
Abstract
Gas-phase clustering of nonionizable iodylbenzene (PhIO2) is attributed to supramolecular halogen bonding. Electrospray ionization results in the formation of ions of proton-charged and preferably sodium-charged clusters assignable to [H(PhIO2) n ]+, n = 1–7; [Na(PhIO2) n ]+, n = 1–6; [Na2(PhIO2) n ]2+, n = 7–20; [HNa(PhIO2) n ]2+, n = 6–19; [HNa2(PhIO2) n ]3+, n = 15–30; and [Na3(PhIO2) n ]3+, n = 14–30. The largest cluster detected has a supramolecular mass of 7147 Da. Electronic structure calculations using the M06-2X functional with the 6-311++G(d,p) basis set for C, H, and O, and LANL2DZ basis set for I and Na predict 298 K binding enthalpies for the protonated and sodiated iodylbenzene dimers and trimers are greater than 180 kJ/mol. This is exceptionally high in comparison with other protonated and sodiated clusters with well-established binding enthalpies. Strongly halogen-bonded motifs found in the crystalline phases of PhIO2 and its derivatives serve as models for the structures of larger gas-phase clusters, and calculations on simple model gas-phase dimer and trimer clusters result in similar motifs. This is the first account of halogen bonding playing an extensive role in gas-phase associations.

ᅟ
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The halogen bond describes strong noncovalent interaction and charge transfer between a Lewis basic electron-pair donor and a halogen atom acceptor site (Figure 1). Awareness of the importance of this interaction as a mechanism in supramolecular architectures has heightened in recent years, in fields as diverse as crystal engineering and pharmokinetics [1, 2]. The strength of this noncovalent interaction varies according to the particular halogen element (X), charge, oxidation state, and halogen bond acceptor. In addition, halogen bonds, like hydrogen bonds, are directed [1, 3]. Bond energies span a wide range, from 5 to 180 kJ/mol [3], and a typical interaction between neutral halogen compounds and neutral heteroatoms (N, O) has a strength in the range of 23–35 kJ/mol [4, 5], similar to the strength of hydrogen bonds [6]. Solution experiments have shown that halogen bonding can outcompete hydrogen bonding [7]. Among the halogen atoms, iodine forms the strongest halogen-bonding interactions, and the tendency decreases in the order I > Br > Cl > F [3, 4]. The nucleophilic partner in a noncovalent halogen bond can be any Lewis base, including other polarizable halogen atoms. Electronic structure calculations indicate that halogen-bond interactions consist primarily of a σ-hole bond [8] in which a covalently bonded atom of groups 14-17 noncovalently interacts with a lone pair of a halogen atom/ion. From calculations, the strengths of halogen-bonded interactions depend strongly on the orientations of the σ-hole bonds [9, 10].

The halogen bond. B = Lewis base, E = any element, X = F, Cl, Br, or I
Although these supramolecular interactions are well established in the solid [11,12,13] and solution [5, 14] states, little attention has been paid to their functionality in the gas phase. The most significant gas-phase experimental work is the rotational spectroscopy performed by Legon [15], who showed that dihalogens, XaXb (both homohalide and interhalide species), and Lewis bases, B (PH3, NH3, H2S, C2H4, C2H2, H2O, HCN, CO, N2, Ar), form 1:1 gas-phase adducts that are stabilized by halogen bonds to form B⋯XaXb moieties rather than BXa +⋯Xb - ion pairs, which would be expected if complete electron transfer had occurred.
Our attention was drawn to the potential for halogen bonding being a powerful mechanism for noncovalent interactions in the gas phase by the unexpected observation of a highly reactive iron complex in electrospray ionization (ESI) mass spectra. Uniquely this catalytically competent complex, [Fe(tpena)(OIPh)]2+, where tpena- is N,N,N'-tris(2-pyridylmethyl)ethylenediamine-N'-acetate, incorporates the terminal oxidant, iodosylbenzene (PhIO), coordinated to the iron atom, and significantly this complex was detected only when associated with Cl- or ClO4 -. The monocationic ions [Fe(tpena)(OIPh)X]+ (X is Cl- or ClO4 -) were observed [16]. We therefore concluded that the chloride and perchlorate anions impart stability to [Fe(tpena)(OIPh)]2+ and postulated that the association is more specific than that expected from Coulombic forces only (aka ion pairs). Actual iron coordination by Cl- or ClO4 - was dismissed because the coordination number (seven) was already high and the ligands were bulky. Rather the association might better be described by supramolecularly associated “adducts” formed through directive Cl-⋯I(OFe)Ph and ClO4 -⋯I(OFe)Ph halogen-bonding interactions. This proposal was supported by the fact that [Fe(tpena)(OIPh)]2+ forms strongly self-associated halogen-bonded dimers {[Fe(tpena)(OIPh)]2}4+ in the crystalline solid state [16]. Two of the six I⋯O interactions between the monomers show the shortest interatomic distances for noncovalently bonded I and O atoms (2.555(3) Å) in the Cambridge Structural Database [17]. Remarkably, this distance is shorter than some covalent I–O single bonds. X-ray absorption near-edge structure spectroscopy verified that significant electron density transfer occurs with a consequent decrease in the oxidation state of the originally hypervalent (+3) iodine compound PhIO by about 1.4 units [18]. These discoveries inspired us to study the gas-phase behavior of a simpler and less reactive analyte for the purpose of establishing halogen-bonding functionality in the gas phase. We can now report that mass spectrometric studies of the neutral nonionizable iodylbenzene (PhIO2) demonstrate that supramolecular halogen bonding is an intermolecular force that should be routinely considered as a mechanism for gas-phase associations.
