
Abstract
Methanobactin (Mb) from Methylosinus trichosporium OB3b is a member of a class of metal binding peptides identified in methanotrophic bacteria. Mb will selectively bind and reduce Cu(II) to Cu(I), and is thought to mediate the acquisition of the copper cofactor for the enzyme methane monooxygenase. These copper chelating properties of Mb make it potentially useful as a chelating agent for treatment of diseases where copper plays a role including Wilson’s disease, cancers, and neurodegenerative diseases. Utilizing traveling wave ion mobility-mass spectrometry (TWIMS), the competition for the Mb copper binding site from Ag(I), Pb(II), Co(II), Fe(II), Mn(II), Ni(II), and Zn(II) has been determined by a series of metal ion titrations, pH titrations, and metal ion displacement titrations. The TWIMS analyses allowed for the explicit identification and quantification of all the individual Mb species present during the titrations and measured their collision cross-sections and collision-induced dissociation patterns. The results showed Ag(I) and Ni(II) could irreversibly bind to Mb and not be effectively displaced by Cu(I), whereas Ag(I) could also partially displace Cu(I) from the Mb complex. At pH ≈ 6.5, the Mb binding selectivity follows the order Ag(I)≈Cu(I)>Ni(II)≈Zn(II)>Co(II)>>Mn(II)≈Pb(II)>Fe(II), and at pH 7.5 to 10.4 the order is Ag(I)>Cu(I)>Ni(II)>Co(II)>Zn(II)>Mn(II)≈Pb(II)>Fe(II). Breakdown curves of the disulfide reduced Cu(I) and Ag(I) complexes showed a correlation existed between their relative stability and their compact folded structure indicated by their CCS. Fluorescence spectroscopy, which allowed the determination of the binding constant, compared well with the TWIMS analyses, with the exception of the Ni(II) complex.

ᅟ
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
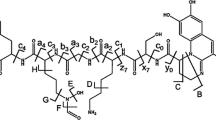
Methanotrophs are methane oxidizing bacteria that synthesize methanobactins, which are modified oligopeptides thought to be part of the methanotrophs copper-acquisition scheme [1]. Copper is a cofactor for the particulate methane mono-oxygenase enzyme, which converts methane to methanol, and methanotrophs will secrete methanobactins in response to low copper availability in their environment [2, 3]. Each of the structurally characterized methanobactins has a distinct bidentate metal ion coordination system consisting of enethiol with a five-member ring (oxazolone, pyrazinedione, or imidazolone) [3] that prefers Cu(I) over other metal ions [4,5,6,7]. All methanobactins will reduce Cu(II) to Cu(I) upon binding [6, 8] and have catalytic redox activities [5, 9]. Methanobactin (Mb) isolated from Methylosinus trichosporium OB3b (MW = 1154.26 Da, Scheme1) is the best characterized methanobactin [2, 10], and has a chemical formula of C45H58N10O16S5. Mb includes seven amino acid residues with two modified enethiol-oxazolone rings and the turning residue, pyrrolidine, with an intramolecular disulfide bond between Cys3 and Cys6, creating a closed loop structure (Scheme 1) [2]. The two enethiol-oxazolone rings and the amino acid tyrosine are chromophores that can be monitored by spectroscopic methods [6, 7]. In order to obtain Mb, a one-step purification method is used that includes cultivation of methanotrophs at low copper concentrations, cell-free spent media separation, and a resin extraction [3]. The sample contains primarily Cu-free Mb but a small amount of Mb-Cu(I) is present due to cultivation media containing about 1 μM Cu(II). Also present is an Mb that does not contain the Met7 (Mb-Met) at the C-terminus, but terminates with the Cys6 instead. A two-step method has also been developed that uses analytical and semi-preparation scale high performance liquid chromatography to separate these two Mb species from one another [11]. The crystal structures of both the Mb-Cu(I) and Mb-Met-Cu(I) complexes show the Cu(I) is coordinated by the two enethiol-oxazolone (2N-2S) groups in a distorted tetrahedral geometry [11].

(a) Structure of the methanobactin (Mb) from Methylosinus trichosporium OB3b. (b) The fragmentation nomenclature used for disulfide-reduced Mb for identifying the product ions from collision-induced dissociation

Geometry optimized gas-phase structures of (a) [Mb-2H+Cu(I)]−, and (b) [Mb-3H+Zn(II)]− using the B3LYP/LanL2DZ level of theory
Wilson’s disease is an autosomal, recessively-inherited disorder of copper metabolism caused by ATPase copper-transporting β (ATP7B) gene mutations [12]. The purpose of this ATPase is to mediate the copper excretion into the bile and the functional loss causes the inability of hepatocytes to maintain copper equilibrium, yielding a persistent copper accumulation [12]. Wilson’s disease is characterized by elevated copper levels in the liver, eyes, and parts of the brain; Cu(I/II) produces reactive oxygen species, causing neurological and hepatolenticular degeneration [13]. Current chelation therapies are penicillamine (cuprimine, a small thiol amino acid) and triethylene-tetramine (Syprine, an EDTA-like chelator). When the copper chelating efficiency of Mb was studied in a rat model of Wilson’s disease, Cu(I)-bound Mb directed excretion of the high copper levels in the liver into the bile [14]. In vitro experiments confirmed that Mb removed the copper from liver cytosol containing high concentrations of copper metallothionein [14]. The use of Mb as a copper chelation therapy is further studied here by examining competition from other metal ions for the 2N-2S binding site, which may disrupt the copper binding ability and whether Mb potentially could be a useful chelation therapy for other metal associated diseases.
In the absence of Cu(I/II), Mb will bind with Ag(I), Pb(II), Co(II), Fe(II), Mn(II), Ni(II), and Zn(II) [7]. Many of these metal ions are essential to many life processes mediated by metal-containing, redox-active enzymes. Metalloenzyme activity depends on the correct metal ion occupying the catalytic center with selectivity being controlled, in part, by the availability of the metal ion when the enzyme is synthesized and by the selective binding to the ligands available in the catalytic center of the enzyme. Metal ion properties that influence the selectivity include [15]: (1) net charge, providing greater charge–dipole or charge–charge interactions between it and enzyme ligands; (2) radius and relative charge density; (3) preferred coordination number or geometry related to the number of available coordinating ligands (i.e., Cu(I) 2-4, Cu(II) 4-6, Zn(II) 4-6, Pb(II) 3 or 5-8); (4) interactions with second-shell ligands, such as π–cation interactions with imidazole or tyrosyl groups of His or Tyr, respectively; and (5) lower desolvation energy, which favors displacing water with new ligands and may also play a part in complex formation during the electrospray process. The general affinity of a divalent metal ion for ligands is correlated with the Irving-Williams series [16], which ranks the relative stability of the complexes as follows: Mg2+ < Mn2+ < Fe2+ < Co2+ < Ni2+ < Cu2+ > Zn2+.
In 2006, Choi et al. [6] studied the spectral, kinetic, and thermodynamic properties of Cu(I) binding to Mb using UV-Vis spectroscopy, fluorescence, circular dichroism, and isothermal titration calorimetry. The results indicated that the mechanism of the coordination of Cu(I/II) changes as a function of the molar equivalents (M eq ) of Cu to Mb. The study also presented the first possibility of Mb forming oligomers during its binding of Cu(I/II). In a further study [7], Mb was shown to bind other metal ions, and it was proposed that the metal ions were bound by two different pathways, which included tetrameric, dimeric, and monomeric Mb binding in a sequence that depended on the M eq of the metal ion availability.
These binding mechanisms were further explored by the present study using traveling wave ion mobility-mass spectrometry (TWIMS), which identified whether the formation of oligomers occurred during the metal ion titrations of Mb. Our group’s previous TWIMS study of Mb [17] included comparison of the arrival distributions of the different charge states of Mb and Mb-Cu(I) complexes with either a reduced or intact disulfide bond. Collision-induced dissociations of the reduced and intact Mb and Mb-Cu(I) complexes also explored the changes in the fragmentation pattern. The results showed that Mb and Mb-Cu(I) complexes existed in multiple conformational charge states and the disulfide bond did not play a key role in the binding of Cu(I) or Mb’s redox activity [17]. We use similar methods [18, 19] in the current study, and also develop our TWIMS titration methods for characterizing the metal ion binding as a function of pH [20,21,22], metal ion concentration [23], and conformational state [24, 25].
Experimental
Chemical Reagents
Freeze-dried methanobactin [3, 4, 7] from Methylosinus trichosporium OB3b was obtained using the one-step purification method from Dr. DongWon Choi, Texas A&M University Commerce–Department of Biology. MnCl2, CoCl2, NiCl2, CuCl2, Cu(NO3)2 99.99% or higher trace metal grade, DL-Dithiothreitol (≥99.0%), and poly-DL-alanine were purchased from Sigma-Aldrich (www.sigmaaldrich.com/). AgNO3, ZnCl2, FeCl3; ACS grade, 98%+ were purchased from Alfa Aesar (www.alfa.com/en/). PbCl2; ACS grade, was purchased from Mallinckrodt (currently Avantor, www.avantormaterials.com). Solutions were prepared using deionized (DI) water (>18.2 MΩ cm, MilliQ, Millipore). NH4OH, trace metal grade, CH3COOH, Optima grade, and CH3OH, Optima LC/MS grade, were purchased from Fisher Scientific (http://www.Fishersci.com).
