
Abstract
Frustrated by worse than expected error for both peak area and time-of-flight (TOF) in matrix assisted laser desorption ionization (MALDI) experiments using samples prepared by electrospray deposition, it was finally determined that there was a correlation between sample location on the target plate and the measured TOF/peak area. Variations in both TOF and peak area were found to be due to small differences in the initial position of ions formed in the source region of the TOF mass spectrometer. These differences arise largely from misalignment of the instrument sample stage, with a smaller contribution arising from the non-ideal shape of the target plates used. By physically measuring the target plates used and comparing TOF data collected from three different instruments, an estimate of the magnitude and direction of the sample stage misalignment was determined for each of the instruments. A correction method was developed to correct the TOFs and peak areas obtained for a given combination of target plate and instrument. Two correction factors are determined, one by initially collecting spectra from each sample position used and another by using spectra from a single position for each set of samples on a target plate. For TOF and mass values, use of the correction factor reduced the error by a factor of 4, with the relative standard deviation (RSD) of the corrected masses being reduced to 12-24 ppm. For the peak areas, the RSD was reduced from 28% to 16% for samples deposited twice onto two target plates over two days.

Graphical Abstract
Similar content being viewed by others

Introduction
When using a time-of-flight (TOF) mass spectrometer (MS) for mass analysis, the measured flight times of the ions are recorded and must be converted to m/z values through the process of calibration [1, 2]. Typically, the flight times for multiple calibrants of known m/z are collected for a given set of instrument parameters and a calibration curve of a desired order relating measured TOF to m/z is calculated from the data to perform the conversion. When used for a matrix-assisted laser desorption ionization (MALDI) experiment, the calibrants and analytes can be combined in a single sample spot and analyzed simultaneously in the case of internal calibration, or located in separate sample spots on the target plate surface in the case of external calibration [3].
Desirable characteristics of good calibrants are that the compounds chosen must be non-reactive, non-interacting with the analytes of interest (i.e., ideally leading neither to signal suppression nor enhancement), and the peaks observed must be well-resolved from other peaks observed in the spectra [3, 4]. The success of both mass spectral imaging and micro-organism identification experiments, two of the most important applications of the MALDI TOFMS technique today, rely on the accurate and precise measurement of both the m/z value and the intensity of the observed peaks [5,6,7,8,9]. More accurate m/z values are generally obtained in the case of internal calibration compared with external calibration. However, a particular problem with internal calibration is that the intensity of the calibrant peaks, which is dependent on the amount of calibrant present in the sample, must be matched to the intensity of analyte peaks so that both sets of peaks can be co-detected with good peak shape [3, 4].
The requirement for peak intensity matching is removed by using external calibration, however, lower mass accuracy is generally observed [10, 11]. For best results, the calibration sample must be as similar as possible to that containing the analyte, as any differences between the samples can affect the measured TOF. The composition of the samples (especially the choice of matrix) should be matched to ensure similar initial velocities of the analyte and calibrant ions [12, 13], and the spectra should be collected as close in time as possible as the applied voltages can also vary with time. Most importantly, it is known that the samples should be deposited close to each other to ensure that the initial positions of the calibrants and analytes are as similar as possible in case the planarity and alignment of the target plate with respect to the rest of the source is non-ideal [4, 14]. This results in many calibrant spots being required to cover a large number of analyte spots, as each analyte spot requires a calibrant spot in close proximity for best results.
The excellent TOFMS tutorial written by Guilhaus [14] is used here to understand the effect of initial position on overall flight time in a TOF instrument. Using the axes defined in Figure 1, in the case of external calibration, the samples containing calibrants and analytes would be located at different positions in the x-y plane corresponding to the surface of the target plate (e.g., for the Bruker 384 MTP plate shown in Figure 1b). Most simply, any difference in the initial sample position along the z axis (i.e., the axis towards the detector) will result in different pathlengths for ions originating from the samples, resulting in differences in flight times and decreased mass accuracy. Additionally, d1, the distance between G0 (the surface of the target plate) and G1 (the first grid or plate in the source as defined in Figure 1a) will also vary as a result of different z positions of the surface of the target plate, leading to changes in the magnitude of the electric field formed in this ion acceleration region. As the z position of the surface of the target plate varies at different sample positions, these small differences in source geometry in terms of distance and applied electric field both contribute to a decrease in mass accuracy [14]. Considering a non-ideal (i.e., not perfectly flat) target plate, the z position can vary linearly in the x-y plane (termed a tilt in this work) or non-linearly (termed a warp in this work). The effect of these small changes in the initial z position on the ion’s TOF are the major contributor to the decreased mass accuracy when external calibration is used compared with internal calibration [14]. Note that both G1 and G2 (the second grid or plate in the source) are fixed in position with no moving parts, resulting in the distance between then, labeled as d2 in Figure 1a, remaining constant as the sample location changes.

(a) Schematic presentation of the TOFMS source (side view), in which G0 is defined by the surface of the sample target plate depicted in (b). (b) Photograph of a Bruker 384 MTP target plate. Samples applied by ESD were located in positions 1-8 and by dry drop were located in positions A-E. The axes (x, y, and z) defined above are used throughout the text
The photograph in Figure 2 shows the target plate holder and X/Y translation stage used on the Bruker Autoflex TOFMS instrument used in this work. The target plate is introduced into the instrument through a vacuum lock (from the front left as shown in the figure); ridges on the base of the target plate slide into the grooves in the target plate holder shown in the right side of the figure. Once inserted into the holder the sample location is selected by moving the plate using the stepper motors/linear slides forming the translation stage. Note that the target plate holder is mounted to its base plate using three adjustment screws; these screws are used to set the d1 distance and plate planarity. Small errors in setting the planarity of the sample holder result in the sample stage alignment error investigated in this work.