Methods
Experimental
PhIO2 was synthesized as previously described [19] with a yield of 45%, and all other chemicals were purchased from Sigma-Aldrich. All ESI mass spectra were recorded in high-resolution positive-ionization mode with a Bruker microTOF-QII mass spectrometer. For spectra scanned from m/z 200 to m/z 1500, the sample solutions were injected with a Dionex UltiMate 3000 high-performance liquid chromatography system (flow rate 0.1 mL/min), and for spectra scanned from m/z 300 to m/z 3000, a KD Scientific syringe pump (flow rate 180 μL/h) was used. Nitrogen was used as the drying gas with a temperature of 180 °C and a flow rate of 4.0 L/min. The capillary voltage was 4500 V and the end plate offset was -500 V. The nebulizer pressure was either 0.3 bar (m/z 200–1500) or 0.4 bar (m/z 300–3000), and the collision cell retention time was either 325 V peak-to-peak (m/z 200–1500) or 917 V peak-to-peak (m/z 300–3000). A 10 mM NaI solution in acetonitrile–water (50:50 w/w) was used for external calibration.
PhIO2 was dissolved in either water or dimethyl sulfoxide (DMSO; 1 mg/mL) and diluted to 10 μg/mL with acetonitrile. Specific experiments were performed with addition of NaCl (67 μg/mL), acetic acid (1%), CaCl2 (17 μM), and extra DMSO (see the electronic supplementary material). Collision-induced dissociation (CID) experiments were performed with collision energies from 1 to 30 eV, and the molecular ions of m/z 967, 1321, and 1360 were used for collision with argon gas. The data were acquired in Bruker’s Compass DataAnalysis and processed with mMass 5.4.1 [20,21,22], where the five most intense spectra were summed. The peak marked by an asterisk at m/z 425 in Figure 2a is due to an impurity derived from the septum and this is present in all samples.

a Section (m/z 200–2000) of the electrospray ionization mass spectrum of PhIO2 in water–acetonitrile (1:100). Assignments for selected peaks are shown (for full assignment, see Table S1). b Expansion showing the isotope pattern for the m/z 966.721 ion (black). Calculated isotope patterns for [Na(PhIO2)4]+ (red) and [Na2(PhIO2)8]2+ (green). The green line and the black line are completely superimposed for the peaks at m/z 967.222 and 968.227. c Expansion showing the isotope pattern for the m/z 1438.587 ion (black). Calculated isotope patterns for [Na(PhIO2)6]+ (red), [Na2(PhIO2)12]2+ (green), and [Na3(PhIO2)18]3+ (blue). d Expansion showing the isotope pattern for the m/z 1910.429 ion (black). Calculated isotope patterns for [Na2(PhIO2)16]2+ (green) and [Na3(PhIO2)24]3+ (blue). The asterisk denotes a systematic impurity
Theoretical
For all calculations, molecular geometries of (PhIO2) n , [H(PhIO2) n ]+, and [Na(PhIO2) n ]+ were obtained by generation of starting structures by means of chemical intuition. For large ionic clusters, at least 20 possible starting structures were generated manually. Molecular geometries were first optimized with a relatively computationally efficient level of theory and basis set (B3LYP/BS1, where BS1 corresponds to 6-31G(p) for C, H, and O, and LANL2DZ for Na and I). From benchmarking studies of the dissociation of 51 halogen-bonded dimers, the M06-2X hybrid functional is relatively accurate owing to the high amount of exact exchange, and is often used for systems with a large amount of charge transfer (e.g., halogen bonding) [10]. Thus, molecular geometries were subsequently optimized with M06-2X/BS2, where BS2 corresponds to 6-311++G(d,p) for C, H, and O, and LANL2DZ for I and Na. Vibrational frequencies were calculated to ensure that all reported structures correspond to ground-state minima. Sequential bond dissociation enthalpies were calculated by inclusion of a correction for zero-point vibrational energy.
