
Abstract
Rheumatoid arthritis (RA) is an extremely painful autoimmune disease characterized by chronic joint inflammation leading to the erosion of adjacent cartilage and bone. Rheumatoid arthritis pathology is primarily driven by inappropriate infiltration and activation of immune cells within the synovium of the joint. There is no cure for RA. As such, manifestation of symptoms entails lifelong management via various therapies that aim to generally dampen the immune system or impede the function of immune mediators. However, these treatment strategies lead to adverse effects such as toxicity, general immunosuppression, and increased risk of infection. In pursuit of safer and more efficacious therapies, many emerging biomaterial-based strategies are being developed to improve payload delivery, specific targeting, and dose efficacy, and to mitigate adverse reactions and toxicity. In this review, we highlight biomaterial-based approaches that are currently under investigation to circumvent the limitations of conventional RA treatments.
Graphical abstract

Similar content being viewed by others
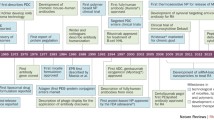

Avoid common mistakes on your manuscript.
Introduction: a brief background, etiology, and epidemiology of rheumatoid arthritis
Rheumatoid arthritis is a debilitating autoimmune disease characterized by chronic inflammation and immune cell infiltration of the joint synovium leading to articular bone and cartilage destruction [1]. Development of rheumatoid arthritis (RA) is complex and driven by various layers of immunity working in concert to promote joint pathology [2]. The pathogenesis of RA is poorly understood, though some clues help demystify the process. Several risk factors for RA have been identified. Sex seems to play a determinative role, as women are 3 times more likely to develop RA than men. Additionally, familial penetrance of RA is up to 50% [3]. Several studies have also linked poor quality of diets to RA. Patients with RA tend to eat more saturated fats and sugars and fewer fruits, vegetables, and whole grains [4]. The most implicated genes conferring risk of RA are (1) the shared epitope (SE), which is encoded by a set of alleles located within the major histocompatibility (MHC) locus, and (2) the PTPN22 polymorphism [3, 5], which encodes an abnormal form of lymphoid protein tyrosine phosphatase, an enzyme that inhibits T cell activation [6]. Antigen-presenting cells (APCs) harboring the SE mutant possess enhanced efficiency for presenting citrullinated peptides in their MHC class II molecules [7], whereas T cells harboring the PTPN22 mutant have impaired self-regulation and are prone to remaining in an active state [8]. Additionally, the wild-type form of PTPN22 normally inhibits intracellular citrullination. However, the PTPN22 polymorphism is unable to do so, resulting in hypercitrullination within immune cells [9]. For the most part, environmental risk factors largely introduce states of inflammation at the mucosal level [10]. For example, smoking, obesity, air pollution, and periodontitis have all been implicated in the development of RA [3, 10], and the presence of both genetic and environmental risk factors significantly increases the likelihood of RA [3]. There is currently no cure for RA, and it continues to be a major health burden affecting ~ 1% of the population [11]. If left unchecked, the chronic systemic inflammation may cause extra-articular complications in the brain, liver, lungs, and heart [2]. Mortality rates in patients with RA is increased by 1.5 times (compared to those without RA), largely due to cardiovascular and lung disease likely arising from or complicated by systemic inflammation [12]. Moreover, those afflicted with RA suffer tremendous economic costs due to expensive treatments, impaired work performance, disability, and early retirement [13]. Further exacerbating their situation are enormous indirect costs through psychological stress, deformity, and progressive decline of self-worth [14]. Current treatment options are suboptimal as they vary in efficacy [15]. Long-term methotrexate is the prescribed anchor drug for RA. However, the PREMIER study, a multicenter, randomized double-blind clinical study, revealed that only 46% of RA patients taking methotrexate experienced a 50% improvement in RA symptoms [16]. Therefore, methotrexate is often combined with other biologics in pursuit of symptom alleviation and remission [15]. Unfortunately, these treatments are accompanied with a multitude of side effects, making adherence to a dosing schedule challenging [17]. An overarching strategy used to treat RA is disruption of inflammatory mediators using targeted drugs [15]. However, these drugs render the patient vulnerable to infection [17], and also are associated with increased risk of cancer [18, 19], which may be through impairing immunosurveillance of oncogenic precursors [20]. Additionally, major concerns have recently arisen regarding the use of immunosuppressants amid the SARS-CoV-2 pandemic [21,22,23]. As such, there is a critical need for uncovering new strategies for the treatment of RA.
RA immunopathogenesis
The quintessential biomarkers for RA are the presence of two autoantibody types: (1) rheumatoid factors (RFs), which are IgM, IgA, or IgG antibodies against the fragment crystallizable (Fc) region of IgG antibodies, and (2) anti-citrullinated protein antibodies (ACPAs), which consist of IgM, IgA, or IgG antibodies that bind peptides which have undergone citrullination, a post-translational modification mediated by the enzyme peptidylarginine deiminase (PAD) [24, 25], though the absence of these autoantibodies does not exclude you from developing RA (seronegative RA), which accounts for about one-third of RA cases [26]. Intriguingly, naïve B cells specific for citrullinated peptides have been detected in the periphery prior to RA manifestation [27], which suggests that autoreactive B and T cells are able to evade central and peripheral tolerance [28]. In support of this, longitudinal studies have revealed that RF and ACPAs can be present in the circulation years presaging the manifestation of RA [29, 30], and the presence of both autoantibody types are associated with elevated levels of inflammatory cytokines and accelerated onset of RA [31], suggesting induction of autoantibodies is an early step in the gradual progression towards development of RA and may require additional factors to trigger disease. This window characterized by presence of autoantibodies and absence or minimal joint inflammation is described as the pre-RA stage [32]. This lag between the detection of RFs and ACPAs and onset of RA may be explained by the evolution and accumulation of multiple ACPA reactivities, which requires time for endogenous antibody maturation and epitope-spreading mechanisms to occur [27, 33, 34]. Additionally, ACPAs develop against various peptide targets including α-enolase, fibronectin, type II collagen, fibrinogen, vimentin, and histones [35], and also exhibit cross-reactivity between citrullinated peptides [36]. Taken altogether, immune cells working cooperatively to accumulate antibodies against various citrullinated epitopes may be the factor that tips the scale in favor of RA onset.