Stock Solution Preparation
Methanobactin was prepared at 12.5 mM in DI H2O in 50 μL portions and frozen at –80 °C until analysis. Samples were thawed and diluted to a final concentration of 8.75 μM. The purity of the Mb sample was determined by the Cu(II) titration of Mb (Supplementary Figure S1) to be 35.0% and have a 1:1 binding stoichiometry. This purity correction was applied to all the titrations taking place shown in this paper. Metal ion stock solutions were prepared at 125 mM and diluted further as needed for each experiment.
Titrations and Collision-Induced Dissociation Experiments
Molar equivalent (M eq ) metal ion titrations were performed by preparing individual samples for each titration point. Each sample was made by first adding the Mb and then diluting to the appropriate volume, so after the addition of the metal ion solution the final volume was 1.00 mL. The pH metal titrations were analyzed with 1:1 M eq of Mb and metal ion (8.75 μM each) with ammonium hydroxide used to buffer the pH at specific points. The pH was measured on the calibrated Orion 98 micro pH electrode. Metal replacement titrations were also performed at 1:1 M eq with Cu(II) added for each prepared sample in increasing concentrations of 0.100 M eq . For the Ag(I) to Cu(I) metal replacement, Cu(NO3)2 was used to avoid AgCl precipitation. For all titrations, each sample was incubated at room temperature for 5 min before TWIMS analysis. Collision-induced dissociation experiments were performed on 1:1 M eq (8.75 μM) Mb:metal samples. After 5 min incubation, a 50-fold excess of DL-dithiothreitol was added to the sample (total volume of 1.00 mL) and incubated at 60 °C for 10 min to reduce the intramolecular disulfide bond of Mb.
Waters Synapt High-Definition Mass Spectrometer (HDMS)
The samples were analyzed using the Waters Synapt high-definition mass spectrometer (G1) equipped with an electrospray ionization (ESI) source in a quadrupole-ion mobility-orthogonal time-of-flight configuration [26]. The data were collected in negative ion mode, with some experiments collected in positive mode to observe if positive ions were formed (Supporting Information for detailed tune conditions, Supplementary Table S1). Samples were directly injected into the ESI source at a flow rate of 10 μL/min. Before injection, the inlet line was flushed with 10% NH4OH (in H2O, v/v) and 10% acetic acid (in H2O, v/v) to remove any metal ion and counter-ion contaminates.
Data Analysis of Metal Ion Binding
The TWIMS identified the arrival time and m/z for each Mb species, including any coincidental m/z species. The negatively charged species were identified based from their m/z and isotope patterns, and their arrival time distributions (ATD) were separated and extracted using Driftscope 2.1. MassLynx 4.1 software was used to determine the centroid of each ATD, for calculating the average collision cross-sections (CCS), and the area under the ATD curve to measure the proportions of the various Mb species. The summation of the integrated ATD for all extracted species at the titration point was used to normalize the relative intensities to the percent scale. The reproducibility of the titration results on the final percent scale were made by conducting three separate experiments on different days that revealed the standard deviations of individual titration points were between 1.3% and 7.9%.
Collision Cross-Sections
A calibration method was used to determine the collision cross-sections of the various Mb species using previously published collision cross-sections in helium (ΩHe) for negative poly-DL-alanine ions measured in a radio-frequency (rf) confining drift cell [27,28,29]. A study [30] of ΩHe measurements of human insulin using the Synapt G1 and G2 (which yields improved mobility resolution relative to the G1) with comparison to absolute ΩHe measurements made using a rf confining drift-tube, concluded accurate ΩHe measurements could be made using both G1 and G2 instruments. The method provided ΩHe for the Mb species with estimated 2% relative uncertainties and 4% absolute errors based on previous analyses of the calibration procedure with the Synapt instrument [27]. Details of the procedure are given in the Supporting Information and Supplementary Figure S2.
Computational Methods
The B3LYP/LanL2DZ level of theory containing the Becke three parameter hybrid functionals [31] with the Dunning basis set [32] and electron core potentials [33,34,35] from Gaussian 09 [36] was used to locate the lowest energy, geometry optimized, gas-phase structures of the [Mb-2H+Cu(I)]− and [Mb-3H+Zn(II)]− complexes. Their theoretical ΩHe were calculated using the ion size scaled Lennard-Jones (LJ) method from the Sigma program, which was developed and written by the Bowers group from the Department of Chemistry and Biochemistry at the University of California–Santa Barbara [37]. The LJ values are derived from the mean and standard deviation from 10 repeated calculations.
Breakdown Curves of Mb Complexes
The energetics relating to the stability of the complexes in the gas phase were determined via the 50% crossover point of the breakdown curves using a Thermo Finnigan (San Jose, CA, USA) LCQ mass spectrometer [19]. Helium was used as the collision gas and the instrument was tuned onto the complex ion by optimizing voltages and gas flows for the best sensitivity and resolution. Each spectrum was an average of 750 microscans of 50 ms each. During the MS/MS experiments, parent ions were isolated and CID spectra were taken over the 2%–3% normalized collision energy (NCE) range in intervals of 0.1% to produce breakdown graphs. The NCE scale was converted to the amplitude of resonance excitation (tickle amplitude) scale using the following equation: tickle amplitude = (NCE/30%) (parent ion mass × tickle amp slope + tick amp intercept) in which tickle amp slope and tick amp intercept are constants with the values 0.001347 V/u and 0.407147 V, respectively.
Fluorescence Quenching
The fluorescence quenching analysis was based on the relationship between the fluorescence intensity during the titration of Mb with the static quenching metal ion [38]. Details of the method are shown in the Supporting Information. Briefly, oxazolone-1 of Mb was the fluorophore with λex= 452 nm and λem= 525 nm, and the intensity of fluorescence was measured during the titration of Mb with the metal ion quencher. The binding constant K was determined using the relationship between the fluorescence intensity and the concentration of unbound Mb [38].
Results and Discussion
Characterization of the Mb Sample
The charge states of Mb observed during TWIMS analyses [22, 23] are closely related to the charge states expected to exist in the solution phase [17]. Mb is tetraprotic because it contains the C-terminus (pKa < 1.7) [39], two enethiol-oxazolone groups (pKa = 5.0 and 9.7) [39], and the Tyr group (pKa = 10.6) [39], which determines that both the Cu(I)-bound and Cu(I)-free Mb species only form negatively charged ions [17]. At pH 6.5 (Figure 1a), the Mb is observed primarily as [Mb–3H]3–, [Mb–2H]2– and [Mb–H]–, consistent with charge sates expected in solution. The other species present in the sample are Mb without the Met at the C-terminus (Mb-Met), which the methanotrophs synthesize as well as the full length Mb [11]. There are also Na+ and K+ adducts of Mb, some small fragment ions from [Mb–3H]3– and [Mb–2H]2–, and a siderophore species at 998 m/z, the structure of which is unknown (Supplementary Figure S3). The Mb-Cu(I) complex (Figure 1b) exists in two charge states, [Mb–2H+Cu(I)]– and [Mb–3H+Cu(I)]2–, consistent with Mb deprotonated at the two enethiolates, which are the binding sites of Cu(I), and deprotonation of the C-terminus for the doubly charged species. Only trace amounts of positively charged species were observed during the analyses, and these were mainly K+ or Na+ adducts.

(a) The identities and charge states of the Mb, Mb-Met, and siderophore in the Mb sample obtained by the one-step purification method at pH 6.5, and (b) after 0.571 molar equivalents of Cu(II) are added to the Mb sample. Peaks labeled * are Na+ and K+ adducts or fragment ions
Metal Ion Titrations
Individual metal ion titrations were performed to determine the effect of metal binding to free Mb. Figure 2a compares the formation of Mb–metal complex from all the metal ion titrations studied here. The comparison allows the metal ions to be grouped into three major categories based on the apparent binding selectivity of Mb: (1) Cu(I) and Ag(I); (2) Zn(II), Ni(II), and Co(II); and (3) Fe(II), Pb(II), and Mn(II). For group 1, the Mb binding for Cu(I) reached 100% at 1.0 M eq , and the Ag(I) complex reached 100% formed at ≥1.1 M eq . Replicate 1.0 M eq titration points had a standard deviation of 1.3%, which indicates the differences between Cu(I) and Ag(I) in the titration at 1.0 M eq are within the uncertainty of the results. Both Cu(I) and Ag(I) share equivalent valence electronic configurations, and preferred coordination numbers and binding geometries of distorted square planar or tetrahedral, which explains their binding similarity [40, 41]. The Shannon-Prewitt effective ionic radii [42] of four coordinate Cu(I) or Ag(I) are 60 pm and 100 pm, respectively, giving a charge density of Cu(I) > Ag(I).