Results and Discussion
A striking feature of ESI mass spectra of PhIO2 in water–acetonitrile (1:100) is the presence of ions that can be associated with impressive cluster formation, Figure 2a (m/z 200–1500) and Fig. S1 (m/z 2000–2500), with ions containing up to 30 monomeric PhIO2 units. These are assigned to [H(PhIO2) n ]+, n = 1–7; [Na(PhIO2) n ]+, n = 1–6; [Na2(PhIO2) n ]2+, n = 7–20; [Na3(PhIO2) n ]3+, n = 14–30; [HNa(PhIO2) n ]2+, n = 6–19; and [HNa2(PhIO2) n ]3+, n= 15–30 (full assignments in Table S1). The isotope distributions of [Na(PhIO2) n ]+, [Na2(PhIO2)2n ]2+, and [Na3(PhIO2)3n ]3+ overlap for n = 5 and n = 6. Calculated isotope patterns were fitted to the experimental data for the assignment of the fraction of each ion. The monocationic ions are detected only at relatively low m/z values, whereas the tricationic ions were detected only at relatively high m/z values. For example, the isotopologues for [Na(PhIO2)4]+ and [Na2(PhIO2)8]2+ both contribute to the peak at m/z 966.721 (Figure 2b), and there is no trace of [Na3(PhIO2)12]3+. By contrast, the peak at m/z 1438.587 (Figure 2c) shows only a minor contribution from the monocationic [Na(PhIO2)6]+, and the dicationic [Na2(PhIO2)12]2+ and tricationic [Na3(PhIO2)18]3+ ions dominate. At higher m/z values, monocationic ions are not detected at all, and the tricationic ions are the major contributors; for example, the peaks at m/z 1910.458 and 2146.391 (Figures 2d and S2).
CID experiments on the m/z 967 ion show that neutral PhIO2 units can be stripped off and monocationic ions containing one to three PhIO2 units (i.e., [Na(PhIO2) n ]+, n = 1–3) are predominantly observed as products. Equations 1, 2, and 3 (Figure 3a) show the fragmentation observed for [Na(PhIO2)4]+. Higher intensities of the peak at m/z 494 (Eq. 2, Fig. S3) compared with the peaks at m/z 730 and 258 obtained with collision energies from 1.0 to 10 eV suggest a propensity for stripping two PhIO2 units simultaneously. Collision energies greater than 10 eV are needed before the Na+-charged monomer of m/z 258 begins to dominate CID spectra of the m/z 967 ion. At these energies the m/z 730 ion is completely absent (Fig. S3). The m/z 967 precursor ion contains [Na2(PhIO2)8]2+, which cannot be separated from [Na(PhIO2)4]+ for a selective CID experiment. However, it is probable that very little energy is needed to charge-separate this ion into two [Na(PhIO2)4]+ ions since no product ions that could directly result from [Na2(PhIO2)8]2+ and not [Na(PhIO2)4]+ were observed; for example, the doubly charged product ions ([Na2(PhIO2) n ]2+, n = 1–7) and the higher m/z ions in Figure 3b. The doubly and triply charged ions of m/z 1321 due to [Na2(PhIO2)11]2+ and m/z 1360 due to [Na3(PhIO2)17]3+ were also selected for CID because their signals do not overlap with signals from clusters with different stoichiometries but the same m/z. In these cases, products formed by charge separation will be distinguishable from the starting precursor; that is, [Na2(PhIO2)11]2+ → [Na(PhIO2)6]+ + [Na(PhIO2)5]+ and [Na3(PhIO2)17]3+ → [Na2(PhIO2)12]2+ + [Na(PhIO2)5]+. Consistent with the outcome when [Na2(PhIO2)8]2+ is used, the higher m/z products of potential charge-separation reactions are not observed under CID, and the same singly charged ions, [Na(PhIO2) n ]+, n = 1–4, that were observed on CID of the m/z 967 ion are produced. This suggests that stripping neutral PhIO2 from ions with higher m/z (e.g., [Na(PhIO2) n ]+, n = 5–7) is facile, and hence they are not observed under the conditions we used. Only one doubly charged product ion [Na2(PhIO2)7]2+ in a tiny yield (1% of base peak) due to the loss of the mass equivalent of four PhIO2 units was found from the CID experiments on [Na2(PhIO2)11]2+.

a Fragmentation of the [Na(PhIO2)4]+ ion during collision-induced dissociation (proposed neutral PhIO2 fragments are unobservable). b Possible charge-separated products (without concurrent neutral PhIO2 losses) from the doubly charged m/z 967 ion
The only reasonable explanation for the high degree of self-association exhibited by PhIO2 can be the presence of I⋯O interactions. Hydrogen-bonding mechanisms are eliminated by positive controls in which acid is added (see later). Strong π−π interactions as a clustering mechanism are presumably not viable. In this respect it is worth noting that clustering is not typically observed in routine ESI mass spectra of nonionizable substituted benzene compounds (e.g., benzaldehyde). For good measure, however, we checked this by recording the ESI mass spectrum of benzaldehyde under conditions identical to those under which we had recorded the spectra of PhIO2. No clusters were observed. This is consistent with their absence in the NIST database [23]. Our conclusion is that the presence of both a halogen-bond acceptor (iodine) and a halogen-bond donor (oxygen) in PhIO2 must be the origin of the extensive gas-phase homocluster formation.