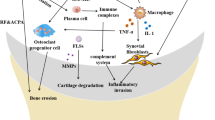
In the periphery, dendritic cells (DCs) harboring the SE variant possess enhanced ability to present citrullinated peptides within their MHC class II molecules [37, 38], and upon activation, the chemokine receptor CCR7 is upregulated to facilitate DC migration towards lymphoid organs rich in CCL19 and CCL21 to initiate an adaptive immune response [39] (Fig. 1(a)). Within the synovium, DCs exposed to inflammatory cytokines such as interferon gamma (IFN-γ) respond by making high levels of interleukin (IL)-12, which, in turn, promotes a Th1 response [40]. Critical to RA pathology, DCs drive autoreactive T cell expansion through citrullinated peptide presentation, co-stimulation, and cytokine production (Fig. 1(b)) [35]. Following this, autoreactive B cell activation is able to receive T cell help to initiate germinal center reactions to produce ACPA-secreting plasma cells (Fig. 1(c)–(e)). The joints possess an abundance of citrullinated epitopes, which provide a natural propensity for ACPAs to accumulate in articular tissue and induce pathology [24]. This stage of established disease, denoted as “early RA”, is characterized by immune dysregulation and inflammation within the joints [32]. Within the joints, following ACPA ligation and RF deposition, immune complex (IC) formation activates macrophages, osteoclasts, neutrophils, and DCs through FCγRIII and complement receptors to initiate an inflammatory response [24]. In comparison to a healthy joint normally devoid of immune cells (Fig. 2a), synovitis in a rheumatic joint is perpetuated by infiltration and coordination of immune cells [40]; synovial infiltration of T helper (Th) 17 cells, Th1 cells, B cells, macrophages, neutrophils, and DCs perpetuates an intensely inflamed environment as long as the antigen is present, eventually leading to adjacent bone and cartilage erosion [2, 35, 39, 41]. Upon cytokine stimulation, activated synoviocytes release the chemokines CCL2 and CX3CL1 to recruit monocytes from circulation and polarize them towards an M1 phenotype or differentiation into osteoclasts [42]. M1 macrophages further promote inflammation through production of the pro-inflammatory cytokines, including tumor necrosis factor (TNF)-α, IL-1β, IL-6, IL-8, and IL-12, as well as [43, 44] granulocyte–macrophage colony stimulating factor (GM-CSF) and monocyte chemoattractant protein-1 (MCP-1) [43, 44], while upon IFN-γ stimulation, Th1 effector cells produce IFN-γ and TNF-α [40]. TNF-α helps activate T cells, synoviocytes, and stromal cells, which in turn release receptor activator of nuclear factor-kB ligand (RANKL), the primary growth factor that drives osteoclastogenesis [45] and osteoclast-mediated bone resorption [42]. Additionally, activated Th17 cells secrete IL-17 and IL-22, which upregulate RANKL expression in synoviocytes [46, 47]. Citrullinated actin and vimentin are expressed on the cellular surface of DCs, and ACPA ligation promotes transdifferentiation into osteoclasts [48]. Osteoclasts express vimentin, and ACPA ligation upregulates TNF-α production [49]. Furthermore, the inflamed joint is abundant in ACPA-secreting plasma cells, B cell aggregates [50], and activated T cells, which secrete CXCL13 and CCL20 to recruit B cells to the synovium [50, 51]. Moreover, functional germinal centers have been detected within the synovium itself [50], which provides a local source of newly minted autoreactive plasma cells. Neutrophils in the circulation become primed and infiltrate the synovium by sensing high levels of IL-8, complement component 5a (C5a), and leukotriene B4 (LTB4) [52]. Upon exposure to IL-17a, TNF-α, and ACPAs within the joint space, neutrophil extracellular trap (NET) extrusion is strongly induced, releasing citrullinated antigens from within the neutrophil into the synovium [53]. This NET formation in turn activates local synoviocytes to produce IL-6 and IL-8, further activating neutrophils [53]. Activated synoviocytes also secrete matrix metalloproteinases (MMPs), which contribute to the remodeling of adjacent cartilage [54]. Neutrophils also participate in cartilage breakdown through secretion of MMP-8, MMP-9, elastase, cathepsin G, and proteinase 3 [54]. There is emerging evidence that distinguishes late RA from early RA by the enhanced contribution of synoviocytes to disease, which may explain why immunotherapy is less successful at this stage [32]. In late stage RA, these synoviocytes display a bona fide transformed phenotype towards fibroblast-like synoviocytes and are highly proliferative and also resistant to apoptosis [32]. Within the joint, these pathological immune cell and stromal cell interactions are descriptive of established RA and are depicted in Fig. 2b.

Immunopathological induction of anti-citrullinated protein antibodies. a Dendritic cells encounter and phagocytose citrullinated proteins in the tissue, and process them into citrullinated peptides presented within MHC II molecules. b The mature dendritic cell migrates to the lymph nodes to activate the citrullinated peptide-specific naïve T cell, inducing differentiation and expansion of citrullinated peptide-specific Th1, Th17, and Tfh subsets. c A naïve B cell undergoes B cell receptor–mediated uptake of citrullinated protein, and processes them into citrullinated peptides presented within MHC II molecules. The newly activated B cell then receives co-stimulatory help from a citrullinated peptide–specific CD4 T cell to initiate germinal center reactions. d Directed evolution of the B cell receptor within the germinal center increases its affinity to citrullinated proteins. e Differentiation of citrullinated protein–specific memory B cells and ACPA-secreting plasma cells

Immunopathogenic mechanisms within a rheumatic joint. a A healthy joint is devoid of immune cell infiltrates. b An inflamed rheumatic joint. ACPA ligation and ensuing immune complex formation activate local innate cells to produce cytokines and chemokines, resulting in migration and activation of additional immune cells from the circulation. TNF-α and RANKL induce osteoclast-mediated resorption of bone. IL-17, IL-22, and TNF-α induce proliferation of fibroblast-like synoviocytes and pannus formation. MMP secretion from neutrophils and fibroblast-like synoviocytes degrades cartilage
Standard clinical practice for RA therapy and associated adverse effects
Upon clinical diagnosis of RA, the standard guideline calls for treatment with a disease-modifying anti-rheumatic drug (DMARD) which include hydroxychloroquine, sulfasalazine, leflunomide, and methotrexate [55]. The first drug regimen typically defaults to methotrexate in combination with glucocorticoids (GCs) such as prednisone or prednisolone [56]. The mechanism of methotrexate is, putatively, through the elevation of anti-inflammatory adenosine to paracrine levels [57, 58], which inhibits neutrophil activation, promotes macrophage polarization towards an M2 phenotype, dampens T cell activation, promotes regulatory T (Treg) cell induction, and promotes endothelial cell tight junction integrity [58]. Additionally, methotrexate is a folate inhibitor, which results in purine depletion, cell cycle arrest, and eventually apoptosis in rapidly proliferating cells [59]. Glucocorticoids are robustly anti-inflammatory through dampening of innate immune cell signaling and promoting of the apoptosis of lymphocytes [60]. While effective, great caution must be considered when prescribing GCs due to side effects such as loss of bone density [61], insomnia, depression [62], and also long-term toxicities [63]. If the prescribed therapy is successful in reducing RA symptoms, then treatment is tapered until remission is sustained at the smallest dose possible [64]. However, if the therapy is ineffective, then the treatment strategy is continuously modified by adding or substituting available biological or targeted synthetic DMARDs until remission is achieved, which is more likely with combinatorial therapies [56]. The biological DMARDs consist of biologically manufactured molecules that have been harnessed for their effect in treating RA; these include TNF inhibitors (such as adalimumab, infliximab, and etanercept), the immune checkpoint protein cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) mimic (abatacept), anti-CD20 antibody (rituximab), anti-IL-6 antibodies (tocilizumab) [35], and histone deacetylase (HDAC) inhibitors (trichostatin A, givinostat, or valproic acid) [65]. Inhibition of TNF-α removes a pan-activating cytokine that is highly elevated in the inflamed synovium [66]. While the toxicity of TNF inhibitors is largely absent, common side effects include injection site reaction as well as increased risk of infection [67]. The CTLA-4 mimic, abatacept, works by competitively binding to co-stimulatory molecules, CD80 and CD86, on the surface of APCs, which prevents the T cell CD28 co-receptor from docking and providing necessary T cell activation signals [68]. Patients receiving abatacept commonly experience headaches and nausea [68], and more severe adverse effects such as serious infection, cardiovascular events, cancer, or death occur at 6.4/100 patient-years [69]. The anti-CD20 antibody, rituximab, works by depleting B cells and, thus, the source of ACPAs [70]. With rituximab, severe adverse effects of infection, cardiovascular events, cancer, or death also occur at a rate of 7.3/100 patient-years [69]. Targeted, synthetic DMARDs are molecules that have been fabricated to target specific pathways in RA pathology, for instance, the Janus kinase (JAK) inhibitors — tofacitinib and baracitinib [71]. Many inflammatory cytokines that contribute to RA pathology such as IL-6, interferons (IFNs), granulocyte colony-stimulating factor (G-CSF), and GM-CSF rely on the mediator proteins, JAK, to transmit intracellular cascades required for their production; thus, JAK inhibition serves to suppress inflammatory cytokine levels [72]. Long-term administration of tofacitinib revealed a rate of serious adverse effects (infection, cardiovascular events, cancer, or death) at 6/100 patient-years [69]. Depending on the disease activity measure used to determine remission, a multinational cross-sectional meta-analysis revealed that remission was achieved at rates between 8.6 and 19.6% in RA patients [73]. The rate of relapse (1) for ongoing treatment was 15.8%; (2) for remission and 50% dose reduction, 38.9%; and (3) for stopping treatment, 51.9%, as determined in a multicenter, randomized controlled, parallel-group phase 3 trial [74].