(a) The percentage of formation of the Cu(I), Ag(I), Zn(II), Ni(II), Co(II), Mn(II), Pb(II), or Fe(II) complexes observed during the individual metal ion titrations of methanobactin. (b) The percentage of reactants and products observed during the Cu(II) titration of methanobactin. (c) The percentage of reactants and products observed during the Zn(II) titration of methanobactin. (d) The percentage of reactants and products observed during the Mn(II) titration of methanobactin
Comparison of Mb Complexes Formed During the Metal Ion Titrations
Each metal ion exhibited a unique metal binding trend that allowed for the interpretation of differences in binding properties. Figure 2b, c, d show how the relative percent of the individual reactants and products changed during the Cu(II), Zn(II), and Mn(II) titrations of Mb. At the beginning of the titrations, the three charge states of [Mb-H]−, [Mb-2H]2−, and [Mb-3H]3− were observed, which progressively converted into a range of Mb complexes as the titrations progressed. The Cu(II) titration (Figure 2b) resulted in a steep increase in the complex [Mb-3H+Cu(I)]2− and a more gradual increase of [Mb-2H+Cu(I)]−. There were no Mb oligomers or multiple Cu(I/II)-binding species observed in either negative or positive mode, indicating Mb only binds Cu(I) in a 1:1 stoichiometry. Previous works [7, 17, 39] have proposed the formation of oligomers as part of the reaction mechanisms of Mb-Cu(I/II) binding, but those conclusions are not supported here. Similar behavior for the formation of [Mb-3H+Ag(I)]2−and [Mb-2H+Ag(I)]− was observed for the Ag(I) titration of Mb (Supplementary Figure S4).
Group 2 included the Zn(II) complex which reached 96% bound, the Ni(II) complex which reached 89% bound, and Co(II), which reached 84% bound, all by 1.4 M eq , indicating the Mb selectivity for these metal ions follows the order Zn(II) > Ni(II)>Co(II) (Figure 2a). Figure 2c shows the Zn(II) titration of Mb resulted with the formation of [Mb-3H+Zn(II)]− and [Mb-4H+Zn(II)]2− both initially increasing together over 0.1 to 0.6 M eq with [Mb-3H+Zn(II)]− reaching 48% and [Mb-4H+Zn(II)]2− reaching 26% of the observed species. Above 0.5 M eq the two chloride adducts, [Mb-3H+Zn(II)+Cl]2−and [Mb-2H+Zn(II)+Cl]−, were also observed, with [Mb-3H+Zn(II)+Cl]2− increasing to 29% and corresponding to the chloride adduct of [Mb-3H+Zn(II)]−, showing the deprotonation of Mb at three sites resulted in the most prolific Zn(II) complexes. The most likely deprotonation sites of Mb are the two enethiolates, deprotonated for metal ion binding, and the carboxylate of the C-terminal because of its low pKa < 1.7 [39]. For the [Mb-4H+Zn(II)]2− complex, the fourth site could be the phenol group of Tyr (pKa 10.6) [39]. The lowest abundant product [Mb-2H+Zn(II)+Cl]− indicates a neutral charged complex [Mb-2H+Zn(II)] may also be formed during the titration with the Zn(II) replacing the two protons at the two enethiolate sites. Comparable formations of complexes were also observed during the Ni(II) and Co(II) titrations of Mb, although there were additional formations of the dimer, [2Mb-6H+2M(II)]2−, where M = Ni or Co (Supplementary Figure S4), which was resolved by IM from the isobaric monomer. The formation of the dimer followed the same relative increase as the isobaric monomer, [Mb-3H+M(II)]−, increasing with M eq to reach a maximum at >1.0 M eq , which suggests the formation of the two species are closely related.
The Mn(II), Pb(II), and Fe(II) of Group 3 where all <50% bound by 1.4 M eq , with Mn(II) 46% bound at 1.4 M eq , followed by Pb(II) at 45%, and Fe(II) at 27% (Figure 2a). Figure 2d shows the results of the Mn(II) titration of Mb over the extended range of 0.0 to 4.4 M eq . The three charged species of Mb are converted above 0.1 M eq into [Mb-4H+Mn(II)]2−, [Mb-3H+Mn(II)]−, and the dimer [2Mb-6H+2Mn(II)]−, and above 0.5 M eq into the chloride adducts [Mb-3H+Mn(II)+Cl]2−and [Mb-2H+Mn(II)+Cl]−. Three of the complexes had three deprotonation sites for each Mb, showing this was the most likely deprotonation state of the complex. Similar results were observed for the Fe(II) titration of Mb, which was also slightly affected by the sample containing the siderophore (also synthesized by the methanotrophs). By the first titration point of 0.29 M eq Fe(III), the siderophore was completely Fe(III) bound, with 2.5% of Mb also showing Fe(II) binding (Supplementary Figures S3 and S4). This indicates the amount of the siderophore in the sample was <2.5 nmol compared with the 8.75 nmol of Mb. However, the siderophore did not seem to influence the Fe(II) titration of Mb significantly because the titration steps that followed also exhibited relatively small increases in the percent of Mb bound to Fe(II) even though the siderophore was completely Fe(III) bound by the first 0.29 M eq Fe(III) addition. This also makes sense because the methanotrophs are synthesizing Mb to specifically bind Cu(I) and the siderophore to bind Fe(III), and, consequently, the Mb would have a low functionality for binding Fe(II). The Pb(II) titration of Mb (Supplementary Figure S4) produced the three complexes, [Mb-4H+Pb(II)]2−, [Mb-3H+Pb(II)]−, and [Mb-3H+Pb(II)+Cl]2−, but not the chloride adduct [Mb-2H+Pb(II)+Cl]− or the dimer [2Mb-6H+2Pb(II)]−. Based on these metal titration results, the apparent binding selectivity for Mb follows the order Ag(I) ≥ Cu(I) > Zn(II) ≥ Ni(II) ≥ Co(II) >> Mn(II) > Pb(II) > Fe(II). This order is similar to the transition metals ranked by the Irving-Williams series [16]: Cu(II) > Zn(II) ≥ Ni(II) > Co(II) > Fe(II) > Mn(II), with the exception of the switching order of Mn(II) and Fe(II) and the oxidation state of Cu(I/II).
Cu(II) Titrations of the Metal Ion Complexes and Replacement of the Metal Ion by Cu(I)
To determine how specifically Mb can act as a copper chelation therapy, a study was conducted to observe whether Cu(I) could efficiently displace one of the other metal ions from the Mb complex. Figure 3a shows the summary of these results and indicates that Ni(II) and Ag(I) were the most resistant for being displaced by Cu(I/II) during the 0.1 to 1.5 M eq Cu(II) titration. This suggests that the majority of the Ni(II) and Ag(I) ions were tightly bound to the same 2N-2S sites of Mb that Cu(I/II) binds and as a result Cu(I) could not effectively displace them. The Ni(II) shares the preferred coordination properties with Cu(I) and can coordinate via distorted square planar or tetrahedral geometries [43, 44], although the greater charge and smaller effective ionic radii of 55 pm for Ni(II) will result in a greater charge density than either of Cu(I) or Ag(I) [42]. The Co(II) was only partially resistant to displacement by Cu(I), with the complexes of Co(II) decreasing from 77% to 15% during the 0.1 to 1.5 M eq Cu(II) titration. Co(II) may also bind to the 2N-2S sites with a tetrahedral coordination [45], and has an effective ionic radius of 58 pm, which is similar to Cu(I), 60 pm, but because of the higher charge will result in a higher charge density [42]. The Cu(II) titration of the Mb complexes of Fe(II), Pb(II), and Mn(II) all resulted with the metal ions being displaced by Cu(I) by the 1.0 M eq point. However, this Cu(I) displacement indicates that these weakly bound metal ions are also dependent on the 2N-2S binding sites, otherwise complexes with both Cu(I) and the Fe(II), Pb(II) or Mn(II) would of expected to be observed, and this was not the case.