The solid-state structures of PhIO2 demonstrate definitively the tendency for the iodyl groups of PhIO2 to self-associate via halogen bonding. Two polymorphs, α-PhIO2 [24] and β-PhIO2 [18], with distinct crystal packing structures are known, and both show the presence of strong I⋯O halogen bonding (Figure 4). The structure of α-PhIO2 shows a lamellar arrangement with the iodyl groups forming layers of iodine and oxygen atoms between two layers of phenyl rings. By contrast, the iodine and oxygen atoms in β-PhIO2 form columns separated by the phenyl rings. Notably the distance between the phenyl groups in both structures (4.0 Å for α-PhIO2 and 6.4 Å for β-PhIO2) is too long for π−π interactions: This otherwise well-established noncovalent interaction is completely overshadowed by stronger supramolecular halogen bonding. The solid-state structure adds credence to our conclusion that π−π interactions are not the cause of gas-phase association of PhIO2 monomers.

The charging of the clusters can potentially alter structure; however, similarly to our analysis of the solid-state structures of PhIO2, plausible hydrogen-bonding, coordination-bonding, halogen-bonding, π−π, and CH–π interactions that might be responsible for the gas-phase clustering of PhIO2 can be addressed by an analysis of the 24 known crystal structures of the substituted derivatives of PhIO2 (Figs. S4, S5, S6, and S7 and Table S2). One or more strong I…O interactions between adjacent units are seen in all structures (Figure 5). In turn, these motifs associate with surrounding molecules with marginally weaker halogen bonding. Iodine atoms are typically found in a sphere of halogen-bonding donors (Fig. S5). The I2O2 rhomb motif is a very common feature and is interesting with respect to the tendency toward concomitant stripping of two PhIO2 units in CID reactions. The homo-intermolecular I⋯O interactions outcompete possible interactions with co-crystallized Lewis bases (solvents and anions; Figs. S6 and S7), and likewise co-crystallized Brønsted and Lewis acids do not disrupt the structures. This structural analysis of PhIO2 and its derivatives demonstrates clearly that halogen bonding is capable of overriding other plausible supramolecular interactions.

Typical intermolecular halogen-bonding I⋯O(sp 2) motifs obtained from analysis of the crystal structures of 24 iodylbenzene-containing compounds (substitution of the phenyl ring, not shown, has been allowed). The noncovalently bonded distances (⋯) fall between 2.5 and 3.5Å (the sum of the van der Waals radii for I and O atoms). For simplicity, not all of the halogen bonding associated with the two molecules is shown. See Table S2 for further details
A preference for cluster charging by adventitious sodium ions rather than more readily available protons from the water cosolvent is apparent. Mixed [HNa(PhIO2) n ]2+ and [HNa2(PhIO2) n ]3+ ions show lower signal intensities compared with the exclusively sodiated dicationic and tricationic ions, and no ions due to [H2(PhIO2) n ]2+, [H3(PhIO2) n ]3+, or [H2Na(PhIO2) n ]3+ are detected (Figure 6). Illustrated also is the fact that ions containing a low number of PhIO2 units and high charge or high number of PhIO2 units and low charge are not observed (absence of ions at the bottom left and top right of Figure 6). Experiments in which samples were doped with acetic acid and/or NaCl (Figs. S8 and S9) emphasize the propensity of sodiation for charging the clusters. Despite a higher availability of protons in samples containing acetic acid, no ions charged by protons are present, suggesting that hydrogen bonding via a proton, which would seem to be otherwise an entirely feasible mechanism for association between PhIO2 units, cannot outcompete aggregation due to halogen bonding. This is consistent with the observations that halogen bonding can outcompete hydrogen bonding in the solution state [7]. When NaCl is deliberately added, charging by protonation is completely eliminated. It is possible to charge clusters with higher numbers of PhIO2 monomers per metal ion if CaCl2 is added. [Ca(PhIO2) n ]2+ ions, n = 2–12, are observed (Fig. S10; Na+ and Ca2+ have similar radii: 102 and 100 pm, respectively). More than one sodium ion is not associated when there are fewer than six PhIO2 units, and this suggests favored cluster stoichiometries. Pertinent to this speculation is the presence of coordination bonds with sodium and potassium in two of the structures found in our aforementioned analysis of pertinent crystal structures [25, 26]. Notably in these crystal structures, coordination of a neutral iodyl oxygen to sodium ion and potassium ion occurs even in the presence of negatively charged oxoanionic groups (Table S2). In summary, this solid-state structure analysis reveals that various substituents, ions, and co-crystallized solvents have only a minor impact on intermolecular halogen-bonding distances despite substantially lower degrees of freedom in a crystal lattice compared with the gas phase.