Photograph of the target plate holder and X/Y sample stage used in the Bruker Autoflex III TOFMS
Electrospray deposition (ESD) has been shown previously to produce sets of samples with better within-sample and between-sample reproducibility compared with those prepared using the dry drop technique [15,16,17,18]. Due to this decreased error attributable to the sample preparation, ESD sample preparation is used here to allow for the easier detection and study of other sources of error in measuring TOF (and resulting m/z values) as well as intensity.
Experimental
Materials
All chemicals were used as received from the supplier. Angiotensin I (human acetate salt hydrate form, >90%), bradykinin (acetate salt, >98%), and formic acid (~98%) were obtained from Sigma-Aldrich (St. Louis, MO, USA). The MALDI matrix alpha-cyano-4-hydroxycinnamic acid (CHCA, >98%) was also obtained from Sigma-Aldrich. Methanol (HPLC grade, 99.9%) and acetonitrile (HPLC grade, 99.9%) were obtained from Fisher Scientific (Waltham, MA, USA). Distilled water was purified in the lab using a Barnstead E-pure system with conductivity of 18 megaohm-cm.
Sample Preparation and Deposition
An analyte solution was created consisting of angiotensin I at a concentration of 0.5 mg/mL in water. CHCA matrix solution was prepared at a concentration of 60 mM in methanol. The solutions were combined in a 10:1 v:v (matrix:analyte) ratio producing a solution of 1650 matrix-to-analyte (M/A) ratio.
For dry drop samples, 1 uL of solution was applied to the target plate and allowed to dry to produce a solid sample. For ESD samples, the solution was applied to the target plate using the ESD apparatus described previously [16]. In preliminary experiments, Bruker (Bremen, Germany) Multiprobe and 384 MTP ground steel target plates were used. After the preliminary experiments described in the Results and Discussion section were completed, the following method was used to prepare all ESD samples. Prior to loading the solution to be sprayed, the apparatus was cleaned by passing 200 uL of 50:50 v:v acetonitrile:0.1% formic acid in water, 200 uL of methanol, and 100 uL of the solution to be sprayed through the spray needle and connected tubing. Samples were then deposited onto specific locations of the Bruker 384 MTP ground steel target plate in a random order using a spray height of 2.0 cm, a flow rate of 5.0 uL/min, a spray time of 1 min, and an applied voltage of 5.9–6.0 kV. The applied voltage was selected to produce a stable cone jet mode as monitored by an oscilloscope for all samples [19]. The sample locations and corresponding labels used throughout the discussion are shown in Figure 1b.
Collection of Mass Spectra
Three different Bruker Autoflex MALDI TOFMS instruments were used to collect mass spectra; instrument A (at Drexel University) and B were an Autoflex III MALDI TOFMS, while instrument C was an Autoflex Speed MALDI TOFMS. For each instrument, FlexControl ver. 3.4 was used to collect spectra in reflectron mode using the Smartbeam II Nd:YAG laser operating with a wavelength of 355 nm, a repetition rate of 200 Hz, and with a diameter of 50 um. Two hundred laser shots were collected over 20 raster positions, with 10 laser shots collected per raster position. When collecting spectra from a set of samples on a given plate during a single insertion of the target plate into the instrument, all samples were sampled in a random order, with five spectra being taken from each location. Without ejecting the target plate, an additional set of five spectra was collected from each location, again in a random order. This results in two sets of replicate spectra for each insertion. The analysis order of the plates was randomized within each set of experiments. If the same plate was present twice sequentially in the random order, the plate was ejected, the top plate and the base plate were separated, remounted together, and re-inserted into the instrument. Peak areas of the protonated peak of angiotensin I were obtained using FlexAnalysis ver. 3.4 using the same parameter file.
Physical Measurement of Target Plates
An Ames (Waltham, MA, USA) 412 depth gage with precision of 2.5 um (1/10,000′′) was used to measure the physical shape of the target plates. Note that the 384 MTP target plates used in this work are composed of two pieces, a top plate where samples are deposited and a base plate onto which the top plates are mounted. The combination of a top plate and a base plate is referred to here as the “target plate”; the physical measurements presented here were made on the combined target plate. In this work the same base plate was used for all measurements. Data from a total of seven different top plates were collected; only the data from two top plates labeled A and B are presented here as they are representative of the shapes of the plates available in our lab.
Results and Discussion
Preliminary Experiments
While attempting to perform quantitative work on the Drexel MALDI TOFMS (instrument A) using ESD prepared samples, both the within-sample and between-sample reproducibility of the peak areas was found to be low compared with work done on previous projects in our lab. A series of experiments to determine the source of this irreproducibility was undertaken, with sample preparation, sample deposition, instrument, and data analysis parameters all being investigated. The accuracy and precision of the concentrations of the solutions prepared were evaluated and uncertainties were determined to not be significant compared with the high observed variability in the measured peak areas. The solution composition parameters investigated included the final sprayed sample solution consisting of pure methanol or 90:10 v/v methanol:water, the absolute concentration of the initial matrix solution varying between 30 mM and 60 mM, the M/A ratio of the final sample solution varying from 1000 to 1500, and using bradykinin as the analyte in place of angiotensin I; changes in none of these solution parameters were found to produce an improvement in the precision of the peak areas obtained from multiple samples.