(a) The percentage of Cu(I), Ag(I), Zn(II), Ni(II), Co(II), Mn(II), Pb(II), or Fe(II) complexes during the titration of the complex with increasing molar equivalents of Cu(II). (b) The percentage of the reactants and products observed during the Cu(II) titration of 1:1 M eq solution of Ag(I) and methanobactin. (c) The percentage of the reactants and products observed during the Cu(II) titration of 1:1 M eq solution of Ni(II) and methanobactin. (d) The change in collision cross-section (CCS) as the [Mb-3H+Zn(II)]− complex was titrated with 0.1 to 1.5 molar equivalents of Cu(II). The CCS shows the conversion from the 237 Å2 of [Mb-3H+Zn(II)]− to the 230 Å2 of [Mb-2H+Cu(I)]−. The titrations determine how effectively Cu(I) can displace the metal ion from the Mb complex
Comparison of Mb Complexes Observed During the Cu(II) Metal Replacement Titrations
Figure 3b shows the individual species observed during the Cu(II) titration of the 1:1 M eq solution of Ag(I) and Mb, with [Mb-3H+Ag(I)]2− and [Mb-2H+Ag(I)]− decreasing by about 20% and 8%, and [Mb-3H+Cu(I)]2− and [Mb-2H+Cu(I)]− increasing by 25% and 3%, respectively. The results show the main change was Cu(I) displacing Ag(I) from the doubly charged [Mb-3H+Ag(I)]2− species. The reverse Ag(I) titration of the Mb-Cu(I) complex was also studied and resulted in approximately 50% of Cu(I) being replaced by Ag(I) by the 1.0 M eq stage, showing Mb has about equal selectivity for Ag(I) and Cu(I) (Supplementary Figure S5). During the Cu(II) titration of Ni(II) complexes, [Mb-4H+Ni(II)]2−, [Mb-3H+Ni(II)]−, and [2Mb-6H+2Ni(II)]2− all decreased moderately (Figure 3c). The main Cu(I) products were [Mb-3H+Cu(I)]2−, which increased to 27% by 1.5 M eq , and [Mb-2H+Cu(I)]−, which increased to 6%. The reverse Ni(II) titration of Mb-Cu(I) showed that Cu(I) was not displaced by Ni(II) by the 1.0 M eq stage, consistent with Mb’s higher selectivity for Cu(I) (Supplementary Figure S5). The principle Ag(I), Ni(II), and Cu(I) complexes observed in the Cu(II) titrations were all deprotonated at three Mb sites, e.g., [Mb-3H+Ag(I)]2−, [Mb-3H+Ni(II)]−, and [Mb-3H+Cu(I)]2−, which coincides with the two enethiolates and the C-terminus. Another major Ni(II) complex that increased over the titration (Figure 3c) was [Mb-3H+Ni(II)+Cl]2−, which related to the Cl− adduct of [Mb-3H+Ni(II)]−, along with the minor [Mb-2H+Ni(II)+Cl]− species, due the titrant CuCl2. Analogous complexes and Cl− adducts were also observed for the Cu(II) titration of the Co(II), Mn(II), Fe(II), and Pb(II) and the individual channels are shown in Supplementary Figure S6.
The Cu(II) titration of the Zn(II) complex provided a unique problem because of the overlapping isotope pattern for the [Mb-3H+Zn(II)]− and [Mb-2H+Cu(I)]− complexes (Supplementary Figure S7). Instead, the titration was monitored as a function of the change in the size of the collision cross-section (CCS) as the complex converted from the significantly larger CCS of [Mb-3H+Zn(II)]− (237.4 ± 0.9 Å2 as determined from 17 separate measurements, Supplementary Table S2) to the smaller CCS of [Mb-2H+Cu(I)]− (230.0 ± 2.0 Å2 as determined from 99 measurements). Figure 3d shows the result of the titration, which exhibits an almost linear conversion from the size of the [Mb-3H+Zn(II)]− to the size of the [Mb-2H+Cu(I)]− by the 1.2 M eq point of the titration, indicating that Zn(II) was displaced by Cu(I) more efficiently than either Ag(I) or Ni(II).
pH Titration of Metal Ion Complexes
The relative stability of the metal ion complexes were studied as a function of pH, and Figure 4a shows a summary of how the metal ion complex was affected by pH for all the titrations. The pH behavior indicates the same ordering of the Mb selectivity for these metal ions as determined from the metal ion titrations of Mb, e.g., (1) Ag(I), Cu(I); (2) Ni(II), Zn(II), Co(II); and (3) Mn(II) Pb(II), Fe(II). Group 1’s Ag(I) complex exhibited no pH dependence across the entire pH 6.5 to 10.5 range, remaining 100% Ag(I) bound, whereas Cu(I) showed 97% Cu(I) bound during pH 6.2 to pH 7.7, but then decreased to 91% at pH 8.8, and 87% at pH 10.4. The results indicate that Mb selectivity for Cu(I) decreased above pH 8 but the selectivity for Ag(I) was not affected. The two [Mb-3H+Cu(I)]2− and [Mb-2H+Cu(I)]− complexes decreased by 8% and 2% over the pH titration as shown in Figure 4b, whereas [Mb-3H]3− and [Mb-2H]2− increased by the same 8% and 2%, respectively. The results of the Ag(I) complex, shows [Mb-3H+Ag(I)]2− increased slightly as [Mb-2H+Ag(I)]− decreased (Supplementary Figure S8). Two previous studies [11, 39] that investigated the pH dependence on the binding constant of Mb and Cu(I/II) determined the binding constant increased as pH increased, when measured in ionic solutions of 0.01 to 0.1 M NaCl. The ionic strength of our solutions are lower and mainly influenced by the addition of the ammonium hydroxide < 2.5 × 10−4 M, and indicates the selectivity of Mb for Cu(I) is affected by competition from unreactive or insoluble metal hydroxides.

(a) The percentage of Cu(I), Ag(I), Zn(II), Ni(II), Co(II), Mn(II), Pb(II), or Fe(II) complexes formed during the pH titration of 1:1 M eq solution of metal ion and methanobactin. (b) The percentage of reactants and products observed during the pH titration of 1:1 M eq solution of Cu(I) and methanobactin. (c) The percentage of reactants and products observed during the pH titration of 1:1 M eq solution of Zn(II) and methanobactin. (d) The percentage of reactants and products observed during the pH titration of 1:1 M eq solution of Pb(II) and methanobactin
The Ni(II), Zn(II), and Co(II) complexes of Group 2 were all adversely affected by pH (Figure 4a). The most resilient was the Ni(II) complex that decreased only moderately from 93% to 86% from pH 6.4 to pH 8.8, but then decreased sharply to 35% by pH 10.3. The Co(II) complex was also relatively stable over pH 6.3 to pH 7.5 only decreasing from 81% to 78%, but then decreased to 29% by pH 10.3. The Zn(II) complexes was the most adversely affected, decreasing from 90% formation at pH 6.4, to only 9% by pH 10.4. The decrease of the individual Zn(II) complexes over the duration of the titration are shown in Figure 4c, with [Mb-3H+Zn(II)]−, [Mb-4H+Zn(II)]2−, and [Mb-3H+Zn(II)+Cl]2−decreasing 38%, 21%, and 17%, respectively, with corresponding increases for the three Mb charge states, chiefly [Mb-3H]3−. Initially, the [Mb-4H+Zn(II)]2− complex showed a small 3% increase over pH 6.5 to pH 7.7 before decreasing. The pH behaviors of the Ni(II) and Co(II) complexes (Supplementary Figure S8) were comparable to Zn(II) although both exhibited the dimer, [2Mb-6H+2M(II)]2−, and greater increases in the formation of [Mb-4H+M(II)]2− before it decreased with the other complexes.
Group 3 complexes of Fe(II), Pb(II), and Mn(II) exhibited <45% formation at the lowest pH, and all decreased to 0% formation (Figure 4a). For example, the Mn(II) complex was 44% at pH 6.5 but decreased almost linearly to only free-Mb by pH 10.4 (see Supplementary Figure S8 for the individual channels). Similarly, the Pb(II) complex was 33% at pH 6.4 but then decreased to only free-Mb by pH 10.4 (Figure 4a). The individual channels for Pb(II) (Figure 4d) show the two complexes [Mb-3H+Pb(II)]− and [Mb-4H+Pb(II)]2− decrease from 28% and 12% over the duration of the pH titration to 0%, with [Mb-3H]3− increasing significantly from 23% to 58%. The Fe(II) complex indicated the lowest percent formation over the pH titration, exhibiting 22% at pH 5.7, which decreased to only 1% by pH 7.6 and only free-Mb by pH 9.0 (see Supplementary Figure S8 for the individual channels). The first point in each of the pH titrations (Figure 4a) was conducted in just DI water and replicates the 1:1 M eq point of the metal ion titration (Figure 2a), and the first point in the Cu(II) replacement titration (Figure 3a). Comparison of these data points indicates that within the reproducibility of the experiment, the selectivity of Mb for the pairs Ag(I) and Cu(I), Ni(II) and Zn(II), Pb(II) and Mn(II) could not be distinguished at pH ~6.5 and the Mb selectivity order is: Ag(I) ≈ Cu(I) > Ni(II) ≈ Zn(II) > Co(II) >> Mn(II) ≈ Pb(II) > Fe(II). However, during pH 7.5 to 10.4, the Mb selectivity order changed to: Ag(I) > Cu(I) > Ni(II) > Co(II) > Zn(II) > Mn(II) ≈ Pb(II)>Fe(II).