Relationship between the numbers of PhIO2 monomers in the clusters, the charge, and the relative abundance based on the spectrum in Figure 2a
PhIO2 is generally most soluble in Lewis basic solvents probably because these can outcompete self-association by halogen-bonding interactions. The electron-pair donor strengths (D s) for acetonitrile, water, and DMSO are 12, 17, and 27.5, respectively [27], and these solvents all have the potential to act as electron-pair donors. In the crystalline solid state, supramolecular I⋯OS(CH3)2 interactions are commonly observed [28,29,30,31], and in cases where solid-state materials are solvated with both DMSO and water molecules, it is always the DMSO molecule that is more strongly interacting with hypervalent iodine compounds [32] (Fig. S6). A preference for forming halogen bonds between iodyl groups apparently takes precedence over potential acetonitrile–iodyl interactions (Fig. S7) [33, 34]. In this context, it is worth noting that the experiments described here were performed in acetonitrile–water mixtures, and varying the ratios of these solvents did not change clustering patterns. We therefore speculated that I⋯OS(CH3)3 interactions, not uncommonly observed in the solid phase, might be capable of overriding self-association processes in solution and during ionization. Experiments where DMSO was used to dissolve PhIO2 before dilution with acetonitrile (1:100) were performed but DMSO adducts were not observed and the stoichiometry of the clusters was unchanged compared with that of water-pretreated samples. Abundance trends of specific ions are altered slightly: in general, the ions of higher m/z and the multiply charged ions display lower signal intensities or the signals are absent (Fig. S11). For example, the [Na3(PhIO2)18]3+ ion contributes less to the isotope pattern for the signal starting with m/z 1438.59 compared with [Na(PhIO2)]6+ when DMSO is used as the solvent before dilution with acetonitrile (Fig. S12). These observations support the expectation that DMSO interacts more strongly with PhIO2 compared with water and acetonitrile in solution and thus effects a slightly greater solution-state deoligomerization. In turn this attenuates the extent of clustering ultimately observed in the gas phase. The addition of five times and ten times larger quantities of DMSO did not further change the spectra.
Electronic structure calculations for (PhIO2) n , [H(PhIO2) n ]+, and [Na(PhIO2) n ]+ (n = 1–3) were performed to investigate the strength of noncovalent interactions in ionic and neutral clusters. To find different conformations, starting structures were generated manually and molecular geometries were optimized with a computationally efficient level of theory and basis set (B3LYP/BS1; Figs. S13, S14, and S15). For the ionic trimers, between 15 and 20 unique ground-state minimum structures were systematically identified through this process, whereas for the dimers, five unique ground-state structures were found. Subsequently, unique ground-state geometries were optimized with M06-2X/BS2 (Figs. S13, S14 and S15). M06-2X is relatively accurate for calculating the binding strength of 51 halogen-bonded dimers (with an average root-mean-square error of less than 5 kJ/mol) [10].
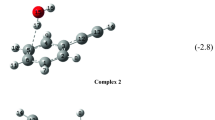
The lowest-energy optimized structures for [H(PhIO2) n ]+ and [Na(PhIO2) n ]+ (n = 2 and 3) are shown in Figure 7. For each cluster, extensive halogen bonding is evident. For example, the three iodine atoms are bridged by two oxygen atoms in the protonated iodylbenzene dimers and trimers, and this motif is found also in the aforementioned solid-state crystal structures. Terminal oxygen atoms are favored by more than 25 kJ/mol and 32 kJ/mol for the site of protonation rather than the oxygen atoms bridging two of three iodine atoms for all stable ground-state minima identified for the protonated dimers and trimers. For the sodiated clusters, the sodium ion is surrounded by iodyl groups such that a coordination sphere of oxygen atoms is favored. The iodyl groups are linked through halogen bonds. Removal of the halogen-bonded linkage of the iodyl groups and preservation of only the coordination to the sodium ion increases the energy dramatically (e.g., by 56.8 kJ/mol for [Na(PhIO2)2]+ compared with the lowest-energy optimized model structure; Figure S14c). The intermolecular I⋯O bonds in the optimized geometries of (PhIO2) n , [H(PhIO2) n ]+, and [Na(PhIO2) n ]+ (n = 2 and 3) (Table S3) show distances in the range of 2.44–2.72 Å. The only outliers are the shortest I⋯O distances of [H(PhIO2) n ]+ (n = 2 and 3), which are 2.193 Å and 2.115 Å, respectively, which indicates that the structures in these geometries have the possibility to take full advantage of the energy stabilization furnished by halogen bonding. The geometries with the sodium ions have to adjust more owing to the presence of a larger positively charged ion. The intermolecular I⋯O bond distances in the lowest-energy optimized geometries are on average slightly shorter than those found in the crystalline solid state for iodyl-containing materials (Table S2), which are in the range of 2.5–3.2 Å. The crystal structure of sodium 2-iodyl-4-nitrobenzoate hydrate [26] shows that even in the presence of sodium ions and a carboxylate group, halogen-bonded iodyl groups are present (Fig. S16) and that pleasingly the coordination sphere around the sodium ion is very similar to that of the lowest-energy optimized gas-phase model structures.