Biomaterial-based immunotherapies for RA
Existing drugs inherently possess limitations in terms of half-life, dosage, efficacy, and toxicity. For instance, the half-life of methotrexate is approximately 6 h [75]. The incorporation of biomaterials into drug design, or rather drug delivery technology, blunts conventional drug limitations, permitting augmentations in a myriad of ways. Biomaterials are any materials not considered food or drugs that come into contact with tissue [76] and are being routinely investigated in many therapeutic applications including cardiovascular disease [77], neurodegenerative disease [78], cancer [79], vaccines [80], and autoimmunity [81,82,83]. Using biomaterials to engineer drug delivery systems across different scales offers numerous advantages. For example, encapsulation of the drug protects it from environments that may cause degradation [76] such as changes in pH or exposure to enzymes. This also extends the drug half-life, which allows for less frequent and smaller doses, thereby lowering the risk of toxic side effects [76]. Manipulation of physicochemical properties of the biomaterials also allows extensive versatility in engineering particulate systems. For instance, passive targeting describes a circumstance where nano- or microparticles can accumulate in target tissues through their innate characteristics [84]. Nanocarriers smaller than 5 µm are readily engulfed and thus passively target and accumulate within phagocytes [84]. Moreover, nanocarrier uptake by DCs also tend to accumulate within lymph nodes (LNs) due to DC ability for intranodal trafficking [84]. Another example of passive targeting is through exploitation of the enhanced permeability retention (EPR) effect [85]. The EPR effect describes a phenomenon where nanoparticles tend to accumulate within tissues with leaky vasculature, such as sites of inflammation, or within tumors [85]. Membrane surfaces of the nanocarriers can also be customized through ligation of antibodies, peptides, and receptors, or incorporation of environmentally sensitive bonds [86] to facilitate active targeting of specific tissue compartments [84]. Nanocarriers can also function as drug depots with programmable release kinetics [84]. In this review, we will discuss the employment of liposomes, nanoparticles (NPs), microparticles (MPs), scaffolds, and hydrogels as versatile drug carriers as well as systems to directly modulate immune responses for treating RA.
Current therapeutics for RA fall prey to acute and chronic toxicities and many other adverse effects [17]. Utilizing liposomes as a drug carrier helps to mitigate these unfavorable outcomes [87]. Liposomes as drug carriers boast many advantages with their poor immunogenicity, versatility, and lack of toxicity [88]. In addition, by nature of hydrophobic interactions, the liposome constituents self-assemble into particles with the lipid bilayer segregating the internal enclosures into a lipophilic region encircling an internal lipophobic core, allowing incorporation of both lipophilic and lipophobic drugs and ease of combinatorial delivery [89, 90]. Discovery of polyethylene glycol (PEG) has also served to increase circulatory retention of liposomes by mitigating off-target engulfment [91]. Additionally, the liposome surface is modifiable, as charge alterations and functionalization with antibodies, peptides, and ligands have all been incorporated to improve specific targeting [90]. It follows that liposomes have garnered considerable interest in improving RA therapies. Ren et al. [92] have comprehensively characterized optimal liposome parameters that favor accumulation within the inflamed synovium. The group fabricated different liposomes with altered surface charges, diameters, PEG chain length, and lipid composition, and tested the biodistribution of different formulations via intravenous (i.v.) injection in the collagen-induced arthritis (CIA) mouse model [92]. They determined that a 100-nm, slightly negative liposome composed of 10% 5-kDa PEG chains yielded the highest accumulation within the diseased joint [92]. The data obtained in this study serve as a valuable reference in fabrication of liposomes for articular targeting. Largely, liposomes have been investigated as vehicles for delivery of anti-rheumatic drugs to sites of inflammation. To this end, several groups have demonstrated encapsulation of bioactive molecules within liposomes is an effective strategy for enhancing articular targeting and potency. For example, dexamethasone [93, 94], glutathione [95], and tofacitinib [96] have been encapsulated within liposomes and shown to be more efficacious in comparison to the unencapsulated drugs in various models of inflammatory arthritis [93, 95,96,97].
Harnessing endogenous immunologic pathways to treat rheumatic disease is one strategy that has gathered significant interest of late. Nano- and microparticles are well suited for various immunomodulatory applications by design and offer a myriad of benefits. The size of the particles, which ranges from nanometers to microns, allows for both active and passive targeting to sites of interest [84], and also alters the rate of particle degradation to modify release kinetics to be either slower or more rapid [98]. They are uniquely equipped to modulate immune cell function due to their aptitude for direct cytosolic or surface delivery [84, 99]. Notably, cytosolic delivery of a peptide within an APC allows for endogenous biomachinery to cross-present the peptide within MHC class I molecules [100, 101]. This allows for encapsulation of a target peptide within a particle that will promote presentation within both MHC class I and class II molecules of an APC, allowing for modulation of both CD8 and CD4 T cells, respectively [100]. Moreover, the potential for customizability is vast. Altering the material used for fabrication of NPs and MPs can bestow dynamic characteristics to the drug carrier system that allows for targeting of different environmental cues or specific disease states [86], or even potentially conferring intrinsic characteristics into the particle itself [102]. For example, many gut-derived molecules exert immunosuppressive effects and are garnering interest for their potential to be fabricated into particles that possess tolerance-inducing effects [102], and poly-conjugated aspirin particles have been used to confer innate anti-thrombus activity into the particle itself [103]. Additionally, particles that incorporate pH-sensitive bonds such as aliphatic monomers incorporating cleavable acetal links can facilitate preferential cargo delivery into specific environments where the pH is acidic [86]. Other environmental attributes can be targeted for precision-based delivery of the payload through incorporation of bonds that are sensitive to various environmental cues, such as temperature, enzymes, light, and hypoxia [86]. A comprehensive review encompassing many different stimulus-responsive biomaterials can be found here [86]. Additionally, the membrane surface of NPs and MPs can be functionalized with a variety of ligands, such as peptides, receptors, or antibodies, further allowing precise delivery to specific immune cell subsets or tissues [83, 84, 104]. Microparticles can also act to provide safe intracellular delivery of a broad range of biological agents including peptides and nucleic acids [84]. As such, design of novel NPs and MPs is being heavily investigated to specifically alter aberrant immune cell behavior to treat various immune-dysregulated maladies [82,83,84]. Unsurprisingly, researchers have begun investigating the use of biomaterial-based systems to treat RA, given its pathogenesis being highly immune cell–driven [81]. In addition to this, in order to meet the nutrient and oxygen demands of the infiltrating immune cells, the inflamed synovium becomes a site of angiogenesis and vascular remodeling [105], which under the context of inflammation results in loose endothelial cell junctions and leaky vasculature [106]. As such, RA is a disease that benefits from the EPR effect, allowing nano-sized particles intrinsic ability to preferentially extravasate and accumulate within the joint synovium [107]. Below we will discuss novel biomaterial-based interventions for RA that are currently under investigation, organized based on immune cell target, which are concisely summarized in Table 1. Our discussion spans the fabrication, characterization, and composition of these biomaterial-based therapies, as well as their respective indicated mechanisms of RA interruption.