Molecular Modeling
Starting with the crystal structure of Mb-Cu(I) [11], a survey of gas-phase structures of [Mb-2H+Cu(I)]− and [Mb-3H+Zn(II)]− was conducted using the B3LYP/LanL2DZ level of theory. The search located two alternative gas-phase structures (Scheme 2a, b) that bound the metal ion via the 2N-2S enethiolate-oxazolone sites but differed because [Mb-3H+Zn(II)]− also exhibited an extra metal binding interaction with the deprotonated carboxylate group of the C-terminal (Scheme 2b). This interaction was predicted by B3LYP/LanL2DZ method to lower both the energy of the complex by 80 kJ/mol and the CCS of the complex by 8 Å2 compared with the Zn(II) complex coordinated by only the two enethiol-oxazolones. The theoretical CCS for the [Mb-2H+Cu(I)]− (2a) and [Mb-3H+Zn(II)]− (2b) are 252 ± 3 Å2 and 245 ± 3 Å2, respectively. It is possible that other arrangements of the substituent groups could relate to new minima structures not found by this study. Chemical dynamics simulations that couple classic and electronic structure theory could further sample the conformational space of these complexes [46].
Collision Cross-Sections and Breakdown Curves of Methanobactin Complexes
Monitoring the CCS of the Mb complexes allows for the comparison of the metal’s binding coordination to the overall size of the complex. Figure 5a compares the average CCS of singly- and doubly charged species determined from replicate measurements, with the complete set of CCS shown in Supplementary Tables S2 and S3. Based on average and standard deviation of these measurements, the metal-free Mb exhibited a 17 Å2 change in CCS between the single charged [Mb–H]– (227 ± 1 Å2) and doubly charged [Mb–2H]2– (244 ± 4 Å2), indicating significant unfolding of the [Mb–2H]2– structure. Considering the pKas of Mb, the unfolding is probably due to Columbic repulsion between the carboxylate of the C-terminal and the enethiolate-oxazolone-1 located near the modified N-terminus of Mb [39]. The removal of aqueous solvent that normally solvates these charge sites is likely to accentuate the unfolding due to the charge repulsion. Most of the singly charged Mb complexes have larger CCS than [Mb–H]– and do not exhibit significant CCS increases from the singly to doubly charged complex because the enethiolates and possibly the carboxylate are coordinated to the metal ion, which restrains the unfolding. The exception was the Cu(I) complex, the doubly charged species of which was 4 Å2 larger than the singly charged species and coincides with the deprotonation of the C-terminus.

(a) Comparison of the collision cross-sections for the single and double negatively charged Mb and Mb complexes. (b) The crossover points from the breakdown curves of the singly charged disulfide reduced Mb complexes as a function of the tickle amplitude used to dissociate the complexes
The singly charged Cu(I) and Ag(I) complexes, 230 ± 2 Å2 and 232 ± 1 Å2, respectively, were significantly smaller than the metal(II) complexes, which ranged from 237 ± 1 Å2 to 242 ± 1 Å2. For an overall single-charge, the metal(I) or metal(II) complexes must have two or three negative charge sites on Mb, including the two enethiolate-oxazolone sites, where the metal ion is bound, and the carboxylate of the C-terminal. Two electrostatic interactions the carboxylate could form are either with the metal ion, as shown in Scheme 2b, or hydrogen bond with hydroxyl group of Tyr4. The doubly charged Mb complexes exhibit significantly smaller CCSs than [Mb–2H]2–, with the exception of Fe(II) and Mn(II). The CCS for [Mb–3H+Cu(I)]2– (234 ± 1 Å2) and [Mb–3H+Ag(I)]2– (232 ± 1 Å2) are generally smaller than the metal(II) complexes range of 237 ± 1 Å2 to 242 ± 2 Å2, with the exception of [Mb–4H+Pb(II)]2– (235 ± 1 Å2). The smaller CCS of the Ag(I) and Cu(I) complexes indicate they have the most tightly coordinated structures, which correlates to the higher Mb selectivity for these two metal ions as observed from the metal and pH titrations.
A breakdown study was conducted to observe whether greater relative stability of the complex, as measured by a higher crossover point, were proportional to the Mb selectivity for the metal ions, with consideration that these are gas-phase measurements and may not necessarily represent the stability of the complex in solution. Figure 5b compares the 50% crossover point exhibited by the breakdown curves from CID of the disulfide reduced Mb complexes (Supplementary Figure S9). Both Cu(I) and Ag(I) complexes exhibited the highest crossover points, 1.49 and 1.43 V, respectively, which coincides with their high selectivity by Mb. There is also a correlation between the relative stability, measured as a function of the crossover point, and the CCS of the Cu(I) and Ag(I) complexes, which indicated stability derived from their compact folded structures. The Mn(II) and Fe(II) complexes exhibited the next highest crossover points of 1.41 and 1.42 V, respectively, indicating they have a higher stability than the Zn(II), Ni(II), and Co(II) complexes, which exhibited crossover points of 1.33, 1.38, and 1.36 V, respectively. These latter crossover points do not coincide with Mb’s apparent higher selectivity for Zn(II), Ni(II), and Co(II) over Mn(II) and Fe(II). However, as we will see in the next section, the Mn(II) and Fe(II) complexes exhibited the fewest products ions, including metal-bound products from the CID, which may have contributed to their apparent high crossover points. The Pb(II) complex exhibited the second lowest crossover point of 1.36 V with the Co(II) complex, which coincides with Mb’s low binding selectivity for Pb(II).
Collision-Induced Dissociation
The CID patterns of the singly charged Mb complexes were compared to determine whether they would give information of the Mb binding specificity or the binding sites of the metal ion. The disulfide bridge of the Mb complex was reduced before CID to aid in identifying sequence ions [17]. The product channels identified from each Mb complex are shown in Supplementary Table S4, and the labeled mass spectra of each CID are shown in Supplementary Figures S10 to S17. Similar product ions were observed from the CID of doubly charged complexes (data not shown). The results reveal that the Mb complexes produced a wide range of metal-bound fragments that were identifiable to contain both a, b, c-type and x, y, z-type sequences. The smallest metal bound a, b, c-type fragment was b5+M, which was observed for Ag(I) and Cu(I) complexes, contained the enethiol-oxazolone-1 site and the reduced Cys3 thiol site (Scheme 1b). If both (ene)thiol sites are deprotonated, the overall charge of the b5+Ag(I) or b5+Cu(I) would be a single negative charge, as observed; however, if the metal(II) ion was attached to the sequence, the overall charge would be neutral and not detected by the TWIMS analyses. The larger b6 ion was observed from the CID of Ni(II) and Co(II) complexes and b6+M and/or b6+M-H2S from the CID of Cu(I), Ag(I), Zn(II), Ni(II), and Co(II) complexes. The b6 ion included the Tyr4 residue which may of been deprotonated to give another negative charge to make it possible to detect the b6+M fragment with either Zn(II), Ni(II), or Co(II). The sites of thiolate at Cys3, phenolate at Tyr4, and enethiolate-oxazolone-1 may also be binding sites for the metal ion. The b6+M-H2S includes the loss of the sulfur from Cys [47], leaving the enethiolate-oxazolone-1 and phenolate to bind the metal(II) ion. Whether the phenolate was an original binding site or became a surrogate site during CID rearrangement, is a question that chemical dynamic simulations may be able to answer [46]. A series of metal-free a7 ions, accompanied by additional H2O and/or H2S loss, were observed for the Pb(II) complex. The a7+M was observed from all the complexes, whereas the a7+M-H2S was also observed from all metal(II) complexes except Mn(II), which exhibited a7+M-H2CO instead. The a7+M ion corresponds to the dissociation of the bond between the enethiol and oxazolone-2 moieties and was the principal product for the Cu(I) and Ag(I) complexes. The Cu(I) and Ag(I) complexes did not exhibit a7+M-H2S that suggests the stability of the a7+M fragment was dependent of the thiolate site of Cys3, which could become a binding site for Ag(I) or Cu(I) after reduction of the disulfide bond or during CID rearrangement. The metal(II) complexes exhibited both a7+M and a7+M-H2S, which indicates that either the metal(II) ion promoted loss of H2S from Cys3 and/or Tyr4 provided a binding site for the metal(II) ion. The exception was the Mn(II) complex, which exhibited a7+M-H2CO instead of a7+M-H2S, which corresponds to loss of H2CO from the Ser2 side group [48, 49].
The smallest x, y, z-type fragments were the y2+M exhibited by Zn(II), Ni(II), Co(II), and Mn(II), and the x2+M exhibited by Ni(II) and Co(II) complexes. The potential binding sites in the y2+M fragment include the carboxylate of the C-terminal, thioether of Met7, thiolate of Cys6, and possibly NH− at the dissociation site, which gives three negative charges and overall negative charge to the y2+M(II) complexes. The x2+M ion contains the additional carbonyl group at the dissociation site and suggests that the formation of an oxazolone ring is possible during CID rearrangement, although this may restrict the flexibility for the binding sites discussed above to coordinate Ni(II) or Co(II). Another possibility is that the metal ion was anchored to either the thioester or C-terminus and includes binding to two sequentially deprotonated N along the fragments backbone [44]. The y2+M and x2+M do not contain either of the two enethiol-oxazolone sites, which suggests that the Zn(II), Ni(II), Co(II), and Mn(II) were binding to the C-terminus, as located by the B3LYP modeling of the [Mb-3H+Zn(II)]− complex (Scheme 2b).