Lowest-energy optimized model structures of [H(PhIO2) n ]+ and [Na(PhIO2) n ]+ (n = 2 and 3) calculated with M06-2X/BS2
Table 1 lists the sequential ligand binding enthalpies for the formation of neutral, protonated, and sodiated iodylbenzene dimers and trimers. Owing to the large conformational space for the trimer clusters, additional lower-energy structures may be possible; that is, these calculated values correspond to upper limits. Thus, the binding enthalpies could be more exothermic for addition of the third ligand. For the protonated iodylbenzene clusters, formation of the dimer and trimer clusters is exothermic by more than 200 kJ/mol (Table 1). For the sodiated iodylbenzene clusters, the formation of the dimer and trimer is exothermic by more than 180 kJ/mol. In contrast, the sequential binding enthalpies for the formation of the neutral iodylbenzene dimer and trimer are much lower, likely owing to reduced ligand polarization for neutral clusters compared with ionized clusters. To our knowledge the calculated binding enthalpies required to form the protonated iodylbenzene dimer and trimer are significantly larger than the binding enthalpies of all A+L2 and A+L3 species (A is H and Na) reported in the NIST database [23] of ion-clustering reactions. The highest binding enthalpies found in the NIST database for A+L2 (where A+ is any singly charged ion) are for Cu+(C2H4)2 (174 kJ/mol) [35] and Cu+L2 (L is 1,2-dimethoxyethane; 180 kJ/mol) [36]. Solvated protonated dimers and alkali metal ions tend to have sequential binding enthalpies that are less than 100 kJ/mol. Overall, the ionic clusters of PhIO2 are calculated to have exceptionally high ligand binding enthalpies compared with any known ionic clusters.
Conclusions
Gas-phase water [37,38,39,40,41,42,43,44] and amino acid [45,46,47,48,49,50] clusters are well documented and demonstrate the strength of hydrogen bonding under the high-vacuum conditions of a mass spectrometry experiment. Using the prototype halogen-bonding donor–acceptor analyte PhIO2, we have shown that, similarly, halogen bonding is a mechanism that can be considered for rationalizing gas-phase associations. Electronic structure calculations of model protonated and sodiated iodylbenzene dimers and trimers indicate that halogen bonding within these clusters is extensive and can result in exceptionally high ligand binding enthalpies (more than 180 kJ/mol). Formulations of [Nan(PhIO2) m ]n+ (n = 1, m = 1–6; n = 2, m = 7–20; n = 3, m =14–30) are typical under the experimental conditions we used. Calculations suggest that charging by protonation should be slightly more favorable; however, this is not borne out by experiment, which shows that sodiation is favored over protonation for cluster charging. Coordination bonds with sodium formed by supramolecular chelates of iodyl oxygen atoms would seem a highly likely mechanism. The potential for halogen bonding and coordination bonding is supported by the analysis of all known relevant crystal structures of PhIO2-derived compounds, which illustrates not only the same supramolecular motifs as predicted by calculation but also the impotency of potentially competing supramolecular interactions such as π−π stacking and association with other potential halogen-bond donors and acceptors that are present in the materials. Dimers based on (O=I⋯O=I)2 rhombs are a prevalent motif in the crystalline solid state, and our results suggest that a pairwise fragmentation of (PhIO2) n clusters may be favored in gas-phase disintegrations.
References
Catalano, L., Cavallo, G., Metrangolo, P., Resnati, G., Terraneo, G.: Halogen bonding in hypervalent iodine compounds. Top. Curr. Chem. 373, 289–309 (2016)
Zheng, Y.-Z., Wang, N.-N., Zhou, Y., Yu, Z.-W.: Halogen-bond and hydrogen-bond interactions between three benzene derivatives and dimethyl sulphoxide. Phys. Chem. Chem. Phys. 16, 6946–6956 (2014)
Cavallo, G., Metrangolo, P., Milani, R., Pilati, T., Priimagi, A., Resnati, G., Terraneo, G.: The halogen bond. Chem. Rev. 116, 2478–2601 (2016)
Riley, K.E., Merz, K.M.: Insights into the strength and origin of halogen bonding: the halobenzene-formaldehyde dimer. J. Phys. Chem. A. 111, 1688–1694 (2007)
Beale, T.M., Chudzinski, M.G., Sarwar, M.G., Taylor, M.S.: Halogen bonding in solution: thermodynamics and applications. Chem. Soc. Rev. 42, 1667–1680 (2013)