Targeting innate immune cells for modulation of rheumatic pathology
Neutrophils
Neutrophil migration into the inflamed joint exacerbates and sustains joint pathology through cytokine production, NET extrusion, and MMP secretion [52, 108]. To reduce pathological neutrophil activity, Zhang et al. [109] fabricated doxorubicin-conjugated, albumin NPs that incorporated pH-sensitive bonds, allowing doxorubicin liberation upon neutrophil engulfment. This resulted in doxorubicin-mediated neutrophil apoptosis, curbing neutrophil-mediated inflammation in murine models of sepsis and cerebral ischemia/reperfusion [109]. Depletion of neutrophils using cytotoxic NPs may interrupt chronic joint inflammation and protect against neutrophil-mediated degradation of cartilage. Neutrophils are the most abundant circulating cells and possess inherent ability to rapidly migrate to inflamed regions [110]. Harnessing these unique aspects of neutrophil biology, Lyu et al. [111] fabricated human serum albumin NPs (HSA-NPs) decorated with a mannose ligand to provide neutrophil targeting via ligation to neutrophil surface mannose receptors (MRs). This group postulated that NPs can hitch a ride on a synovium-bound neutrophil, allowing for subsequent accumulation of the payload within the inflamed joint [111]. Using a rat model of CIA, Lyu and the group encapsulated methotrexate within the mannose-decorated HSA-NPs and, upon i.v. administration, showed that there was an in increase in uptake within neutrophils as well as preferential accumulation within arthritic limbs, compared to the same NP lacking a mannosylated surface [111]. This neutrophil-mediated delivery of a methotrexate-encapsulated NP resulted in significantly greater alleviation of inflammatory arthritis pathology when compared to unencapsulated methotrexate, and even restoration of bone structure to that of a healthy rat [111]. In similar design, Hu et al. [112] also sought to borrow the neutrophil’s innate ability for migration into inflamed tissue. The group fabricated a liposome decorated with sialic acid and encapsulated dexamethasone palmitate in its core [112]. Neutrophils express high levels of the sialic acid receptor, CD62L (also known as L-selectin), which is the receptor that mediates neutrophil slow rolling and subsequent transendothelial migration [113]. The group demonstrated in a rat model of adjuvant-induced inflammatory arthritis that i.v. administration of this sialic acid–conjugated NP laden with dexamethasone palmitate was able to preferentially accumulate within the arthritic limbs, and significantly reduce serum levels of IL-1β and TNF-α, and alleviate joint histopathology, when compared to the vehicle control [112]. Both groups successfully showed that neutrophil-targeting mechanisms could be used to transform the neutrophil into a Trojan horse to efficiently deliver anti-rheumatic drugs directly into the inflamed synovium. Zhang et al. [114] devised another clever strategy to harness the innate attributes of neutrophils for alleviation of synovial inflammation. This team isolated the plasma membrane from activated human neutrophils and used them to coat poly(lactic acid-co-glycolic acid) (PLGA) NPs [114]. These PLGA NPs effectively masqueraded as activated neutrophils and acted as cytokine sinks reducing serum levels of IL-1β and TNF-α [114]. In using this neutrophil-like NP, the group demonstrated a significant reduction in synovial chondrocyte activation in the CIA mouse model, as well as a transgenic humanized TNF-α mouse model of inflammatory arthritis [114]. The overall strategies described here aim at selective apoptosis of neutrophils, or exploiting neutrophil migration, to aid in cargo delivery to the inflamed joint.
Macrophages
Stymieing the contribution of macrophages to RA pathogenesis is another promising avenue of intervention, due to the central role they play in causing and sustaining joint pathology via cytokine production and participation in osteoclastogenesis [44]. Activated macrophages have been shown to upregulate expression of the folate receptor β (FRβ) [115], which is more sparse on non-activated macrophages [116]. Therefore, macrophages expressing FRβ can preferentially identify inflammatory macrophages and, thus, have amassed keen interest as a selective therapeutic target for treating RA [115]. To this end, ligation of folate to the surface membrane of a biomaterial-based carrier has emerged as an effective strategy for incorporating a targeting component specific for pro-inflammatory macrophages. Verma et al. [117] engineered a double-layered liposome, where the inner liposome encapsulated both prednisolone and methotrexate, while the outer membrane was decorated with folate [117]. Following i.v. administration of this double liposome into the CIA rat model, the group observed preferential accumulation of both prednisolone and methotrexate within the inflamed joint, compared to the non-inflamed joint [117]. The transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) is a master regulator of a pro-inflammatory response in macrophages [118]. Duan and Li [119] used folate-functionalized liposomes to co-deliver methotrexate alongside a NP containing small interfering RNA (siRNA) against NF-κB (Fig. 3a) into the diseased joint. They hypothesized that macrophage uptake of this liposome would result in NF-κB silencing, impairing activation and production of pathogenic cytokines. The group performed i.v. administration of this liposome into the CIA mouse model and observed amelioration of overt inflammatory arthritis symptoms (Fig. 3b), as well as a fourfold decrease in serum levels of IL-1β (Fig. 3c) and a twofold decrease in TNF-α (Fig. 3d), compared to the same liposome without folate conjugation [119], demonstrating the effectiveness of the folate targeting system. In efforts to further refine a delivery system that excels at preferentially targeting activated macrophages, Chen et al. [120] incorporated a catalase within the folate-conjugated liposome. In addition to upregulation of FRβ, activated macrophages also express high levels of intracellular reactive oxygen species (ROS) in the form of hydrogen peroxide (H2O2) [120]. As such, Chen and co-authors took advantage of this secondary characteristic of activated macrophages by adding catalase into their delivery system. Once the liposome is engulfed by the macrophage, H2O2 diffuses into the liposomal core and is exposed to catalase, breaking it down into H2O and O2, which leads to destabilization of the liposome and rapid release of the methotrexate payload [120]. Incorporation of two targeting components further enhances the rigor in selectively targeting only the inflammatory macrophages, and contributes to mitigating off-target effects such as regulatory macrophages. In vitro studies showed that in ROS-abundant conditions, encapsulation of catalase improved the cumulative release of methotrexate from the liposome by a fourfold increase, compared to the same liposome without catalase [120]. Additionally, i.v. delivery of this catalase- and folate-functionalized liposome laden with methotrexate into CIA mice exhibited greater inflammatory arthritis disease remission, compared to the folate-conjugated liposome containing methotrexate only, demonstrating that incorporation of catalase improves the efficacy of this carrier system in treating inflammatory arthritis [120]. In a similar fashion, Ni et al. [121] formulated a folate-conjugated NP out of 4-phenylboronic acid pinacol ester-conjugated α-cyclodextran (Oxi-αCD), another strategy to incorporate a ROS-sensitive component into the carrier system, and they subsequently encapsulated dexamethasone into its core [121]. This also yielded a ROS-reactive nanocarrier armed with two attributes that target inflammatory macrophages [121]. Administration of this NP into the CIA mouse model resulted in accumulation within the inflamed joint and downregulation of TNF-α and B cell-activating factor (BAFF), and alleviated inflammatory arthritis symptoms within the paw [121]. Ligation of macrophage nuclear retinoic acid receptors (RARs) to its ligand, all-trans-retinal, promotes polarization from an inflammatory M1 phenotype towards a regulatory M2 phenotype [122]. In efforts to re-educate pro-inflammatory synovial macrophages, Li et al. [123] incorporated three components into a pH-sensitive NP, (1) all-trans-retinal, (2) galactose, and (3) triptolide, an anti-inflammatory compound, into a dextran NP containing hydrazone bonds [123]. This NP served to target macrophages via the galactose-type lectin (MGL) receptor, and subsequent intracellular delivery of all-trans-retinal and triptolide would coerce polarization towards a regulatory phenotype [123]. Administration of this NP into CIA mice achieved a reduction of TNF-α, IL-6, and IL-1β within the synovium [123]. On the topic of re-polarization, silver NPs have been shown to increase intracellular Ag+, promoting macrophage polarization towards an M2 phenotype through inhibition of phosphoinositide 3-kinase (PI3K)/Akt signaling, decreasing expression of glucose transporter expression, and inducing autophagy [124]. In a similar macrophage re-educating strategy, Yang et al. [125] functionalized a silver NP with folate and successfully induced the death of inflammatory macrophages, or their polarization towards an M2 phenotype in the CIA murine model [125]. Taken altogether, folate-functionalized nanoparticles provide an effective and customizable framework for targeting problematic macrophages. The common and effective strategies employed were either depletion of inflammatory macrophages or re-polarization towards a regulatory phenotype, which were both effective in alleviating inflammatory arthritis symptoms.

Adapted from Duan and Li [119]. Open access
In vivo targeting of macrophages using a folate-conjugated liposome/nanoparticle hybrid encapsulating NF-κB siRNA and methotrexate. a Schematic diagram of the liposome/nanoparticle hybrid carrier. b Folate-functionalized carrier encapsulating NF-κB siRNA and methotrexate was the most efficacious at reducing the arthritis score in the collagen-induced arthritis (CIA) mouse. c Forty-four days post-treatment of a CIA mouse, the folate-functionalized carrier encapsulating NF-κB siRNA and methotrexate reduced serum levels of IL-1β fourfold and d TNF-α by twofold compared to the same carrier lacking folate-functionalization.