The next sized fragments x3+M-H2CO and x3+M were CID products for all the complexes, except Ag(I) and Pb(II), and the x3+M-H2CO was the main product ion for Zn(II), Co(II), and Mn(II) complexes. The x3+M-H2CO ion includes the Ser5 residue and its side group is the source of the H2CO loss [48, 49]. The x3+M-H2CO also contains the metal binding enethiolate group from enethiolate-oxazolone-2 with the other potential binding sites being the C-terminus, thioether, thiolate, or deprotonated N from the backbone. The y4+M, which is produced by dissociation of the peptide bond between pyrrolidine and Tyr4, was another common product ion from all the complexes, except Pb(II), with the y4+M-H2S ion also present. The y7+M-H and a variety of y8+M ions, with additional losses of H2S and CO, were also products from all complexes except from the weakest bound metals of Mn(II), Fe(II), and Pb(II). The Ag(I) and Cu(I) complexes produced the x8+M ion and the metal(II) complexes produced x8+M-H, by dissociation of the bond between the enethiolate and oxazolone-1 moieties, indicating all metal ions studied here will bind to this enethiolate site.
The CID of the Cu(I) and Ag(I) complexes produced exactly the same product ions, except for one channel (x3+M-H2CO), and were almost identical in their CID spectra (Supplementary Figures S10 and S11). The Zn(II) complex exhibited eight of the same channels as the Cu(I) and Ag(I) complexes but differed by producing more channels with H2S loss. The Ni(II) complex dissociated into the highest number of identifiable product ions and yielded the most product ions that retained the metal ion, indicating Ni(II) was binding to a greater variety of sites than the other metal ions. The Co(II) complex exhibited the second highest number of identifiable product ions and its CID spectra produced many of the same product ions as the Ni(II) complex, although its primary product ion was x3+M-H2CO, and it did not exhibit y8+M-CO that was the main product for Ni(II). The CID of Mn(II), Fe(II), and Pb(II) complexes resulted in the lowest number of identifiable products and the lowest number of metal-bound fragments, which coincides with these metal ions having the lowest apparent Mb selectivity. Of these metal ions, Pb(II) was the most labile metal ion and produced more Pb(II)-free than Pb(II)-bound product ions.
The two most common metal-bound product ions from all complexes, a7+M and x3+M-H2CO, were products related by the dissociation of the double bond between the enethiol and oxazolone-2 moieties. The Mb has the highest selectivity for Ag(I) and Cu(I), which were predominantly retained by a7+M that contained the oxazolone-2 and enethiol-oxazolone-1 groups. The singly charged Ag(I) and Cu(I) complexes were primarily deprotonated at only the two enethiol sites (Scheme 2a) and, therefore, had no strong interaction between the metal(I) ion and C-terminus, which limited the formation of the x3+M-H2CO. The Zn(II), Co(II), and Mn(II) complexes, however, included the deprotonated carboxylate, which coincided with an increased interaction between the metal ion and the C-terminus (Scheme 2b) and the formation of the x3+M-H2CO ion. This behavior suggests that CID can produce information related to the Mb binding selectivity, but also substantial rearrangement of the initial conformation can occur. In the future, direct dynamic simulations may shed further light on the rearrangement mechanisms implied by these results [50, 51].
Comparison of Apparent Selectivity Determined by TWIMS and Fluorescence Quenching
To compare gas-phase with solution-phase results, the binding constants for the selected metal ion complexes were estimated using a fluorescence quenching model that has been previously applied to the binding of TiO2 to DNA [38] and flavonoids to bovine serum albumin [52]. Fluorescence quenching was observed during the titrations of Mb with all the metal ions studied here except Zn(II), which exhibited an increase in fluorescence. The results and model fits are shown in Supplementary Figures S19 and S20. The binding constants determined by fluorescence quenching analyses are compared with the binding selectivity order determined by the TWIMS analyses and the one-site and two-site binding constants previously measured by Choi et al. using isothermal titration calorimetry (ITC) in Table 1.
There is reasonable agreement between the binding selectivity determined by TWIMS and the binding constants determined by the fluorescence quenching with the exception of Ni(II) and the switching of the order of Fe(II) and Pb(II). The fluorescence quenching fit of the Ni(II) titration of Mb gave the lowest R2 = 0.845 value and the highest n = 2.01 value, which relates to the stoichiometry of the metal ion binding to Mb (Supplementary Figure S20c). Removing the first two data points by considering them outliers gave R2 = 0.917 and decreased n = 1.27 and the binding constant to 5.3 × 104 (Supplementary Figure S20d). Considering the values of 6.2 × 107 and 5.3 ×104 as upper and lower limits for the Ni(II) binding constant of Mb is more consistent with the order determined by the TWIMS analyses. Choi et al. [6, 7] published binding constants based on one-site and two-site binding models fitting the ITC curves. Their interpretation was the Mb monomer bound Cu(I) in the one-site model, whereas, the two-site model resulted in the Mb dimer binding Cu(I). Comparison of the binding constants from the one-site model with the fluorescence values show good agreement for Cu(I), but an order of magnitude difference for Co(II), Mn(II), and Pb(II). The first binding constant for the two-site model of Ag(I) and Zn(II) are also an order of magnitude greater, and for Cu(I) approximately100 times greater than the fluorescence binding constants. For Ni(II) and Fe(II) the second binding constants of 1.2 × 107 and 1.7 × 105 from the two-site model are in better agreement with the fluorescence values than the first binding constants, which coincides with the TWIMS analyses observing a second metal ion binding by the Mb dimer for these metal ions. Overall, these ITC values, which are shown bold in Table 1, follow the same order as the TWIMS binding order and are also in reasonable agreement with the binding constants from the fluorescence quenching study.
Conclusions
This study expanded on the research of Choi et al.[7] and our previous work on the TWIMS analysis of Mb.[17] The current study was able to determine the reaction products from the various titrations of Mb with each of the selected metal ions. The TWIMS binding selectivity order compared reasonably well with the order determined by fluorescence quenching and those determined by Choi et al.,[7] with the main exception of Ni(II). A major difference was that Choi et al. categorized the binding of the metal ions into two groups: (1) Ag(I) and Pb(II) that were bound by Mb by a mechanism similar to Cu(I/II) and included reduction of the metal ion; and (2) Mn(II), Fe(III), Co(II), Ni(II), and Zn(II) proposed to bind through a mechanism where the metal was not reduced. However, the TWIMS analyses showed Fe(III) and Cu(II) were both reduced on binding with Mb but not Ag(I) or Pb(II). Moreover, both binding models were proposed to go through a sequence of Mb tetramer to Mb dimer binding of the metal ion at <0.5 M eq . At > 0.5 M eq , group 1 metals were bound by the Mb monomer, whereas group 2 metals were bound by the Mb dimer. These mechanisms are not supported here because only the Mb complexes that were monomers of Mb were detected at <0.5 M eq to bind the metal ion. At >0.5 M eq the Mb complexes of Mn(II), Fe(II), Co(II), and Ni(II) did include the formation of the Mb dimers but these bound two metal ions, as opposed to one as the Choi model predicted, and these species increased with M eq and reached their highest formation at >1.0 M eq . These dimers with two metal ions were isobaric with the more abundant monomers with one metal ion, and the two species behaved similarly in their formation, indicating the dimer and the monomer were related by either shared coordination of the two metal ions or an intermolecular hydrogen bond between the carboxylate of the C-terminus and the hydroxyl group of Tyr4.
The competition from the selected metal ions for the Cu(I) binding site of Mb was mainly from Ag(I) that was able to displace Cu(I) from the complex, and Ag(I) and Ni(II) that could irreversibly bind to Mb and not be effectively displaced by Cu(I). Exposure to soluble forms of silver have been reported to result in kidney and liver damage, bluish-gray discoloration of the skin (argyria) or eyes (argyrosis), irritation of the respiratory and intestinal tract, and changes in blood cells [53]. The work here suggests that at least some of these effects are related to Ag(I) being able to displace Cu(I) from active sites of proteins. Copper(I/II) is a crucial trace element in many redox-active enzymes, such as those involved in mitochondrial respiration (cytochrome oxidase), oxidative protection (Zn,Cu-super-oxide dismutase), collagen cross-linking (lysil oxidase), pigmentation (tyrosinase), and biosynthesis of cathecolamine (dopamine-b-monooxygenase) [13, 54]. Free copper ions can also produce reactive oxygen species under aerobic conditions, when reducing equivalents are available [14, 55]. As a consequence, organisms have developed tightly regulated homeostasis mechanisms to ensure safe copper uptake, distribution, and storage, including chaperone proteins that deliver copper safely to essential cellular sites [56]. The displacement of copper from enzymes or disruption of copper homeostasis by silver(I) or nickel(II) may have direct implications for disease.