Emsley, J.: Very strong hydrogen bonding. Chem. Soc. Rev. 9, 91–124 (1980)
Metrangolo, P., Resnati, G.: Halogen bonding: a paradigm in supramolecular chemistry. Chem. Eur. J. 7, 2511–2519 (2001)
Politzer, P., Murray, J.S., Clark, T.: Halogen bonding and other σ-hole interactions: a perspective. Phys. Chem. Chem. Phys. 15, 11178–11189 (2013)
Huber, S.M., Scanlon, J.D., Jimenez-Izal, E., Ugalde, J.M., Infante, I.: On the directionality of halogen bonding. Phys. Chem. Chem. Phys. 15, 10350–10357 (2013)
Kozuch, S., Martin, J.M.L.: Halogen bonds: benchmarks and theoretical analysis. J. Chem. Theory Comput. 9, 1918–1931 (2013)
Ouvrard, C., Questel, J.-Y.L., Berthelot, M., Laurence, C.: Halogen-bond geometry: a crystallographic data-base investigation of dihalogen complexes. Acta Crystallogr. B59, 512–526 (2003)
Metrangolo, P., Meyer, F., Pilati, T., Resnati, G., Terraneo, G.: Halogen bonding in supramolecular chemistry. Angew. Chem. Int. Ed. 47, 6114–6127 (2008)
Troff, R.W., Mäkelä, T., Topić, F., Valkonen, A., Raatikainen, K., Rissanen, K.: Alternative motifs for halogen bonding. Eur. J. Org. Chem. 9, 1617–1637 (2013)
Erdélyi, M.: Halogen bonding in solution. Chem. Soc. Rev. 41, 3547–3557 (2012)
Legon, A.C.: The halogen bond: an interim perspective. Phys. Chem. Chem. Phys. 12, 7736–7747 (2010)
de Sousa, D.P., Wegeberg, C., Vad, M.S., Mørup, S., Frandsen, C., Donald, W.A., McKenzie, C.J.: Halogen bonding assisted iodosylbenzene activation by a homogenous iron catalyst. Chem., Eur. J. 22, 3810–3820 (2016)
Allen, F.H.: The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr. B58, 380–388 (2002)
Wegeberg, C., Frankær, C.G., McKenzie, C.J.: Reduction of hypervalent iodine by coordination to iron(III) and the crystal structures of PhIO and PhIO2. Dalton Trans. 45, 17714–17722 (2016)
Sharefkin, J.G., Saltzman, H.: Iodoxybenzene. Org. Synth. Coll. 43, 65–66 (1973)
Niedermeyer, T.H.J., Strohalm, M.: mMass as a software tool for the annotation of cyclic peptide tandem mass spectra. PLoS ONE 7, e44913 (2012)
Strohalm, M., Kavan, D., Novák, P., Volný, M., Havlíček, V.: mMass 3: a cross-platform software environment for precise analysis of mass spectrometric data. Anal. Chem. 82, 4648–4651 (2010)
Strohalm, M., Hassman, M., Košata, B., Kodíček, M.: mMass Data Miner: an open source alternative for mass spectrometric data analysis. Rapid Commun. Mass Spectrom. 22, 905–908 (2008)
Shen, V. K., Siderius, D. W., Krekelberg, W. P., Hatch, H.W., Eds.: NIST Standard Reference Simulation Website, NIST Standard Reference Database Number 173, National Institute of Standards and Technology, Gaithersburg MD, 20899, doi:10.18434/T4M88Q (retrieved 01/03/17).
Alcock, N.W., Sawyer, J.F.: Secondary bonding. Part 6. Distorted octahedral geometry in seleninyl dichloride–dioxan(1/1) and iodylbenzene. J. Chem. Soc., Dalton Trans. 115-120 (1980). doi:10.1039/DT9800000115
Yusubov, M.S., Yusubova, R.Y., Nemykin, V.N., Maskaev, A.V., Geraskina, M.R., Kirschning, A., Zhdankin, V.V.: Potassium 4-iodylbenzenesulfonate: preparation, structure, and application as a reagent for oxidative iodination of arenes. Eur. J. Org. Chem. 30, 5935–5942 (2012)
Katritzky, A.R., Savage, G.P., Palenik, G.J., Qian, K., Zhang, Z., Durst, H.D.: The cyclic structure of 2-iodosyl- and 2-iodyl-benzoic acid anions: a basicity and X-ray crystallographic study. J. Chem. Soc. Perkin Trans. 2, 1657–1661 (1990)
Sandström, M., Persson, I., Persson, P.: A study of solvent electron-pair donor ability and lewis basicity scales. Acta Chem. Scand. 44, 653–675 (1990)
Fucke, K., Howard, J.A.K., Steed, J.W.: Overcoming the solvation shell during the crystallisation of diatrizoic acid from dimethylsulfoxide. Chem. Commun. 48, 12065–12067 (2012)
Lieffrig, J., Jeannin, O., Fourmigué, M.: Expanded halogen-bonded anion organic networks with star-shaped iodoethynyl-substituted molecules: from corrugated 2D hexagonal lattices to pyrite-type 2−fold interpenetrated cubic lattices. J. Am. Chem. Soc. 135, 6200–6210 (2013)
Moss, R.A., Bracken, K., Emge, T.J.: 9-Iodoso-10-phenanthroate: structure and phosphorolytic properties of a nonplanar iodoxolone. J. Org. Chem. 60, 7739–7746 (1995)
Ostermeier, M., Berlin, M.-A., Meudtner, R.M., Demeshko, S., Meyer, F., Limberg, C., Hecht, S.: Complexes of click-derived bistriazolylpyridines: remarkable electronic influence of remote substituents on thermodynamic stability as well as electronic and magnetic properties. Chem. Eur. J. 16, 10202–10213 (2010)