For sample deposition by ESD, the order in which samples were deposited was found to have no effect as long as two requirements were fulfilled. First, the ESD apparatus was required to be well flushed with the proper solution by injecting approximately twice the volume from the injection port to the ESD needle. Second, the samples could not be deposited from solution near the end of a loading of the apparatus where mutual diffusion of the analyte solution and the backing solvent causes a change in solvent composition, analyte and matrix concentration, and matrix-to-analyte ratio. The cleaning method employed for the ESD apparatus was altered to include a washing step using a 50:50 v:v acetonitrile:0.1% formic acid in water solution; flushing of the apparatus with this cleaning solution was performed prior to each loading of the ESD apparatus. Several sets of ESD parameters were also evaluated, with spray times of 1–2 min, spray heights of 1.5–3.0 cm, and flow rates of 2.5–5.0 uL/min all producing similar results. In all cases, the resulting samples were checked for complete surface coverage by collecting mass spectra from multiple locations across a sample [20]. It was determined through this work that the use of Bruker Multiprobe plates was the main source of the within-sample error, as the average within-sample relative standard deviations (RSD) were 4%–8% for samples deposited by ESD onto the 384 MTP target plates as opposed to the 20%–35% for samples deposited identically by ESD onto the Multiprobe plates. The increased error is attributed to the less uniform electric fields produced when using the Multiprobe plates, which have holes used for mounting the target plates present, as opposed to the planar and featureless 384 target plates. The lower within-sample RSD values obtained on the 384 target plates were similar to previous results obtained in our lab [15, 16], however, the between-sample RSD values remained high.
Investigating various TOFMS parameters, a set of different lens voltages (from 7.4 to 8.4 kV in reflectron mode and 6.4 to 7.4 kV in linear mode) and pulsed ion extraction (PIE) times (0 ns to 400 ns in reflectron mode and 30 ns to 130 ns in linear mode) were used to collect spectra in both reflectron and linear mode. Several reflectron voltages (from 20.6 to 21.6 kV) and two detector gain voltages (1.65 and 1.70 kV) were also used in reflectron mode. Again, the between-sample RSD of the peak areas collected remained stubbornly over 20%. Using different laser powers, including one near the ion appearance threshold, and multiple laser repetition rates (20 to 200 Hz) did not improve the results obtained. By comparing the measured peak area over time from a sample, it was determined that the length of time in vacuum for a sample also did not affect the obtained peak areas over a period of 3 h. This was supported by an experiment which determined that the sampling order within the mass spectrometer had no effect on the results. It should be noted that this determination was for samples consisting of angiotensin I and CHCA and would be dependant on the composition of the samples. It was also determined that acceptable (~5% RSD) variations in peak areas can be obtained within 1 min of the high voltages being applied to the system (either after switching target plates or toggling the instrument high voltage on and off) unless the instrument has entered sleep mode. If the instrument is coming out of sleep mode, acceptable reproducibility can be attained within 10 min after the instrument has turned on, hypothesized to be due to the power supplies requiring a warm-up period.
Two different mass calibrations were applied to a selected set of mass spectra, with no improvement in the precision of the peak areas observed. Several different peak picking parameters were also applied to a single set of spectra, some of which integrated each isotopic peak separately and others which integrated the full isotopic envelope of the analyte together; however, no improvement in the precision of the peak areas collected was observed for parameter sets which appropriately detected the peaks. Despite the significant improvement in the within-sample reproducibility of the peak areas following these experiments, no set of parameters studied reduced the between-sample reproducibility significantly until a correlation between the peak area, measured TOF and sample position on the target plate was identified, as described below.
Diagnosis of Instrumental Misalignment Resulting in Decreased Mass Accuracy
After the many experiments discussed above demonstrating that the variables studied did not significantly contribute to the observed variability over the ranges studied, it was determined that the cause of the correlation of the shifts in TOF and peak intensity with location on the target plate was actually due to changes in the initial z position of ions along the instrument flight axis, as defined in Figure 1. This effect is due to the initial position along the z axis affecting two of the factors which define the measured TOF. First, the path length ions travel is dependant on the initial position of their creation. Second, the magnitude of the electric field felt by the ion is determined by the distance d1 and voltages applied to the plates as shown in the figure. Third, the kinetic energy imparted to the ions is dependent on their initial position within the ionization region as a result of the use of pulsed ion extraction. Ions will travel a distance into the source, away from the target plate, after their creation under field free conditions during the PIE delay. The absolute distance ions move in this time period should be constant regardless of the distance d1 as shown in Figure 1. However, as the initial position of the ions varies between sample positions, ions will have traversed different fractions of the distance d1 between G0 and G1, since the total distance is defined by the z position of G0, assuming the location of G1 (and the rest of the source) is unchanged. Once the voltages are applied following the PIE delay, these differences in the fraction of the distance traversed will translate to differences in the amount of the applied electric field experienced by the ions as they accelerate out of the source. This in turn results in ions gaining different kinetic energies and having different velocities as they exit the source, depending on the z value of the surface of the target plate for each sample position. Essentially, the z value of the surface of the target plate will not only define the path length ions travel, but also the geometry of the source used to accelerate the ions. Shifts in the path length and the source geometry would cause different TOFs to be measured from different sample positions. In the following discussions, both factors would be responsible for TOF shifts resulting from differences in the initial position of ions produced by the different z values across multiple sample positions.
