hydroxy-cholestane side chain, the group XXI includes asterosaponins with aglycone having 20-hydroxy-cholestan-22,24-diene side chain, and the group XXII includes asterosaponins with aglycone having 20-hydroxy-cholestan-22-ene side chain.
Asterosaponin 194 (group XXIII) exhibited in its (–)MS/MS spectra product ions Y0 at m/z 509.29 [C28H45O6S]−. A weak fragment ion at m/z 411.18 indicates the presence of C-20 hydroxy group. It is probable that AG XXIII has 20-hydroxy-ergost-22-ene side chain.
Conclusions
In this work, a profiling of polar steroid compounds of the starfish L. fusca was successfully performed by nLC/CSI-QTOF-MS/MS. Application of nanoflow liquid chromatography coupled with captive spray ionization increased sensitivity and reduced noise level compared to conventional LC/ESI-MS. Some characteristic diagnostic fragmentations and retention behavior were established from analysis of 43 standards of starfish polar steroids. Based on the obtained data as well as accurate mass measurements, fragmentation behaviors, and retention times, 207 steroid compounds, including 106 asterosaponins, 6 native aglycones of asterosaponins, 81 glycosides of polyhydroxysteroids, and 14 polyhydroxylated steroids, were detected and characterized, and their tentative structures were proposed.
Detected asterosaponins contain different oligosaccharide chains and have 23 types of aglycones, most of them are new aglycone types. From the fragmentation studies on polyhydroxysteroid glycosides, we deduced fragmentation patterns indicating localization of sulfated group, double bonds, and monosaccharide units. Although in many cases stereochemistry and some details of exact structures cannot be deduced from mass spectra, reasonable proposals for new glycoside structures can be given as it was demonstrated for 91 compounds. Polyhydroxysteroid glycosides have sulfated or non-sulfated aglycones with five to seven hydroxy groups and cholestane, ergostane, or stigmastane side chains. Among the detected biosides, only one compound has disaccharide moiety in the side chain, and other biosides have sulfated or non-sulfated pentoses and hexoses in the aglycone and side chain simultaneously.
Unlike steroid metabolome of the previously studied starfish A. japonica, polar steroid compounds of L. fusca were found in sulfated and non-sulfated forms. However, in contrast to P. pectinifera, where non-sulfated compounds were also found, a number of non-sulfated compounds in L. fusca are very limited. The presence of compounds with cholestane, ergostane, and stigmastane side chains indicates that L. fusca uses dietary phytosterols as well as dietary cholesterol of animal origin for the biosynthesis of its polar steroids.
The approach we used provides a clue to the fast and effective evaluation of the complete set of glycosides allowing to dispense with the isolation of pure compounds. The conclusions about tentative structures are based on the obtained data as well as previously published data on the patterns of fragmentation of compounds with exactly established structures and biosynthetic assumptions. This approach can be extensively used to study a wide range of metabolites, including natural steroid compounds from various sources.
References
Minale, L., Riccio, R., Zollo, F.: Steroidal oligoglycosides and polyhydroxysteroids from echinoderms. Fortschr. Chem. Org. Naturst. 62, 75–308 (1993)
Stonik, V.A.: Marine polar steroids. Russ. Chem. Rev. 70, 673–715 (2001)
Stonik, V.A., Ivanchina, N.V., Kicha, A.A.: New polar steroids from starfish. Nat. Prod. Commun. 3, 1587–1610 (2008)
Ivanchina, N.V., Kicha, A.A., Stonik, V.A.: Steroid glycosides from marine organisms. Steroids. 76, 425–454 (2011)
Dong, G., Xu, T., Yang, B., Lin, X., Zhou, X., Yang, X., Liu, Y.: Chemical constituents and bioactivities of starfish. Chem. Biodivers. 8, 740–791 (2011)
Popov, R.S., Ivanchina, N.V., Kicha, A.A., Malyarenko, T.V., Dmitrenok, P.S., Stonik, V.A.: Metabolite profiling of polar steroid constituents in the Far Eastern starfish Aphelasterias japonica using LC–ESI MS/MS. Metabolomics. 10, 1152–1168 (2014)