Dendritic cells
Dendritic cells play significant and opposing roles in promoting autoimmunity [126] or maintaining tolerance [127]. Upon exposure to pathogen- or damage-associated molecular patterns (PAMPs/DAMPS), DCs undergo maturation and upregulate inflammatory cytokines IL-12 and IFN-α, as well as co-stimulatory molecules CD80 and CD86, ultimately orchestrating an inflammatory immune response against the associated pathogen [128, 129]. On the other hand, DCs exposed to immunosuppressive molecules such as IL-10 and TGF-β1 induce tolerogenic dendritic cells (tDCs) which express low levels of IL-12, IFN-α, CD80, and CD86 [129]. Tolerogenic DCs are proficient mediators of tolerance, the mechanism of which is primarily through inducing anergy of autoreactive T cells and induction of regulatory T cells (Tregs) [127, 129, 130]. As such, modulating DCs towards a tolerogenic phenotype has amassed considerable interest for their potential in restoring tolerance in autoimmune diseases [83, 131, 132], including transplant rejection [133], type I diabetes [130, 134], multiple sclerosis [135], and RA [82]. A plethora of biomolecules have been identified that promote induction of tDCs such as 1α,25-dihydroxycholecalciferol (vitamin D3), transforming growth factor-β1 (TGF-β1), IL-10, galectin-1, and glucocorticoids, among others [129]. Of particular interest, vitamin D3 has been extensively studied for its proficiency in DC modulation that is permissive of downstream induction of Treg cells [130, 136]. Autologous delivery of tDCs directly into the arthritic joint has undergone a phase I clinical trial with promising results [137] (NCT01352858, NCT03337165). To harness endogenous tDC mechanisms, Allen et al. [82] devised a novel dual PLGA microparticle system termed “REGvac” to induce generation of tDCs in vivo [82]. This microparticle system consists of (i) a 1-μm phagocytosable microparticle encapsulating type II collagen and vitamin D3 to facilitate uptake by dendritic cells and (ii) a 30-μm unphagocytosable microparticle encapsulating GM-CSF and TGF-β1, to mediate DC chemotaxis into a pro-tolerogenic environment (Fig. 4a) [82]. Subcutaneous delivery of REGvac in the CIA mouse prevented DC maturation (Fig. 4b) ameliorated inflammatory arthritis symptoms (Fig. 4c and d), improved gait, and induced Treg cells within the draining lymph nodes of the inflamed joint [82]. In a similar vein, Galea et al. [138] formulated a liposome encapsulating vitamin D3 (calcitriol) alongside the model antigen ovalbumin (OVA) to investigate the capacity for inducing antigen-specific tolerance [138]. Using the TCR-transgenic OVA-specific T cell line DO11.10, subcutaneous immunization with this vitamin D3/OVA liposome was found to induce expansion of OVA-specific Tregs and induce anergy of OVA-specific non-Treg T cells via upregulation of PD-1 [138]. Under inflammatory conditions where mice were primed with OVA and the adjuvant QuilA, the calcitriol/OVA liposome induced DC downregulation of MHC class II, and upregulated expression of PD-L1, which was found to suppress OVA-specific T cell expansion and induce Tregs in a PD-L1-dependent manner [138]. Next, Galea and co-authors assessed the capability of this antigen/calcitriol liposome for inducing tolerance in a mouse model of proteoglycan-induced inflammatory arthritis, and also type IV collagen α3–induced model of vasculitis, where the group observed suppression of arthritis score and also a reduction in antigen-specific CD4+ T cells [138]. Inhibition of the transcription factor NF-κB has been show to impair DC maturation [139]. Capini et al. [140] showed that DC-targeted NF-κB inhibition alongside autoantigen delivery was able to induce antigen-specific FoxP3+ Treg–mediated suppression of inflammatory arthritis using an antigen-induced arthritis (AIA) mouse model, with methylated BSA (mBSA) as the antigen [140]. In order to achieve this, the group fabricated egg phosphatidylcholine liposomes encapsulating one of three different NF-κB inhibitors: curcumin, quercetin, or Bay11-7082, as well as mBSA in order to facilitate direct co-delivery of the NF-κB inhibitor and mBSA directly into the cytosol of DCs via subcutaneous injection into the mouse tail [140]. Following these promising results, Benham et al. [141] conducted a phase 1 clinical trial using autologous DCs treated with Bay11-7082 and exposed to four different RA-associated citrullinated antigens (termed Rheumavax) to treat RA patients. A single dose of Rheumavax was administered intradermally into the upper thigh and yielded no or minimal adverse effects and was able to reduce RA disease activity [141]. Taken together, the potential for using biomaterial-based strategies to immunoengineer DCs for antigen-specific tolerance induction in RA is robust. Targeting DCs in RA therapy is a sound enterprise given its role as a nucleating factor in the induction of ACPAs [5].

Adapted from Allen et al. with permission [82]
In vivo immunomodulation of dendritic cells using a dual microparticle delivery system in the collagen-induced arthritis (CIA) mouse model. a Schematic of constituents of the microparticle system (REGvac) and proposed cellular targets. REGvac was delivered subcutaneously in the abdominal region, draining to the inguinal lymph nodes. b Twenty-eight days and 56 days post-treatment, spleen, inguinal, and popliteal lymph nodes were harvested and flow cytometry was performed to measure dendritic cell maturation status. c CIA mice were clinically scored for limb erythema and swelling. d Representative photographs of the paws of a REGvac-treated CIA mouse at 14 days and 56 days post-treatment.
Biomaterial-based instruction of adaptive immune elements for correction of RA
T cells
The transcription factor c-Rel is a master regulator for inflammatory cytokine production in Th1 and Th17 cellular responses and has been implicated in various autoimmune diseases [142]. Fan et al. [143] used poly(ethylene glycol)-b-poly(l-lysine)-b-poly(l-leucine) (PEG-PLL-PLLeu) triblock polymers to construct a nanoparticle encapsulating siRNA against c-Rel transcripts [143]. Delivery of the c-Rel-loaded NPs into CIA mice halted disease progression and ameliorated joint pathology [143]. In vivo delivery of c-Rel siRNA NP exhibited a reduction in DC ability to activate T cells, resulting in abrogation of Th1- and Th17-associated cytokines, IFN-γ and IL-17, as well as a reduction in inflammatory cytokines TNF-α, IL-1β, IL-6, IL-12, and IL-23 [143]. Additionally, the c-Rel siRNA NP reduced T cell infiltration into the diseased joint [143]. The antioxidant coenzyme Q10 (CoQ10) inhibits the transcription factor STAT3, a critical regulator for Th17 effector function [144, 145]. Jhun et al. [146] used a liposome and gold hybrid nanoparticle to encapsulate CoQ10 (LGNP-CoQ10) for curbing Th17 activity in the CIA mouse model [146]. These CoQ10 NPs were administered orally, and the authors found a greater reduction in arthritic score, and a reduction in Th17 numbers and cytokine levels, compared to unencapsulated CoQ10 [146]. Regulatory T cells are remarkable mediators of tolerance through production of IL-10 and their ability to competitively inhibit DC-mediated co-stimulatory activation of T cells [28, 126]. Moreover, the absence of Treg cells results in spontaneous autoimmunity [147]. As such, engineered biomaterial strategies to enhance Treg function is an enticing enterprise. Notably, Fisher et al. [148] encapsulated the Treg chemokine, CCL22, within a PLGA NP. Administration of these CCL22 NPs into an allografted rat hind limb resulted in local recruitment of Tregs and mediated long-term graft acceptance [148]. This study provides an interesting concept of exploiting Treg recruitment to a target site that may be applied to rheumatic joints. Activated macrophages and dendritic cells secrete high levels of IL-23 to sustain Th17 activity [46, 47]. Th17 cells produce RANKL themselves, but also produce IL-17 and IL-22 to induce RANKL expression in synoviocytes, ultimately resulting in osteoclast activation [46, 47]. Rather than systemic administration of neutralizing antibodies, Lima et al. [149] fabricated a liposome functionalized with anti-IL-23 neutralizing antibodies to concentrate the site of cytokine neutralization within the diseased joint. The authors tested the anti-IL-23 liposomes on the serum of RA patients and were able to achieve a significant reduction of IL-17 production [149], indicating that IL-23 neutralization successfully impaired Th17 cell ability to make IL-17. Functionalizing particles with antibodies to neutralize cytokines is a promising strategy, as there are many different cytokines within the arthritic joint that sustain inflammation. In addition to cytokines, this strategy may also be employed to neutralize harmful autoantibodies within circulation [150], and given how RA is an autoantibody-driven disease [35], an engineered particle that reduces the serum levels of ACPAs and RF may be an effective therapeutic venture for treating RA.