References
Balasubramanian, R., Rosenzweig, A.C.: Copper methanobactin: a molecule whose time has come. Curr. Opin. Chem. Biol. 12, 245–249 (2008)
Behling, L.A., Hartsel, S.C., Lewis, D.E., DiSpirito, A.A., Choi, D.W., Masterson, L.R., Veglia, G., Gallagher, W.H.: NMR, mass spectrometry and chemical evidence reveal a different chemical structure for methanobactin that contains oxazolone rings. J. Am. Chem. Soc. 130, 12604–12605 (2008)
Bandow, N.L., Gallagher, W.H., Behling, L., Choi, D.W., Semrau, J.D., Hartsel, S.C., Gilles, V.S., Dispirito, A.A.: Isolation of methanobactin from the spent media of methane-oxidizing bacteria. Methods Enzymol. 495, 259–269 (2011)
Choi, D.W., Bandow, N.L., McEllistrem, M.T., Semrau, J.D., Antholine, W.E., Hartsel, S.C., Gallagher, W., Zea, C.J., Pohl, N.L., Zahn, J.A., DiSpirito, A.A.: Spectral and thermodynamic properties of methanobactin from γ-proteobacterial methane oxidizing bacteria: a case for copper competition on a molecular level. J. Inorg. Biochem. 104, 1240–1247 (2010)
Choi, D.W., Semrau, J.D., Antholine, W.E., Hartsel, S.C., Anderson, R.C., Carey, J.N., Dreis, A.M., Kenseth, E.M., Renstrom, J.M., Scardino, L.L., Van Gorden, G.S., Volkert, A.A., Wingad, A.D., Yanzer, P.J., McEllistrem, M.T., de la Mora, A.M., DiSpirito, A.A.: Oxidase, superoxide dismutase, and hydrogen peroxide reductase activities of methanobactin from types I and II methanotrophs. J. Inorg. Biochem. 102, 1571–1580 (2008)
Choi, D.W., Zea, C.J., Do, Y.S., Semrau, J.D., Antholine, W.E., Hargrove, M.S., Pohl, N.L., Boyd, E.S., Geesey, G.G., Hartsel, S.C., Shafe, P.H., McEllistrem, M.T., Kisting, C.J., Campbell, D., Rao, V., de la Mora, A.M., Dispirito, A.A.: Spectral, kinetic, and thermodynamic properties of Cu(I) and Cu(II) binding by methanobactin from Methylosinus trichosporium OB3b. Biochemistry. 45, 1442–1453 (2006)
Choi, D.W., Do, Y.S., Zea, C.J., McEllistrem, M.T., Lee, S.W., Semrau, J.D., Pohl, N.L., Kisting, C.J., Scardino, L.L., Hartsel, S.C., Boyd, E.S., Geesey, G.G., Riedel, T.P., Shafe, P.H., Kranski, K.A., Tritsch, J.R., Antholine, W.E., DiSpirito, A.A.: Spectral and thermodynamic properties of Ag(I), Au(III), Cd(II), Co(II), Fe(III), Hg(II), Mn(II), Ni(II), Pb(II), U(IV), and Zn(II) binding by methanobactin from Methylosinus trichosporium OB3b. J. Inorg. Biochem. 100, 2150–2161 (2006)
Bandow, N., Gilles, V.S., Freesmeier, B., Semrau, J.D., Krentz, B., Gallagher, W., McEllistrem, M.T., Hartsel, S.C., Choi, D.W., Hargrove, M.S., Heard, T.M., Chesner, L.N., Braunreiter, K.M., Cao, B.V., Gavitt, M.M., Hoopes, J.Z., Johnson, J.M., Polster, E.M., Schoenick, B.D., Umlauf, A.M., Dispirito, A.A.: Spectral and copper binding properties of methanobactin from the facultative methanotroph Methylocystis strain SB2. J. Inorg. Biochem. 110, 72–82 (2012)
Choi, D.W., Antholine, W.E., Do, Y.S., Semrau, J.D., Kisting, C.J., Kunz, R.C., Campbell, D., Rao, V., Hartsel, S.C., DiSpirito, A.A.: Effect of methanobactin on the activity and electron paramagnetic resonance spectra of the membrane-associated methane monooxygenase in Methylococcus capsulatus Bath. Microbiology. 151, 3417–3426 (2005)
Kim, H.J., Graham, D.W., DiSpirito, A.A., Alterman, M.A., Galeva, N., Larive, C.K., Asunskis, D., Sherwood, P.M.: Methanobactin, a copper-acquisition compound from methane-oxidizing bacteria. Science. 305, 1612–1615 (2004)
El Ghazouani, A., Basle, A., Firbank, S.J., Knapp, C.W., Gray, J., Graham, D.W., Dennison, C.: Copper-binding properties and structures of methanobactins from Methylosinus trichosporium OB3b. Inorg. Chem. 50, 1378–1391 (2011)
Lichtmannegger, J., Leitzinger, C., Wimmer, R., Schmitt, S., Schulz, S., Kabiri, Y., Eberhagen, C., Rieder, T., Janik, D., Neff, F., Straub, B.K., Schirmacher, P., DiSpirito, A.A., Bandow, N., Baral, B.S., Flatley, A., Kremmer, E., Denk, G., Reiter, F.P., Hohenester, S., Eckardt-Schupp, F., Dencher, N.A., Adamski, J., Sauer, V., Niemietz, C., Schmidt, H.H.J., Merle, U., Gotthardt, D.N., Kroemer, G., Weiss, K.H., Zischka, H.: Methanobactin reverses acute liver failure in a rat model of Wilson disease. J. Clin. Invest. 126, 2721–2735 (2016)
Tisato, F., Marzano, C., Porchia, M., Pellei, M., Santini, C.: Copper in diseases and treatments, and copper-based anticancer strategies. Med. Res. Rev. 30, 708–749 (2010)
Summer, K.H., Lichtmannegger, J., Bandow, N., Choi, D.W., DiSpirito, A.A., Michalke, B.: The biogenic methanobactin is an effective chelator for copper in a rat model for Wilson disease. J. Trace Elem. Med. Biol. 25, 36–41 (2011)
Dudev, T., Lim, C.: Competition among metal ions for protein binding sites: determinants of metal ion selectivity in proteins. Chem. Rev. 114, 538–556 (2014)
Irving, H., Williams, R.J.P.: Order of stability of metal complexes. Nature (London). 162, 746–747 (1948)
Choi, D., Sesham, R., Kim, Y., Angel, L.A.: Analysis of methanobactin from Methylosinus trichosporium OB3b via ion mobility mass spectrometry. Eur. J. Mass Spectrom. (Chichester, Eng.) 18, 509–520 (2012)
Raja, U.K.B., Injeti, S., Culver, T., McCabe, J.W., Angel, L.A.: Probing the stability of insulin oligomers using electrospray ionization ion mobility mass spectrometry. Eur. J. Mass Spectrom. 21, 759–774 (2015)
Utley, B., Angel, L.A.: Effects of transition metal ion identity and π–cation interactions in metal-bis(peptide) complexes containing phenylalanine. Eur. J. Mass Spectrom. 16, 631–643 (2010)
Vytla, Y., Angel, L.A.: Applying Ion mobility-mass spectrometry techniques for explicitly identifying the products of Cu(II) reactions of 2His-2Cys motif peptides. Anal. Chem. (Washington DC). 88, 10925–10932 (2016)
Wagoner, S.M., Deeconda, M., Cumpian, K.L., Ortiz, R., Chinthala, S., Angel, L.A.: The multiple conformational charge states of zinc(II) coordination by 2His-2Cys oligopeptide investigated by ion mobility-mass spectrometry, density functional theory, and theoretical collision cross-sections. J. Mass Spectrom. 51, 1120–1129 (2016)
Sesham, R., Choi, D., Balaji, A., Cheruku, S., Ravichetti, C., Alshahrani, A.A., Nasani, M., Angel, L.A.: The pH dependent Cu(II) and Zn(II) binding behavior of an analog methanobactin peptide. Eur. J. Mass Spectrom. 19, 463–473 (2013)
Choi, D., Alshahrani, A.A., Vytla, Y., Deeconda, M., Serna, V.J., Saenz, R.F., Angel, L.A.: Redox activity and multiple copper(I) coordination of 2His-2Cys oligopeptide. J. Mass Spectrom. 50, 316–325 (2015)
Angel, L.A.: Study of metal ion labeling of the conformational and charge states of lysozyme by ion mobility mass spectrometry. Eur. J.Mass Spectrom. 17, 207–215 (2011)
Giganti, V.G., Kundoor, S., Best, W.A., Angel, L.A.: Ion mobility-mass spectrometry study of folded ubiquitin conformers induced by treatment with cis-Pd(en)(H2O)22+. J. Am. Soc. Mass Spectrom. 22, 300–309 (2011)
Pringle, S.D., Giles, K., Wildgoose, J.L., Williams, J.P., Slade, S.E., Thalassinos, K., Bateman, R.H., Bowers, M.T., Scrivens, J.H.: An investigation of the mobility separation of some peptide and protein ions using a new hybrid quadrupole/travelling wave IMS/oa-ToF instrument. Int. J. Mass Spectrom. 261, 1–12 (2007)