Popov, R.S., Ivanchina, N.V., Kicha, A.A., Malyarenko, T.V., Dmitrenok, P.S., Stonik, V.A.: LC-ESI MS/MS profiling of polar steroid metabolites of the Far Eastern starfish Patiria (=Asterina) pectinifera. Metabolomics. 12, 21 (2016)
Popov, R.S., Ivanchina, N.V., Kicha, A.A., Malyarenko, T.V., Grebnev, B.B., Dmitrenok, P.S., Stonik, V.A.: LC-MS-based metabolome analysis on steroid metabolites from the starfish Patiria (=Asterina) pectinifera in conditions of active feeding and stresses. Metabolomics. 12, 106 (2016)
Ivanchina, N.V., Malyarenko, T.V., Kicha, A.A., Kalinovskii, A.I., Dmitrenok, P.S.: Polar steroidal compounds from the Far-Eastern starfish Lethasterias fusca. Russ. Chem. Bull. 57, 204–208 (2008)
Ivanchina, N.V., Kalinovsky, A.I., Kicha, A.A., Malyarenko, T.V., Dmitrenok, P.S., Ermakova, S.P., Stonik, V.A.: Two new asterosaponins from the Far Eastern starfish Lethasterias fusca. Nat. Prod. Commun. 7, 853–858 (2012)
Kicha, A.A., Kalinovsky, A.I., Levina, E.V., Stonik, V.A., Elyakov, G.B.: Asterosaponin P1 from the starfish. Tetrahedron Lett. 24, 3893–3896 (1983)
Kicha, A.A., Ivanchina, N.V., Kalinovsky, A.I., Dmitrenok, P.S., Stonik, V.A.: Asterosaponin P2 from the Far-astern starfish Patiria (Asterina) pectinifera. Russ. Chem. Bull. 49, 1794–1795 (2000)
Kicha, A.A., Ivanchina, N.V., Gorshkova, I.A., Ponomarenko, L.P., Likhatskaya, G.N., Stonik, V.A.: The distribution of free sterols, polyhydroxysteroids and steroid glycosides in various body components of the starfish Patiria (=Asterina) pectinifera. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 128, 43–52 (2001)
Kicha, A.A., Ivanchina, N.V., Kalinovsky, A.I., Dmitrenok, P.S., Stonik, V.A.: Sulfated steroid compounds from the starfish Aphelasterias japonica of the Kuril population. Russ. Chem. Bull. 50, 724–727 (2001)
Ivanchina, N.V., Kicha, A.A., Kalinovsky, A.I., Dmitrenok, P.S., Stonik, V.A., Riguera, R., Jiménez, C.: Hemolytic polar steroidal constituents of the starfish Aphelasterias j aponica. J. Nat. Prod. 63, 1178–1181 (2000)
Popov, R.S., Ivanchina, N.V., Kicha, A.A., Malyarenko, T.B., Kalinovskii, A.I., Dmitrenok, P.S.: Minor steroidal glycosides from the far-east starfish Aphelasterias japonica. Chem. Nat. Compd. 49, 286–290 (2013)
Ivanchina, N.V., Kicha, A.A., Kalinovsky, A.I., Dmitrenok, P.S., Prokof’ev, N.G., Stonik, V.A.: New steroid glycosides from the starfish Asterias rathbuni. J. Nat. Prod. 64, 945–947 (2001)
Ivanchina, N.V., Malyarenko, T.V., Kicha, A.A., Kalinovskii, A.I., Dmitrenok, P.S., Mollo, E.: Polar steroidal compounds from the Antarctic starfish Diplasterias brucei. Chem. Nat. Compd. 42, 621–622 (2006)
Kicha, A.A., Ivanchina, N.V., Kalinovskii, A.I., Dmitrenok, P.S., Sokolova, E.V., Agafonova, I.G.: Sulfated steroid glycosides from the Viet Namese starfish Linckia laevigata. Chem. Nat. Compd. 43, 76–80 (2007)
Kicha, A.A., Ivanchina, N.V., Kalinovsky, A.I., Dmitrenok, P.S., Agafonova, I.G., Stonik, V.A.: Steroidal triglycosides, kurilensosides A, B, and C, and other polar steroids from the Far Eastern starfish Hippasteria kurilensis. J. Nat. Prod. 71, 793–798 (2008)
Ivanchina, N.V., Kicha, A.A., Malyarenko, T.V., Kalinovsky, A.I., Menchinskaya, E.S., Pislyagin, E.A., Dmitrenok, P.S.: The influence on LPS-induced ROS formation in macrophages of capelloside A, a new steroid glycoside from the starfish Ogmaster capella. Nat. Prod. Commun. 10, 1937–1940 (2015)
Malyarenko, T., Malyarenko (Vishchuk), O., Ivanchina, N., Kalinovsky, A., Popov, R., Kicha, A.: Four new sulfated polar steroids from the Far Eastern starfish Leptasterias ochotensis: structures and activities. Mar. Drugs. 13, 4418–4435 (2015)
Kicha, A.A., Ivanchina, N.V., Malyarenko, T.V., Kalinovsky, A.I., Dmitrenok, P.S.: Sulfated steroidal glycosides, regulusosides S1 and S2, from the tropical starfish Pentaceraster regulus. Chem. Nat. Compd. 53, 88–92 (2017)
Ivanchina, N.V., Kicha, A.A., Malyarenko, T.V., Ermolaeva, S.D., Yurchenko, E.A., Pislyagin, E.A., Van Minh, C., Dmitrenok, P.S., Granulatosides, D.: E and other polar steroid compounds from the starfish Choriaster granulates. Their immunomodulatory activity and cytotoxicity. Nat. Prod. Res. (2018). https://doi.org/10.1080/14786419.2018