B cells
As the culprits responsible for maturing and generating ACPAs, B cells undeniably play a destructive role in RA pathogenesis and in sustaining chronic disease [27, 33, 34, 151, 152]. As such, depletion of B cells in RA has seen therapeutic effects [65, 153]. The pro-survival factor, BAFF, is produced by APCs and is important for B cell development and maturation [154], and also aids in T cell co-stimulation [155]. Additionally, BAFF overexpression has been linked to autoimmunity [154]. In order to curb RA-associated pathogenic B cell activity, Wu et al. [156] constructed a PEGylated PLGA nanoparticle encapsulating siRNA against BAFF. Administration of this NP i.v. into CIA mice resulted in amelioration of paw pathology, a reduction in circulating anti-collagen antibodies, and a reduction in the number of B cells [156]. In an effort to introduce a targeting component specific for only autoreactive B cells, Pozsgay et al. [157] covalently fused a fibrin peptide, bindable by ACPAs, and a peptide from human immunodeficiency virus type 1 (HIV-1), to the surface of PLGA NPs, termed “bifunctional NPs.” The fibrin peptide provides specificity for ACPA-producing B cells, and the HIV-1 peptide promotes complement-mediated cytotoxicity [157]. B cells were isolated from RA patients and, upon Toll-like receptor 7/8 stimulation, were found to secrete antibodies against the fibrin peptide [157]. Following this, the bifunctional NP was introduced in vitro with the RA-derived B cells, alongside human sera to provide complement components [157]. The authors observed a reduction in fibrin-specific B cells in the presence of both bifunctional NPs and human sera, but not in heat-inactivated human sera [157], suggesting that the bifunctional NPs were indeed inducing complement-mediated death of fibrin-specific B cells. In a similar precision-based design that targets autoreactive B cells, Bednar et al. [158] fabricated a liposome conjugated with (1) a high-affinity glycan ligand for the inhibitory B cell receptor, CD22, and (2) a cyclic citrullinated peptide, to facilitate interaction with B cells specific for this autoantigen. Upon B cells encountering this dual-functionalized liposome, the glycan ligand engages CD22 and induces B cell deletion, subsequently restoring B cell tolerance towards the citrullinated antigen [158]. Bednar and co-authors demonstrated that this liposome was able to induce tolerance in autoreactive B cells isolated from RA patients, as well as in SJL/J mice immunized against cyclic citrullinated peptides [158]. Here, the significance of applying biomaterial-based designs in B cell-centric therapies is enabling the precise targeting of autoreactive B cells, thereby minimizing off-target effects and decreasing toxicities.
Using particles for manipulation of RA-promoting, non-immune cells
In RA, osteoclast generation is highly upregulated and they are the primary cells that facilitate bone damage [159]. Osteoclastogenesis is a process by which RANKL-activated macrophages undergo cell fusion to yield a multinucleated osteoclast [160]. It has been reported that mononuclear precursors gradually crescendo surface expression of mannose and MRs during the process of osteoclastogenesis, and subsequent MR ligation by mannose is important for mediating cellular fusion [161]. Neog et al. [162] have previously demonstrated that the polyphenol p-coumaric acid exerts anti-inflammatory and anti-osteoclastogenic effects in an AIA rat model. In a follow-up study, Neog and co-authors [163] took advantage of osteoclast upregulation of MR and incorporated mannose into the liposomal surface to provide a p-coumaric acid-encapsulated liposome with an osteoclast-targeting component [163]. In the AIA rat model, biodistribution of this mannosylated liposome was demonstrated to preferentially accumulate in the arthritic paw following i.v. administration [163], verifying the efficacy of mannose to guide liposomes to the inflamed paw. Subsequent isolation of primary synovial macrophages from the AIA rat treated with the p-coumaric acid-laden liposome showed impaired ability to perform osteoclastogenesis and bone resorption, compared to methotrexate-treated and untreated synovial macrophages [163]. In another follow-up study from the same group, Sujitha et al. [164] incorporated berberine into the surface of the mannosylated liposome, which has been previously shown to induce silencing of inflammatory genes in macrophages through upregulation of microRNA (miRNA)-23a [165]. Using bone-marrow derived macrophages, the berberine-loaded mannosylated liposome demonstrated in vitro inhibition of osteoclastogenesis and impaired bone resorption ability in existing osteoclasts [164]. Furthermore, various gold nanoparticle formulations have been shown to inhibit osteoclastogenesis and bone resorption [166,167,168,169], likely through exerting intracellular antioxidant effects [166]. Fibroblast-like synoviocytes (FLSs) play key roles within the inflamed joints. In RA, articular inflammation activates FLSs to invade the cartilage space, participate in extracellular matrix (ECM) remodeling via MMP production, and also produce inflammatory cytokines such as IL-6, GM-CSF, and RANKL [170]. Tang et al. [171] formulated a photosensitive PLGA NP by encapsulating indocyanine green (ICG), as well as liquid perfluorocarbon, as an O2 carrier to introduce ROS. Tang and co-authors sought to selectively kill FLSs through a combinatorial intervention of photodynamic and sonodynamic therapy. In this system, introduction of near-infrared (IR) energy followed by ultrasonic irritation allows targetable and triggerable ROS-mediated cytotoxicity within a target site [171]. Tang and co-authors utilized the synoviocyte cell line, MHA7, and demonstrated that in vitro, their photosensitive NP was able to induce robust ROS-mediated death of synoviocytes upon exposure to near-IR and ultrasonic energies [171].
Scaffolds help to build therapy
To this point, our discussion has centered around particulate formulations largely because their unique properties allow researchers to effectively target immune cells. However, bulkier material constructs such as scaffolds and hydrogels are also under intense investigation for applications in therapy of rheumatic disease. Biomaterial-based scaffolds and hydrogels offer numerous benefits for tissue engineering, drug delivery [172], cancer therapy [173], and immunomodulatory applications [174]. Engineered constructs possess a myriad of customizable attributes such as encapsulation, type of polymer, firmness, size of pores, and release kinetics [172]. These biocompatible constructs excel as support depots through localized sustained release of bioactive molecules [172]. They can also be seeded with autologous precursor cells to promote in situ growth of new organs or tissue [175], and can also be used to deliver regenerative cells alongside growth factors and hormones to promote cell proliferation and tissue repair at the target site [176]. In terms of immunomodulatory applications, scaffolds and hydrogels possess unique prowess at modifying the local environment. Encapsulation of immunomodulatory agents within engineered constructs allows these bioactive molecules to concentrate at the site of interest and exert potent effects on the nearby immune cells [174]. For the majority of the disease, rheumatoid arthritis pathology is contained to the joints [35]. This specific localization of diseased sites makes the use of intra-articular implantation of scaffolds and hydrogels an attractive therapeutic strategy. Intra-articular hyaluronic acid (HA) supplementation has been commonly used as an anti-inflammatory agent to treat inflammatory arthritides [177]. Seo et al. [178] incorporated methotrexate into a cross-linked HA depot to promote long-term sustained release within the diseased joint, which helps to mitigate side effects of systemic methotrexate treatment. The group then administered this depot intra-articularly into the CIA rat model and observed sustained retention of elevated HA and methotrexate within the synovium, resulting in alleviation of inflammatory arthritis joint pathology, improved articular cartilage thickness and bone formation, and downregulation of TNF-α and IL-6 [178]. Wu et al. [179] sought to devise a self-assembling hydrogel system by using NPs made of caprolactam–polyvinyl acetate–polyethylene glycol (Soluplus). The Soluplus NPs are thermosensitive, thickening in viscosity at body temperature, allowing for hydrogel formation upon injection into the target site [179]. Wu and co-authors encapsulated tacrolimus, an inhibitor of T cell activation, within the Soluplus NPs, allowing for a self-assembling drug depot upon injection [179]. The group administered the tacrolimus-laden Soluplus NPs via subcutaneous injection adjacent to the arthritic joints, and observed a significant reduction in edema within the arthritic paw over time, compared to the same hydrogel without tacrolimus [179]. Chen et al. [180] formulated a hydrogel composed of Pluronic F127, poly(γ-glutamic acid), and hyaluronic acid, and encapsulated infliximab, an anti-TNF-α antibody. Through incorporation of infliximab into the hydrogel, Chen and co-authors aim to sequester infliximab within the diseased joint, thereby mitigating systemic suppression of TNF-α [180]. In a rabbit model of ovalbumin (OVA)–induced RA, the group injected the hydrogel intra-articularly 1 week post RA induction. After 6 weeks, they observed alleviation of joint pathology and cartilage destruction, as well as a reduction in synovial fluid levels of TNF-α, IL-1β, IL-6, and IL-17, compared to the same hydrogel without infliximab [180]. Zhao and co-authors [181] sought to add a healing component to their strategy in addition to alleviation of joint pathology. The group utilized an infliximab-based hydrogel combined with a 3D-printed porous metal scaffold (3DMS) that was seeded with adipose-derived mesenchymal stem cells (ADSCs), where introduction of the ADSCs would aid in the rebuilding of the articular ECM [180]. This scaffold was implanted intra-articularly within the OVA-induced rabbit model and led to amelioration of RA pathology and promoted restoration of cartilage and bone repair [180]. Similarly, Yin et al. [182] prepared a hydrogel system with F127 and a polyethyleneimine NP encapsulating (1) the non-steroidal anti-inflammatory drug, indomethacin; (2) methotrexate; and (3) siRNA against MMP-9, to halt further damage to the articular ECM. Yin and co-authors administered this hydrogel into the inflamed joint of a CIA mouse and observed significant amelioration of joint pathology as well as a reduction in TNF-α, IL-6, and MMP-9 in serum and in synovial fluid [182]. Remarkably, they also observed joint architecture to return close to baseline [175].