Bush, M.F., Campuzano, I.D.G., Robinson, C.V.: Ion mobility mass spectrometry of peptide ions: effects of drift gas and calibration strategies. Anal. Chem. 84, 7124–7130 (2012)
Allen, S.J., Giles, K., Gilbert, T., Bush, M.F.: Ion mobility mass spectrometry of peptide, protein, and protein complex ions using a radio-frequency confining drift cell. Analyst. 141, 884–891 (2016)
Forsythe, J.G., Petrov, A.S., Walker, C.A., Allen, S.J., Pellissier, J.S., Bush, M.F., Hud, N.V., Fernandez, F.M.: Collision cross-section calibrants for negative ion mode traveling wave ion mobility-mass spectrometry. Analyst. 14, 6853–6861 (2015)
Salbo, R., Bush, M.F., Naver, H., Campuzano, I., Robinson, C.V., Pettersson, I., Jorgensen, T.J.D., Haselmann, K.F.: Traveling-wave ion mobility mass spectrometry of protein complexes: accurate calibrated collision cross-sections of human insulin oligomers. Rapid Commun. Mass Spectrom. 26, 1181–1193 (2012)
Becke, A.D.: Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993)
Dunning Jr., T.H., Hay, P.J.: Gaussian basis sets for molecular calculations. Mod. Theor. Chem. 3, 1–7 (1977)
Hay, P.J., Wadt, W.R.: Ab initio effective core potentials for molecular calculations. Potentials for potassium to gold including the outermost core orbitals. J. Chem. Phys. 82, 299–310 (1985)
Hay, P.J., Wadt, W.R.: Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms scandium to mercury. J. Chem. Phys. 82, 270–283 (1985)
Wadt, W.R., Hay, P.J.: Ab initio effective core potentials for molecular calculations. Potentials for main group elements sodium to bismuth. J. Chem. Phys. 82, 284–298 (1985)
Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H.P., Izmaylov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery Jr., J.A., Peralta, J.E., Ogliaro, F., Bearpark, M., Heyd, J.J., Brothers, E., Kudin, K.N., Staroverov, V.N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Rega, N., Millam, J.M., Klene, M., Knox, J.E., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Martin, R.L., Morokuma, K., Zakrzewski, V.G., Voth, G.A., Salvador, P., Dannenberg, J.J., Dapprich, S., Daniels, A.D., Farkas, Ö., Foresman, J.B., Ortiz, J.V., Cioslowski, J., Fox, D.J.: Gaussian 09, Revision C.01. Gaussian Inc., Wallingford (2012)
Wyttenbach, T., von Helden, G., Batka Jr., J.J., Carlat, D., Bowers, M.T.: Effect of the long-range potential on ion mobility measurements. J. Am. Soc. Mass Spectrom. 8, 275–282 (1997)
Kathiravan, A., Renganathan, R.: Photoinduced interactions between colloidal TiO2 nanoparticles and calf thymus-DNA. Polyhedron. 28, 1374–1378 (2009)
Pesch, M.L., Christl, I., Hoffmann, M., Kraemer, S.M., Kretzschmar, R.: Copper complexation of methanobactin isolated from Methylosinus trichosporium OB3b: pH-dependent speciation and modeling. J. Inorg. Biochem. 116, 55–62 (2012)
Soliman, S.M.: Molecular structure and vibrational spectra of free and coordinated 3-bromoquinoline: unexpected intramolecular C-H···O interactions. J. Mol. Struct. 1017, 135–142 (2012)
Li, Y., Wang, J.-G., Duan, T., Chen, Q., Zhang, Q.-F.: Silver(I) and mercury(II) complexes with N,N'-bis(acetylacetone)-1R,2R-diaminocyclohexane ligands exhibiting three different coordination modes. J. Organomet. Chem. 696, 2294–2298 (2011)
Shannon, R.: Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Section A. 32, 751–767 (1976)
Krause, M.E., Glass, A.M., Jackson, T.A., Laurence, J.S.: MAPping the chiral inversion and structural transformation of a metal-tripeptide complex having Ni-superoxide dismutase activity. Inorg. Chem. 50, 2479–2487 (2011)
Joszai, V., Turi, I., Kallay, C., Pappalardo, G., Di Natale, G., Rizzarelli, E., Sovago, I.: Mixed metal copper(II)-nickel(II) and copper(II)-zinc(II) complexes of multihistidine peptide fragments of human prion protein. J. Inorg. Biochem. 112, 17–24 (2012)
MacColl, R., Eisele, L.E., Stack, R.F., Hauer, C., Vakharia, D.D., Benno, A., Kelly, W.C., Mizejewski, G.J.: Interrelationships among biological activity, disulfide bonds, secondary structure, and metal ion binding for a chemically synthesized 34-amino-acid peptide derived from α-fetoprotein. Bioch. Biophys. Acta -General Subjects. 1528, 127–134 (2001)
Paranjothy, M., Sun, R., Zhuang, Y., Hase, W.L.: Direct chemical dynamics simulations: coupling of classical and quasiclassical trajectories with electronic structure theory. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 3, 296–316 (2013)
Bilusich, D., Brinkworth, C.S., McAnoy, A.M., Bowie, J.H.: The fragmentations of [M-H]- anions derived from underivatised peptides. The side-chain loss of H2S from Cys. A joint experimental and theoretical study. Rapid Commun. Mass Spectrom. 17, 2488–2494 (2003)
Reid, G.E., Simpson, R.J., O'Hair, R.A.J.: Leaving group and gas phase neighboring group effects in the side chain losses from protonated serine and its derivatives. J. Am. Soc. Mass Spectrom. 11, 1047–1060 (2000)
Waugh, R.J., Eckersley, M., Bowie, J.H., Hayes, R.N.: Collision induced dissociations of deprotonated peptides: dipeptides containing serine or threonine. Int. J. Mass Spectrom. Ion Processes. 98, 135–145 (1990)
Homayoon, Z., Pratihar, S., Dratz, E., Snider, R., Spezia, R., Barnes, G.L., Macaluso, V., Martin Somer, A., Hase, W.L.: Model simulations of the thermal dissociation of the TIK(H+)2 Tripeptide: mechanisms and kinetic parameters. J. Phys. Chem. A. 120, 8211–8227 (2016)
Spezia, R., Martin-Somer, A., Macaluso, V., Homayoon, Z., Pratihar, S., Hase, W.L.: Unimolecular dissociation of peptides: statistical vs. non-statistical fragmentation mechanisms and time scales. Faraday Discuss. 195, 599–618 (2016)
Zhang, Y., Shi, S., Sun, X., Xiong, X., Peng, M.: The effect of Cu2+ on interaction between flavonoids with different C-ring substituents and bovine serum albumin: structure–affinity relationship aspect. J. Inorg. Biochem. 105, 1529–1537 (2011)
Drake, P.L., Hazelwood, K.J.: Exposure-related health effects of silver and silver compounds: a review. Ann. Occupat. Hyg. 49, 575–585 (2005)
Ellingsen, D.G., Horn, N., Aaseth, J.: Copper. In: Friberg, L., Nordberg ,G.F., Vouk, V.B. (Eds.). Elsevier Ltd., Amsterdam (2007)
Rubino, J.T., Franz, K.J.: Coordination chemistry of copper proteins: How nature handles a toxic cargo for essential function. J. Inorg. Biochem. 107, 129–143 (2012)
Robinson, N.J., Winge, D.R.: Copper metallochaperones. Ann. Rev. Biochem. 79, 537–562 (2010)
Acknowledgements
The authors thank NSF for instrument support from MRI-0821247 and Welch Foundation T-0014 for scholarship and financial support. The authors also thank Maheshbabu Nasani for determining the binding constant for the Mb-Zn(II) complex.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(DOCX 3533 kb)
Rights and permissions
About this article

Cite this article
McCabe, J.W., Vangala, R. & Angel, L.A. Binding Selectivity of Methanobactin from Methylosinus trichosporium OB3b for Copper(I), Silver(I), Zinc(II), Nickel(II), Cobalt(II), Manganese(II), Lead(II), and Iron(II). J. Am. Soc. Mass Spectrom. 28, 2588–2601 (2017). https://doi.org/10.1007/s13361-017-1778-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-017-1778-9