The sources of the variation in the initial position of ions can be attributed to four factors when using the Bruker Autoflex III instrument with 384 MTP target plates as illustrated in Figure 3. Any stage misalignment (as illustrated in panel A) results in the surface of an ideal flat target plate being non-orthogonal to the flight axis of the instrument. This stage misalignment is expected to be constant for a given instrument unless physically adjusted, but can vary from instrument to instrument. The insertion variation (panel B) is caused by imprecision in the way target plates enter the source, resulting in slightly different z positions for a plate each time it is inserted into the sample holder of an instrument. The remounting variation (panel C) arises from the usage of the two piece MTP 384 target plates. Each time a top plate is mounted to a base plate, the tilt and height of the surface of the top plate may vary. Finally, each top plate will have a particular topography and thickness (panel D) that will vary from plate to plate. Each of these sources of variation can produce a change in initial position for the ions formed along the z axis, resulting in the initial position of ions being dependent on what target plate position the sample is located on. The stage misalignment, the insertion variation, and the mounting variation will create differences that either change the height of the plate surface, the angle of tilt of the surface with respect to the z axis, or a combination of the two. The final factor, different top plate shapes (illustrated in panel D), can cause these variations in addition to having nonplanar defects, such as convex (panel 3D4) or concave (not shown) surfaces.

Sources of variation of the initial ion position: (a) misalignment of the stage, (b) insertion of the plate into the source, (c) mounting the target plate onto the base plate, and (d) the physical shape of the target plates. Note that in each panel the thicker black rectangle on the bottom represents the base plate whereas the thinner black rectangle represents the target plate to which the samples are applied
This effect of sample location on the measured TOF is shown in Figure 4. For each combination of instrument and target plate, a plot is obtained for measured TOF versus the sample location, as shown in panels A through F. The observed patterns are a result of the combination of the effects shown in Figure 3 affecting the measured TOF. Considering that the effect of the stage alignment and the plate shape on the initial position and resulting TOF are constant for a given pair of target plate and instrument, the within position precision (i.e., the error bars on any of the graphs shown in Figure 4) is the result of variations of multiple insertions and mountings of a target plate (Figure 3b and c). Since the within sample position error is smaller than the overall error between sample positions, the variation due to multiple insertions of the plate and mountings of the two pieces of the plate is less than the other two factors affecting initial position, indicating that the major source of error in measuring TOF (and the resulting mass, after calibration) from multiple sample positions, insertions, and/or target plates is mainly the result of the sample stage alignment and target plate shape. Since the differences in the measured TOF are the result of these two factors, if either the misalignment of the stage or the deviations from planarity of the target plates were known, the other can be determined. As described in the “Experimental” section above, the surfaces of the target plates were measured using a high precision depth gage; the measured shapes of plates A and B are shown in Figure 5.

Correlation of TOF to sample position and diagram of the interaction of the stage alignment and target plate shape for: (a) plate A on instrument A, (b) plate A on instrument B, (c) plate A on instrument C, (d) plate B on instrument A, (e) plate B on instrument B, and (f) plate B on instrument C. Error bars on the points represent the 95% confidence intervals

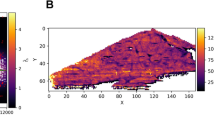
Surface plots of the height measurements of target plates (a) A and (b) B determined using an Ames 412 depth gage. Note that the alphabetical row labels were entered as numbers for plotting, with 1 corresponding to row A and 16 corresponding to row P
Plate A has a convex shape (as illustrated in Figure 3d4); the measured height is similar at the ends of the plate along the x axis (in reference to Figure 1), referred to as the left (corresponding to lower column numbers and sample positions 1 and 5) and right (higher column numbers and sample positions 4 and 8) directions. Note that the analyte TOF measured on both instruments A and C are not simply the result of this shape due to the additional effect of a tilt resulting from the two instrument sample stages being misaligned. On instrument A the TOF is longer on the right side of the plate (Figure 4a) as opposed to on instrument C where the TOF is longer on the left side of the plate (Figure 4c). This is a result of the two instruments having stage misalignments in opposite directions. The situation is illustrated by the schematic located below each plot, where the shape of the target plate and its tilt is illustrated by the black shape; the blue circles represent the sampling locations on the target surface. For plate A in instrument A’s stage shown in Figure 4a, the left side of plate A is higher than the right, resulting in the initial position and the resulting TOF of ions created at positions on the left side of the plate to be more similar. For plate A in the instrument C stage shown in Figure 4c the opposite is true, with the sample positions on the right side of the plate being more similar in height and the resulting TOF being closer than on the left side of the target plate. In both cases, the TOF shifts approximately 3 ns across the eight sample positions used. Conversely, the TOF trend with sample position obtained with instrument B and plate A in Figure 4b reflects the measured shape of plate A more directly than those obtained with the other two instruments. The TOF shift is only 2 ns across the eight positions, indicating the stage of instrument B is closer to planarity than the other two instruments’ stages. It still does introduce a tilt, skewing the TOF values from simply depending on the shape of plate A as expected if only the target plate shape was considered. It should be noted that while the shifts in TOF are dependant on the differences in initial ion position, they are not proportional to the actual change in z value as a result of the focusing of the TOF as a result of the use of a two step source, pulsed ion extraction, and the reflectron.