Future outlooks and conclusion
To date, RA remains a prevalent health burden with no cure and current treatments fail to achieve consistent remission and are limited by toxicities, adverse effects, and, most notably, increased risk of infection [17, 55]. Amid an evolving SARS-CoV-2 pandemic, major concerns regarding the use of general immunosuppressants for autoimmune patients [21, 22] and solid organ transplant recipients [183,184,185] are in contention. It is a tragic dilemma when the life-saving therapy may possibly escalate the risk of a SARS-CoV-2 infection to a life-threatening event [21, 186], especially when these drug regimens generally require lifelong management [55]. The need to develop autoimmune therapies that sidestep broad immunosuppression has never been greater, and application of biomaterial-based designs (liposomes, nanoparticles, microparticles, hydrogels, or scaffolds) to this dilemma offers remarkable promise. Discovery and incorporation of biomaterials into cutting-edge nanomedicines have profoundly expanded the toolbox of therapeutic strategies, granting access to problem-solving from perspectives previously unavailable. While broad immunosuppressive drugs are invaluable and necessary, there is still copious room for improvement. By way of developing new biomaterial-based therapies, the concept of precision has emerged as potentially the most valuable enterprise. This review has revealed that common themes of biomaterial-inspired therapeutic strategies aim to mitigate systemic effects through tactics such as facilitating drug delivery to the target site, or incorporating a targeting component into the delivery vehicle. In this regard, our current understanding of autoimmunity has highlighted the significance of thwarting RA in a specific manner in order to minimize off-target effects. For example, this can be through specific depletion of a problematic cell type or induction of tolerance towards autoantigens, which permits general immunity to still respond appropriately to pathogens, such as SARS-CoV-2. In particular, inducing antigen-specific tolerance is a highly coveted outcome because it does not immunocompromise the patient, whereas conventional drug therapies render the patient immunocompromised and susceptible to infection [187]. To this end, several biomaterial-inspired strategies are being investigated to restore antigen-specific immunological tolerance towards the autoantigens that propagate RA [82, 138, 140, 157, 158]. Pertaining to RA pathogenesis, this implicates DCs, and autoreactive B and T cells for their pivotal roles in bringing forth ACPAs [24], and as such are promising targets in the pursuit of restoring tolerance. For example, our understanding of immunology, progression of RA, and biomaterials has provided the groundwork for using nanocarriers to coerce immune cells into restoring immunological tolerance towards autoantigens, as evidenced by the studies carried out by Allen et al. [82], Galea et al. [138], and Capini et al. [140] where induction of antigen-specific tDCs was able to exert rippling downstream therapeutic effects through induction of antigen-specific Treg cells and anergy of autoreactive T cells. Continued exploration of the mechanisms of tolerance induction during the disease state of RA is very welcome. We believe taking an omic approach to advance knowledge would unlock a multitude of new potential targets that we may use biomaterials to investigate. For example, a multi-omic interrogation of an induced tDC from an inflammatory arthritis disease state will inform us how our biomaterial-based strategies are modulating a specific cell type to induce therapeutic effects. Additionally, cross-referencing differentially expressed genes from RNA-seq, with differentially accessible regions from assay for transposase-accessible chromatin (ATAC)-seq, and gene regulatory motifs from chromatin immunoprecipitation (ChIP)-seq will reveal critical up- or downregulated pathways, transcription factors, or genes that will become attractive targets for future biomaterial-based strategies to induce tolerance in an antigen-specific manner, as we believe this venture to be the most groundbreaking one in the pursuit of new biomaterial-based immunotherapies for RA.
References
Scott DL, Wolfe F, Huizinga TWJ. Rheumatoid arthritis. In: The Lancet [Internet]. Lancet Publishing Group; 2010 [cited 2020 Dec 21]. p. 1094–108. Available from: https://pubmed.ncbi.nlm.nih.gov/20870100/
McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365(23):2205–19.
Deane KD, Demoruelle MK, Kelmenson LB, Kuhn KA, Norris JM, Holers VM. Genetic and environmental risk factors for rheumatoid arthritis. Best Pract Res Clin Rheumatol [Internet]. 2017;31(1):3–18. Available from: https://doi.org/10.1016/j.berh.2017.08.003
Coras R, Murillo-Saich J, Guma M. Circulating pro- and anti-inflammatory metabolites and its potential role in rheumatoid arthritis pathogenesis. Cells [Internet]. 2020 Mar 30 [cited 2021 Jun 20];9(4):827. Available from: https://www.mdpi.com/2073-4409/9/4/827
Holoshitz J. The rheumatoid arthritis HLA-DRB1 shared epitope [Internet]. Vol. 22, Current Opinion in Rheumatology. NIH Public Access; 2010 [cited 2020 Dec 23]. p. 293–8. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2921962/?report=abstract
Burn GL, Svensson L, Sanchez-Blanco C, Saini M, Cope AP. Why is PTPN22 a good candidate susceptibility gene for autoimmune disease? Vol. 585, FEBS Letters. No longer published by Elsevier; 2011. p. 3689–98.
Hill JA, Southwood S, Sette A, Jevnikar AM, Bell DA, Cairns E. Cutting edge: the conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-DRB1*0401 MHC class II molecule. J Immunol [Internet]. 2003 Jul 15 [cited 2020 Aug 29];171(2):538–41. Available from: https://www.jimmunol.org/content/171/2/538http://www.jimmunol.org/content/171/2/538.full#ref-list-1
Sood S, Brownlie RJ, Garcia C, Cowan G, Salmond RJ, Sakaguchi S, et al. Loss of the protein tyrosine phosphatase PTPN22 reduces mannan-induced autoimmune arthritis in SKG mice. J Immunol [Internet]. 2016 Jul 15 [cited 2021 Jan 22];197(2):429–40. Available from: https://www.jimmunol.org/content/197/2/429
Chang H-H, Dwivedi N, Nicholas AP, Ho I-C. The W620 polymorphism in PTPN22 disrupts its interaction with peptidylarginine deiminase type 4 and enhances citrullination and NETosis. Arthritis Rheumatol [Internet]. 2015 Sep 1 [cited 2021 Jun 23];67(9):2323–34. Available from: http://doi.wiley.com/10.1002/art.39215
Lucchino, Spinelli, Iannuccelli, Guzzo, Conti, Di Franco. Mucosa–environment interactions in the pathogenesis of rheumatoid arthritis. Cells [Internet]. 2019 Jul 10 [cited 2020 Dec 28];8(7):700. Available from: https://pubmed.ncbi.nlm.nih.gov/31295951/pmc/articles/PMC7474575/?report=abstract
Scublinsky D, Gonzalez CD. Quantifying disease in challenging conditions: incidence and prevalence of rheumatoid arthritis. J Rheumatol. 2016;43(7):1263–4.
van den Hoek J, Boshuizen HC, Roorda LD, Tijhuis GJ, Nurmohamed MT, van den Bos GAM, et al. Mortality in patients with rheumatoid arthritis: a 15-year prospective cohort study. Rheumatol Int [Internet]. 2017 Apr 1 [cited 2021 Jun 21];37(4):487–93. Available from: https://pubmed.ncbi.nlm.nih.gov/28032180/
Hsieh P-H, Wu O, Geue C, Mcintosh E, Mcinnes IB, Siebert S. Economic burden of rheumatoid arthritis: a systematic review of literature in biologic era. 2020;1–7.