Zhang, Y., Han, J.L., Xu, Y.J., Wei, Y.P.: 2-Iodoxybenzoic acid-mediated transformation of halogenated hydrocarbon in the presence of tetraethyl ammonium bromide. J. Chem. Res. 36, 303–307 (2012)
Zhdankin, V.V., Koposov, A.Y., Netzel, B.C., Yashin, N.V., Rempel, B.P., Ferguson, M.J., Tykwinski, R.R.: IBX amides: a new family of hypervalent iodine reagents. Angew. Chem., Int. Ed. 42, 2194–2196 (2003)
Altermann, S.M., Schäfer, S., Wirth, T.: New chiral hypervalent iodine(V) compounds as stoichiometric oxidants. Tetrahedron 66, 5902–5907 (2010)
Sievers, M.R., Jarvis, L.M., Armentrout, P.B.: Transition metal ethene bonds: thermochemistry of M+(C2H4)n (M=Ti-Cu, n=1 and 2) complexes. J. Am. Chem. Soc. 120, 1891–1899 (1998)
Koizumi, H., Armentrout, P.B.: Collision-induced dissociation and theoretical studies of Cu+-dimethoxyethane complexes. J. Am. Soc. Mass Spectrom. 12, 480–489 (2001)
Meng, C.K., Fenn, J.B.: Formation of charged clusters during electrospray ionization of organic solute species. Org. Mass Spectrom. 26, 542–549 (1991)
König, S., Fales, H.M.: Formation and decomposition of water clusters as observed in a triple quadrupole mass spectrometer. J. Am. Soc. Mass Spectrom. 9, 814–822 (1998)
Lancaster, G.M., Honda, F., Fukuda, Y., Rabalais, J.W.: Secondary ion mass spectrometry of molecular solids. Cluster formation during ion bombardment of frozen water, benzene, and cyclohexane. J. Am. Chem. Soc. 101, 1951–1958 (1979)
Beyer, M.K.: Hydrated metal ions in the gas phase. Mass Spectrom. Rev. 26, 517–541 (2007)
Hartke, B., Charvat, A., Reich, M., Abel, B.: Experimental and theoretical investigation of microsolvation of Na+-ions in the gas phase by high resolution mass spectrometry and global geometry optimization. J. Chem. Phys. 116, 3588–3600 (2002)
Zatula, A.S., Ryding, M.J., Andersson, P.U., Uggerud, E.: Proton mobility and stability of water clusters containing alkali metal ions. Int. J. Mass Spectrom. 330, 191–199 (2012)
Donald, W.A., Leib, R.D., O’Brian, J.T., Holm, A.I.S., Williams, E.R.: Nanocalorimetry in mass spectrometry: a route to understanding ion and electron solvation. Proc. Natl. Acad. Sci. U.S.A. 105, 18102–18107 (2008)
Donald, W.A., Leib, R.D., Demireva, M., O’Brien, J.T., Prell, J.S., Williams, E.R.: Directly relating reduction energies of gaseous Eu(H2O)n 3+, n = 55-140, to aqueous solution: The absolute SHE potential and real proton solvation energy. J. Am. Chem. Soc. 131, 13328–13337 (2009)
Nemes, P., Schlosser, G., Vékey, K.: Amino acid cluster formation studied by electrospray ionization mass spectrometry. J. Mass Spectrom. 40, 43–49 (2005)
Counterman, A.E., Clemmer, D.E.: Magic number clusters of serine in the gas phase. J. Phys. Chem. B. 105, 8092–8096 (2001)
Zhang, D., Wu, L., Koch, K.J., Cooks, R.G.: Arginine clusters generated by electrospray ionization and identified by tandem mass spectrometry. Eur. Mass Spectrom. 5, 353–361 (1999)
Koch, K.J., Gozzo, F.C., Zhang, D., Eberlin, M.N., Cooks, R.G.: Serine octamer metaclusters: formation, structure elucidation and implications for homochiral polymerization. Chem. Commun. 18, 1854–1855 (2001)
Schalley, C.A., Weis, P.: Unusually stable magic number clusters of serine with a surprising preference for homochirality. Int. J. Mass Spectrom. 221, 9–19 (2002)
Nanita, S.C., Cooks, R.G.: Serine octamers: Cluster formation, reactions, and implications for biomolecule homochirality. Angew. Chem. Int. Ed. 45, 554–569 (2006)
Acknowledgements
This work was supported by the Danish Council for Independent Research | Natural Sciences (grant 4181-00329 to C.J.M.). W.A.D. thanks the Australian Research Council for a Discovery Early Career Researcher Award (DE130100424) and funding from the Discovery Projects scheme (DP160102681). Pia Klingenberg Haussman is thanked for help with the mass spectrometry experiments.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1.82 mb)
Rights and permissions
About this article

Cite this article
Wegeberg, C., Donald, W.A. & McKenzie, C.J. Noncovalent Halogen Bonding as a Mechanism for Gas-Phase Clustering. J. Am. Soc. Mass Spectrom. 28, 2209–2216 (2017). https://doi.org/10.1007/s13361-017-1722-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-017-1722-z