In the case of plate B, Figure 5b shows that the surface of plate B is slanted with a decrease in height from left to right (as illustrated in Figure 3d3). The degree of the slant also changes between the top portion of the target plate and the bottom, with the top of the plate exhibiting a smaller slope than the bottom. Note that the TOF trends for each instrument shown in Figure 4d–f are fairly linear, a result of the slanted shape of plate B (as opposed to the convex shape of plate A) interacting with the linearly misaligned sample stages. Figure 4d corresponds to plate B in instrument A’s source, in which both the plate and the source produce higher initial positions on the left side of the plate, resulting in a large TOF shift of 5 ns. For instrument C’s data shown in Figure 4f, the TOF shift is only 2.5 ns, a result of the two sources of the change in height acting against each other, nearly cancelling each other. The stage misalignment must be larger than the measured 48 um change in height due to the plate shape to result in an overall increase in height from left to right and the observed decrease in TOF from left to right. This compensation of stage misalignment and plate shape is also seen when plate B is used in instrument B, as shown in Figure 4e. Here, the top and bottom of the plate (sample positions 1–4 and 5–8, respectively) show opposing trends. This is a result of the stage misalignment being larger than the 28 um height change from the plate shape at the top of plate B but less than the 48 um height change across the bottom of plate B. The stage misalignment overcompensates for the shape of plate B across the top of the plate and undercompensates for the shape at the bottom of the plate. This produces opposing trends for TOF for positions 1–4 and 5–8. Additionally, considering that both instrument A and C impact the TOF obtained for plate A more than instrument B, the degree of misalignment of both instruments A and C are greater than that of instrument B. Considering the TOF trends obtained when using plates A and B on the three instruments and the physical measurements of the two plates, the same conclusions are reached concerning the misalignment of the sample stages.
This understanding of the dependence of the measured TOF on the sample position being attributable to the combination of the shape of a specific target plate and misalignment of the sample stage of a particular instrument allows for an explanation of the mass accuracy obtained on each instrument using plates A and B. The corresponding mass accuracy data is shown in Table 1. Comparing the data collected on plate A for each instrument, the previous conclusions concerning the stage misalignments are supported. The data collected with instrument B has better TOF and mass precision compared with the data collected on instruments A and C, indicating the stage of instrument B is better aligned than that of the other two instruments. Additionally, the data collected on instruments A and C have similar mass errors. This is a result of the shape of plate A shown in Figure 5 and the magnitude of the stage misalignments being similar.
Considering the data obtained from plate B shown in Table 1, errors observed in instruments B and C are similar, whereas the errors observed for instrument A are larger. The misalignment of instrument A is additive to the slanted target plate surface observed for plate B, enhancing the differences in the initial position of ions across the plate and producing larger TOF and mass errors. Conversely, the direction of the misalignment is in the opposite direction on instruments B and C, resulting in the stage compensating for the slanted shape of the target plate and consequently lower TOF and mass errors being observed compared with those obtained with instrument A.
The performance test used by the manufacturer to check the alignment of the sample stage is based on measuring the average mass accuracy of an analyte located at the corners of the target plate, locations labeled A–D in Figure 1. This test omits the center location E used in this work. The inclusion of the center location allows for better determination of the mass error across plates shaped similarly to plate A. It is important to note that this performance test determines the mass accuracy when a particular plate is used with a particular instrument. Using plate A, instruments A and C produce similar mass accuracy values that pass the test (<200 ppm difference). When plate B is used, the target plate shape acts constructively with the misalignment of the source of instrument A, increasing the error in the measured TOF and mass, acting destructively with the misalignment of instrument C’s source, decreasing the error in the measured TOF and mass. If a plate with the inverse shape of B, i.e., with increasing height from left to right, were to be used, the mass error would be decreased on instrument A and increased on instrument C. To characterize either the surface shape of a target plate or the alignment of the source of an instrument, the other must be known. Characterization of the source alignment can be done with a target plate of known shape as described above. Characterization of a plate can be done using mass spectral data obtained on an instrument with a known stage alignment, but likely can be more easily performed by physically measuring the target plate as done here.
It can also be observed that in all cases in Table 1, the TOF and mass error obtained from samples applied by ESD is only approximately 60% of the error obtained from samples prepared by the dry drop method, confirming the benefit in terms of reproducibility of applying samples by ESD. The average RSD within each sample of the TOF and mass for ESD samples, 2.0 ppm and 4.1 ppm, respectively, were also better than corresponding dry drop samples, with the dry drop samples resulting in over twice the error of the TOF and mass within each sample.
Effect of Stage Misalignment on Signal Intensity Reproducibility
While changes in flight time due to changes in the initial ion position caused by non-ideal target plates and sample stages can be easily explained with respect to the well-known equations of ion motion in the TOF MS, changes in the observed ion peak areas with changes in z are harder to explain. Similar to the trends seen when plotting the TOF against the sample positions as shown in Figure 4, consistent trends are observed that correlate to the combination of the target plate shape and stage alignment with the resulting spectral intensity in terms of integrated peak area. These trends are shown in Figure 6. Considering the data obtained on instrument A, peak areas increase from the left to right across both target plates A and B from positions 1 to 4 and again from positions 5 to 8, with the positions corresponding to the locations shown in Figure 1b. When data is taken from the better aligned instrument B, there is no longer an obvious increase in peak area moving left to right across either plate, attributable to the better aligned source stage resulting in a narrower range of heights of the surface of the target plates when loaded into the instrument. The RSD of the mean areas obtained on the two instruments support this, with a 20% RSD being obtained from two insertions each of the two plates on instrument A (the data shown in Figure 6a and b) and 16% RSD being obtained from three insertions each of the two plates on instrument B (the data shown in Figure 6c and d). Analogous experiments were conducted using dry drop deposited samples, but the trends were not observed due to the significantly worse within-sample peak area reproducibility of the dry drop deposited samples (RSD = 20%–50%) compared with the samples applied by ESD (RSD = 2%–7%).