Domon, B., Costello, C.E.: A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconj. J. 5, 397–409 (1988)
Griffiths, W.J.: Tandem mass spectrometry in the study of fatty acids, bile acids, and steroids. Mass Spectrom. Rev. 22, 81–152 (2003)
Huo, F., Bai, Y., Liu, H.: Fragmentation investigation of brassinosteroid compounds by ion trap and quadrupole time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 24, 3325–3334 (2010)
Popov, R.S., Dmitrenok, P.S.: Stereospecific fragmentation of starfish polyhydroxysteroids in electrospray ionization mass spectrometry. J. Anal. Chem. 71, 1368–1376 (2016)
Gonçalves, A.G., Ducatti, D.R.B., Grindley, T.B., Duarte, M.E.R., Noseda, M.D.: ESI-MS differential fragmentation of positional isomers of sulfated oligosaccharides derived from carrageenans and agarans. J. Am. Soc. Mass Spectrom. 21, 1404–1416 (2010)
Minamisawa, T., Hirabayashi, J.: Fragmentations of isomeric sulfated monosaccharides using electrospray ion trap mass spectrometry. Rapid Commun. Mass Spectrom. 19, 1788–1796 (2005)
Hsu, F.-F., Turk, J.: Elucidation of the double-bond position of long-chain unsaturated fatty acids by multiple-stage linear ion-trap mass spectrometry with electrospray ionization. J. Am. Soc. Mass Spectrom. 19, 1673–1680 (2008)
Jones, J.W., Thompson, C.J., Carter, C.L., Kane, M.A.: Electron-induced dissociation (EID) for structure characterization of glycerophosphatidylcholine: determination of double-bond positions and localization of acyl chains. J. Mass Spectrom. 50, 1327–1339 (2015)
Ivanchina, N.V., Kicha, A.A., Malyarenko, T.V., Kalinovsky, A.I., Dmitrenok, P.S., Stonik, V.A.: Biosynthesis of polar steroids from the Far Eastern starfish Patiria (=Asterina) pectinifera. Cholesterol and cholesterol sulfate are converted into polyhydroxylated sterols and monoglycoside asterosaponin P1 in feeding experiments. Steroids. 78, 1183–1191 (2013)
Kicha, A.A., Ivanchina, N.V., Kalinovsky, A.I., Dmitrenok, P.S., Stonik, V.A.: Steroidal monoglycosides from the Far Eastern starfish Hippasteria kurilensis and hypothetic pathways of polyhydroxysteroid biosynthesis in starfish. Steroids. 74, 238–244 (2009)
Sandvoss, M., Weltring, A., Preiss, A., Levsen, K., Wuensch, G.: Combination of matrix solid-phase dispersion extraction and direct on-line liquid chromatography–nuclear magnetic resonance spectroscopy–tandem mass spectrometry as a new efficient approach for the rapid screening of natural products. J. Chromatogr. A. 917, 75–86 (2001)
Kicha, A.A., Ivanchina, N.V., Kalinovsky, A.I., Dmitrenok, P.S., Stonik, V.A.: Alkaloidosteroids from the starfish Lethasterias nanimensis chelifera. Tetrahedron. Lett. 44, 1935–1937 (2003)
Riccio, R., Iorizzi, M., Minale, L.: Starfish Saponins XXX. Isolation of sixteen steroidal glycosides and three polyhydroxysteroids from the Mediterranean starfish Coscinasterias tenuispina. Bull. des Sociétés Chim. Belges. 95, 869–893 (2010)
Malyarenko, T.V., Kicha, A.A., Ivanchina, N.V., Kalinovsky, A.I., Popov, R.S., Vishchuk, O.S., Stonik, V.A.: Asterosaponins from the Far Eastern starfish Leptasterias ochotensis and their anticancer activity. Steroids. 87, 119–127 (2014)
Riccio, R., Iorizzi, M., Minale, L., Oshima, Y., Yasumoto, T.: Starfish saponins. Part 34. Novel steroidal glycoside sulphates from the starfish Asterias amurensis. J. Chem. Soc. Perkin Trans. 1, 1337 (1988)
Acknowledgements
The study was supported by the Russian Science Foundation Grant No. 16-13-10185. We express our sincere gratitude to Dr. Valery G. Voinov and Tatyana Kolchugina for their assistance in editing this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Materials
ESM 1
(DOCX 1443 kb)
Rights and permissions
About this article

Cite this article
Popov, R.S., Ivanchina, N.V., Kicha, A.A. et al. Structural Characterization of Polar Steroid Compounds of the Far Eastern Starfish Lethasterias fusca by Nanoflow Liquid Chromatography Coupled to Quadrupole Time-of-Flight Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 30, 743–764 (2019). https://doi.org/10.1007/s13361-019-02136-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-019-02136-3