Correlation of peak area to sample position for: (a) plate A and (b) plate B on instrument A, and (c) plate A and (d) plate B on instrument B. Error bars represent the 95% confidence interval
We hypothesize that the reason for the correlation between the sample position and the peak area is that the varying height of the target plate when inserted into the instrument affects the alignment of the laser with the ion optics responsible for extracting the ions in order to transmit them through the rest of the instrument to the detector, as depicted schematically in Figure 7. When either the shape of the target plate, the alignment of the source stage, or the combination of the two results in the height of the surface of the plate varying across a target plate, the position that the laser strikes the plate surface will vary. In Autoflex instruments, the laser enters the source and strikes the plate at approximately a 30° angle from the perpendicular. As a result of the varying height of the plate surface and the laser beam angle, the position that the laser strikes the plate surface will move orthogonally (i.e., on the x-y plane) to the flight axis of the instrument (the z axis) in reference to the rest of the instrument, including the ion optics. If the laser is well aligned with the ion optics, ions are transmitted efficiently through the instrument, resulting in large signal. As the target plate is moved by the sample stage in order to sample different sample locations the surface height changes, and the laser position shifts to a new location. As a result, the alignment of the laser with the ion optics is changed. If this change is significant enough to reduce the transmission of ions through the rest of the instrument, the peak area will decrease because of the change of the initial position of ion formation. As the instruments used in this work use gridless sources, ions are effectively transmitted from only a small region of the source compared with analogous gridded sources. This would result in the intensity of signal in gridless systems being more sensitive to the alignment of the laser position with the ion optics than similar gridded systems.

Illustration of the cause of the peak area effect with sample position shown: (a) from a side perspective, and (b) from a top-down perspective. Positions 1 and 2 refer to sampling from different areas of the sample plate
Correction of TOF and m/z Values
As mass spectra are taken from many locations on the target plate, the spread of TOF increases, resulting in a loss of mass accuracy if the data are averaged. This necessitates the use of either internal calibration in which the calibrants are contained in the sample or external calibration in which the location of the standard and unknown containing samples are close in proximity (resulting in similar initial z positions of ions) in order to attain good mass accuracy. By measuring the effect of the sources of variation of initial ion position as done in this work, they can be corrected increasing mass accuracy without the use of either internal or close proximity external calibration. Essentially, improved mass accuracy can be attained with a single calibration spot in one location on a target plate as long as the combination of the sample stage alignment and target plate shape are well characterized beforehand.
To correct the TOF and masses presented in Tables 2 and 3, spectra were collected from locations 1 through 8 in Figure 1b for two sets of samples applied by ESD. Position 2 was selected as the reference position. The first set of replicates from the first insertion were used for both plates as the reference data set. For each measured TOF and mass, information from these reference insertions and positions are used in Equation 1:
where R′ is the corrected value of interest, R is the measured value of interest, Cpos is the sample position correction factor, and Cins is the insertion correction factor. Cpos is determined using Equation 2:
where Ppos,ref is the value of interest from the position of interest from the reference data set selected to make the correction factors, and Pstd,ref is the position from the reference data set selected as the standard (position 2 in this work). Cins is determined using
where PposX is the value of interest from the standard position within the data set to be corrected and PposX,ref is the value of interest from the same standard position within the reference data set, position 2 in this work. Note that three different correction factors may be calculated, one each using the “value of interest” of TOF, mass, or peak area in Equations 1–3. Specifically, Equation 1 can be applied for TOF and mass as long as either TOFs or masses are used through the calculation; similarly, peak areas can be corrected using Equation 1 as shown below as long as peak areas are used throughout the calculation.
Table 2 shows examples of the correction factors for both position (Cpos) and insertion (Cins) used to convert the uncorrected TOFs into corrected TOFs. Since all data shown are from the same plate, for a given sample position, the Cpos values are identical. These values would be expected to change should a different plate be used, as the different plates would have different shapes. The Cins values are the same within a given insertion while changing between insertions, as there is some error involved in bringing the target plate into the source as indicated in Figure 3b. The mounting of the target plate to the base plate can also produce some additional error over insertions if the top plate was separated from the base plate as done in this work, as indicated in Figure 3c. The corrected values show better precision following the correction, with the overall RSD of the TOFs decreasing from 27 ppm to 7 ppm.
RSDs of the measured TOF and calculated mass for the angiotensin I analyte prior to and following the application of the corrections are shown in Table 3. The first set of replicates of insertion 1 of sample sets 1 for plates A and B were used as the reference insertion for each set of replicates collected. For this reason, the RSDs of both the TOF and masses after the corrections are found to be extremely small. Essentially, this would be analogous to using internal calibration, where there is calibrant present at every location from which a spectrum containing an unknown is collected from. Considering the other sets of replicates from sample set 1 in Table 3, the error is reduced by approximately a factor of 10, while there is a factor of four reduction of the RSDs for sample set 2. The larger reduction seen with sample set 1 is a result of the reference set of data being collected from those samples, whereas sample set 2 is from independently prepared samples deposited on a following day. The data obtained from sample set 2 would be more applicable to typical MALDI TOFMS experiments, as these significant reductions in the TOF and mass error would translate to improved mass accuracy without the need for internal calibration, or even close proximity external calibration. Additionally, it is interesting to note that when the correction factors for plate A for the plate shape are applied to data collected from plate B, the variance in the data set decreases, but not as much as when the proper correction factors determined from plate B are applied. The same is true when correction factors determined from plate B are applied to data collected on plate A. This is a result of the impact of the source alignment being corrected with either set of correction factors, as both were found using data collected on instrument A, but the effect of the target plate shapes is corrected only when the proper set of correction factors from the same target plate are applied. This is an indication that if a set of plates were similar enough in shape, the same correction factors could be applied to the entire set of data with good results.
While unique for each combination of target plate and instrument, the set of Cpos (one value for each sample position) does not need to be found for repeated use of a target plate on a particular instrument. Once measured, the Cpos values do not vary as long as the target plate shape and sample stage alignment remain constant. For Cins, a single value is applied to all samples present on a target plate during a given insertion of the plate into the source. As a result, it can be determined from spectra collected from a single sample position. Variations in the TOF and mass caused by differences between initial positions (z position) as a result of sample stage misalignments and target plate shape are corrected by Cpos and variations caused by differences of the initial position are corrected by Cins, improving the results obtained using external calibration, but without the need to apply calibrants in close physical proximity to samples containing unknowns. The reduction of the need for internal calibration, without sacrificing mass accuracy, can significantly increase sample throughput by reducing sample preparation time and decrease the amount of calibrants required. Note that this increase in throughput will only follow the initial investment in time and effort required to measure a reference data set on each plate. Similarly to how a calibration is stored within an experimental method and automatically applied, the correction factors can be measured once, then stored and applied automatically by the data analysis software. This can be facilitated by the current use by Bruker of RFID chips on sample plates without requiring additional hardware. Additionally, the complexity of an analysis is reduced, as the requirement of matching the relative intensities of analyte and calibrants is removed as well as there being no chance of analytes and internal calibrant peaks overlapping with each other. This correction method can be applied to MALDI IMS experiments as well, allowing users to maintain good mass accuracy across an image without the need for applying mass calibrants across the entire sample during sample preparation.
Correction of Analyte Peak Area
Owing to the reproducible effect of the sample position on the peak area as seen in Figure 6, a similar correction can be applied to the peak area as the TOF and mass. This effect is the result of the combination of the sample stage alignment and target plate shape producing a change in the height of the target plate, which results in the alignment of the laser and ion optics varying across a target plate, as discussed above and illustrated in Figure 7. Equation 1 was used to correct the peak areas obtained from a data set consisting of three sets of independently prepared samples deposited on three different days by ESD. For each day, eight samples were deposited in positions 1–8 as shown in Figure 1b on target plates A and B. Spectra were collected from four insertions per plate on instrument A. Example uncorrected peak areas, corrected peak areas, and the correction factors used to do the peak area correction are shown in Table 4. Here two plates were used on instrument A. As a result, the Cpos factors are different for the same position, as the two plates have different shapes as shown in Figure 5. The Cpos values are the same for a given position for a given target plate. Additionally, as shown above with TOFs in Table 2, the Cins values are the same within an insertion but vary across different insertions. This correction improves the precision of the presented peak areas, with the RSD of the data set decreasing from 34% to 17%.
The RSD of the peak areas within additional data sets are shown in Table 5. The within sample precision for the data was excellent due to the use of ESD to create samples, with the mean RSD of each subset of five spectra being 5.2% with a 95% confidence interval of the RSD ranging from 4.9% to 5.4%. The largest increase of peak area precision is seen in the reference data set, with a decrease in RSD that approaches the within-sample reproducibility of the ESD applied samples of 5.2%. This is attributable to the correction factors being calculated from that data set. As a result, any variation between the samples due to sample-to-sample variation created during the application of the samples by ESD would be accounted for. For sample sets 1 and 2 in Table 5, this would not be the case, as the correction factors derived from the reference set were applied to sets 1 and 2, so any sample-to-sample variability would still affect the peak areas. The data obtained from sets 1 and 2 would be analogous to a typical experiment in which the correction factors were previously determined using a reference set of samples. Overall, the peak areas after the correction are more homogenous, resulting in better reproducibility of the peak area measurement even when data is compared across sample positions, target plates, and sets of samples, as the RSD of the corrected peak areas of sample sets 1 and 2 on plates A and B are reduced from 28% to 17%, a significant difference at the 95% confidence level as determined by an f-test. This significant decrease in the error of the peak area is a result of correcting the effect of multiple insertions of the plates into the instrument, multiple mountings of the target plate onto the base plate and multiple target plates, and is achieved without a requirement of an internal standard, as the correction factors can be determined previously from reference samples.
Conclusion
As a result of recognizing the relationship between the sample position and resulting TOF, mass and peak area, as well as obtaining physical measurements of the target plates, the degree of misalignment of the sample stage of three Bruker Autoflex instruments was characterized through the collection of mass spectra. Variations in the initial ion position created by the target plate shape and sample stage alignments resulted in reproducible TOF shifts due to ions traveling different path lengths to the detector and experiencing small differences in acceleration due to small differences in the source geometry. Owing to the laser beam striking the target plate at an angle, these initial z axis ion position differences also resulted in the laser beam and ion optics alignment varying with sample position, producing changes in ion transmission efficiency and, hence, measured ion peak areas.
The TOF and masses collected across target plates were corrected, resulting in a factor of four improvement in mass accuracy. Additionally, the peak areas were corrected, resulting in a reduction of the overall RSD of the peak areas decreasing from 28% to 16%. In each case, internal or close proximity external calibration were not required. Following the initial characterization of a combination of a MALDI TOFMS instrument and a target plate, only a single location would require a calibrant to perform these corrections. If the instrument control software is updated to store and recall these correction factors automatically, the complexity of experiments can be reduced while the throughput can be increased without sacrificing mass accuracy and improving intensity reproducibility.
References
Tanaka, K., Waki, H., Ido, Y., Akita, S., Yoshida, Y., Yoshida, T.: Protein and Polymer analyses up to m/z 100,000 by laser ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2, 151–153 (1998)
Karas, M., Hillenkamp, F.: Laser desorption ionization of proteins with molecular masses exceeding 10,000 Daltons. Anal. Chem. 60, 2299–2301 (1988)
Xiang, B., Prado, M.: An accurate and clean calibration method for MALDI-MS. J. Biomol. Tech. 21, 116–119 (2010)
Moskovets, E., Chen, H., Pashkova, A., Rejtar, T., Andreev, V., Karger, B.L.: Closely spaced external standard: a universal method of achieving 5 ppm mass accuracy over the entire MALDI plate in axial matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 17, 2177–2187 (2003)
Chumbley, C.W., Reyzer, M.L., Allen, J.L., Marriner, G.A., Via, L.E., Barry, C.E., Caprioli, R.M.: Absolute quantitative MALDI imaging mass spectrometry: a case of rifampicin in liver tissues. Anal. Chem. 88, 2392–2398 (2016)
Hamma, G., Bonnela, D., Legouffea, R., Pamelarda, F., Delbosb, J., Bouzomb, F., Stauber, J.: Quantitative mass spectrometry imaging of propranolol and olanzapine using tissue extinction calculation as normalization factor. J. Proteom. 75, 4952–4921 (2012)
Goodwin, R.J.A., Mackay, C.L., Nilsson, A., Harrison, D.J., Farde, L., Andren, P.E., Iverson, S.L.: Qualitative and quantitative MALDI imaging of the positron emission tomography ligands raclopride (a D2 dopamine antagonist) and SCH 23390 (a D1 dopamine antagonist) in rat brain tissue sections using a solvent-free dry matrix application method. Anal. Chem. 83, 9694–9701 (2011)
Pirman, D.A., Reich, R.F., Kiss, A., Heeren, R.M.A., Yost, R.A.: Quantitative MALDI tandem mass spectrometric imaging of cocaine from brain tissue with a deuterated internal standard. Anal. Chem. 85, 1081–1089 (2013)
Wunschel, S.C., Jarman, K.H., Petersen, C.E., Valentine, N.B., Wahl, K.L., Schauki, D., Jackman, J., Nelson, C.P., White, E.: Bacterial analysis by MALDI-TOF mass spectrometry: an inter-laboratory comparison. J. Am. Soc. Mass Spectrom. 16, 456–462 (2005)
Gobom, J., Mueller, M., Egelhofer, V., Theiss, D., Lehrach, H., Nordhof, E.: A calibration method that simplifies and improves accurate determination of peptide molecular masses by MALDI-TOF MS. Anal. Chem. 74, 3915–3923 (2002)
Bantscheff, M., Duempelfeld, B., Kuster, B.: An improved two-step calibration method for matrix-assisted laser desorption/ionization time-of-flight mass spectra for proteomics. Rapid Commun. Mass Spectrom. 16, 1892–1895 (2002)
Spengler, B., Kirsch, D.: On the formation of initial ion velocities in matrix-assisted laser desorption ionization: virtual desorption time as an additional parameter describing ion ejection dynamics. Int. J. Mass Spectrom. 226, 71–83 (2003)
Berkenkamp, S., Menzel, C., Hillenkamp, F., Dreisewerd, K.: Measurements of mean initial velocities of analyte and matrix ions in infrared matrix-assisted laser desorption ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 13, 209–220 (2002)
Guilhaus, M.: Principles and instrumentation in time-of-flight mass spectrometry. J. Mass Spectrom. 30, 1519–1532 (1995)
Hensel, R.R., King, R.C., Owens, K.G.: Electrospray sample preparation for improved quantitation in matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 11, 1785–1793 (1997)
Szyszka, R., Hanton, S.D., Henning, D., Owens, K.G.: Development of a combined standard additions/internal standards method to quantify residual PEG in ethoxylated surfactants by MALDI TOFMS. J. Am. Soc. Mass Spectrom. 22, 633–640 (2011)
Erb, W.J., Owens, K.G.: Development of a dual-spray electrospray deposition system. Rapid Commun. Mass Spectrom. 22, 1168–1174 (2008)
Axelsson, J., Hoberg, A.M., Waterson, C., Myatt, P., Shield, G.L., Varney, J., Haddleton, D.M., Derrick, P.J.: Improved reproducibility and increased signal intensity in matrix-assisted laser desorption/ionization as a result of electrospray sample preparation. Rapid Commun. Mass Spectrom. 11, 209–213 (1997)
Marginean, I., Nemes, P., Vertes, A.: Order-chaos-order transitions in electrosprays: the electrified dripping faucet. Phys. Rev. Lett. 97, 064502 (2006)
Szyszka, R.: Ph.D. Thesis, Drexel University, Philadelphia (2012)
Acknowledgments
The authors thank the National Science Foundation for funding to purchase the Bruker Autoflex III MALDI TOFMS used in this work at Drexel University (NSF grant no. 0840273). The authors also thank their colleagues, who wished to remain anonymous, for generously allowing them to run the performance tests on the colleagues’ instruments, as well as Mark Shiber of the Drexel University machine shop for providing the depth gage used to measure the target plates.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Malys, B.J., Piotrowski, M.L. & Owens, K.G. Diagnosing and Correcting Mass Accuracy and Signal Intensity Error Due to Initial Ion Position Variations in a MALDI TOFMS. J. Am. Soc. Mass Spectrom. 29, 422–434 (2018). https://doi.org/10.1007/s13361-017-1845-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-017-1845-2