Cooper NJ. Economic burden of rheumatoid arthritis: a systematic review. Rheumatology. 2000;39(1):28–33.
Burmester GR, Pope JE. Novel treatment strategies in rheumatoid arthritis. Lancet [Internet]. 2017;389(10086):2338–48. Available from: https://dx.doi.org/10.1016/S0140-6736(17)31491-5
Breedveld FC, Weisman MH, Kavanaugh AF, Cohen SB, Pavelka K, Van Vollenhoven R, et al. The PREMIER study: a multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum. 2006;54(1):26–37.
Wang W, Zhou H, Liu L. Side effects of methotrexate therapy for rheumatoid arthritis: a systematic review. Eur J Med Chem [Internet]. 2018;158:502–16. Available from: https://doi.org/10.1016/j.ejmech.2018.09.027
Walder BK, Robertson MR, Jeremy D. Skin cancer and immunosuppression. Lancet. 1971;298(7737):1282–3.
Howard MD, Su JC, Chong AH. Skin cancer following solid organ transplantation: a review of risk factors and models of care [Internet]. Vol. 19, American Journal of Clinical Dermatology. Springer International Publishing; 2018 [cited 2021 Feb 22]. p. 585–97. Available from: https://doi.org/10.1007/s40257-018-0355-8
Penn I, Starzl TE. Immunosuppression and cancer. Transplant Proc [Internet]. 1973 Mar [cited 2021 Feb 22];5(1):943–7. Available from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2840634/
Askanase AD, Khalili L, Buyon JP. Thoughts on COVID-19 and autoimmune diseases. Lupus Sci Med. 2020;7(1):19–21.
Roongta R, Ghosh A. Managing rheumatoid arthritis during COVID-19 [Internet]. Vol. 39, Clinical Rheumatology. Springer Science and Business Media Deutschland GmbH; 2020 [cited 2020 Dec 28]. p. 3237–44. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7474575/?report=abstract
Ruth Topless BK, Phipps-Green A, Leask M, Dalbeth N, Stamp LK, Robinson PC, et al. Gout, rheumatoid arthritis and the risk of death from COVID-19: an analysis of the UK. medRxiv [Internet]. 2020 Nov 9 [cited 2020 Dec 28];2020.11.06.20227405. Available from: https://doi.org/10.1101/2020.11.06.20227405
van Delft MAM, Huizinga TWJ. An overview of autoantibodies in rheumatoid arthritis. J Autoimmun [Internet]. 2020;110(January):102392. Available from: https://doi.org/10.1016/j.jaut.2019.102392
Van Venrooij WJ, Van Beers JJBC, Pruijn GJM. Anti-CCP antibodies: the past, the present and the future. Nat Rev Rheumatol. 2011;7(7):391–8.
Somers K, Geusens P, Elewaut D, De Keyser F, Rummens JL, Coenen M, et al. Novel autoantibody markers for early and seronegative rheumatoid arthritis. J Autoimmun. 2011;36(1):33–46.
Lu DR, McDavid AN, Kongpachith S, Lingampalli N, Glanville J, Ju CH, et al. T cell–dependent affinity maturation and innate immune pathways differentially drive autoreactive B cell responses in rheumatoid arthritis. Arthritis Rheumatol. 2018;70(11):1732–44.
Rosenblum MD, Remedios KA, Abbas AK. Mechanisms of human autoimmunity. J Clin Invest. 2015;125(6):2228–33.
Rantapää-Dahlqvist S, De Jong BAW, Berglin E, Hallmans G, Wadell G, Stenlund H, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum [Internet]. 2003 Oct [cited 2020 Dec 29];48(10):2741–9. Available from: https://pubmed.ncbi.nlm.nih.gov/14558078/
Nielen MMJ, Van Schaardenburg D, Reesink HW, Van De Stadt RJ, Van Der Horst-Bruinsma IE, De Koning MHMT, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum [Internet]. 2004 Feb [cited 2020 Dec 29];50(2):380–6. Available from: https://pubmed.ncbi.nlm.nih.gov/14872479/
Lingampalli N, Sokolove J, Lahey LJ, Edison JD, Gilliland WR, Holers VM, et al. Combination of anti-citrullinated protein antibodies and rheumatoid factor is associated with increased systemic inflammatory mediators and more rapid progression from preclinical to clinical rheumatoid arthritis. Clin Immunol [Internet]. 2018;195(April):119–26. Available from: https://doi.org/10.1016/j.clim.2018.05.004
Coutant F, Miossec P. Evolving concepts of the pathogenesis of rheumatoid arthritis with focus on the early and late stages. Curr Opin Rheumatol [Internet]. 2020 Jan 1 [cited 2021 Jun 24];32(1):57–63. Available from: https://journals.lww.com/10.1097/BOR.0000000000000664
Kongpachith S, Lingampalli N, Ju CH, Blum LK, Lu DR, Elliott SE, et al. Affinity maturation of the anti–citrullinated protein antibody paratope drives epitope spreading and polyreactivity in rheumatoid arthritis. Arthritis Rheumatol. 2019;71(4):507–17.
Elliott SE, Kongpachith S, Lingampalli N, Adamska JZ, Cannon BJ, Mao R, et al. Affinity maturation drives epitope spreading and generation of proinflammatory anti–citrullinated protein antibodies in rheumatoid arthritis. Arthritis Rheumatol. 2018;70(12):1946–58.
Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet [Internet]. 2016;388(10055):2023–38. Available from: http://dx.doi.org/10.1016/S0140-6736(16)30173-8
Ge C, Xu B, Liang B, Lönnblom E, Lundström SL, Zubarev RA, et al. Structural basis of cross-reactivity of anti–citrullinated protein antibodies. Arthritis Rheumatol [Internet]. 2019 Feb 1 [cited 2021 Jun 23];71(2):210–21. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/art.40698
Holoshitz J. The rheumatoid arthritis HLA-DRB1 shared epitope. Curr Opin Rheumatol. 2010;22(3):293–8.
Ling S, Cheng A, Pumpens P, Michalak M, Holoshitz J. Identification of the rheumatoid arthritis shared epitope binding site on calreticulin. Gay N, editor. PLoS One [Internet]. 2010 Jul 22 [cited 2020 Dec 23];5(7):e11703. Available from: https://dx.plos.org/10.1371/journal.pone.0011703
Nevius E, Gomes AC, Pereira JP. Inflammatory cell migration in rheumatoid arthritis: a comprehensive review. Clin Rev Allergy Immunol [Internet]. 2016;59–78. Available from: https://dx.doi.org/10.1007/s12016-015-8520-9
Mateen S, Zafar A, Moin S, Qayyum A, Zubair S. Clinica Chimica Acta. Understanding the role of cytokines in the pathogenesis of rheumatoid arthritis. Clin Chim Acta [Internet]. 2016;455:161–71. Available from: http://dx.doi.org/10.1016/j.cca.2016.02.010
Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423(6937):356–61.
Kumar RA, Li Y, Dang Q, Yang F. International Immunopharmacology. Monocytes in rheumatoid arthritis: circulating precursors of macrophages and osteoclasts and their heterogeneity and plasticity role in RA pathogenesis. 2018;65(October):348–59.
Iwahashi M, Yamamura M, Aita T, Okamoto A, Ueno A, Ogawa N, et al. Expression of Toll-like receptor 2 on CD16+ blood monocytes and synovial tissue macrophages in rheumatoid arthritis. Arthritis Rheum [Internet]. 2004 May 1 [cited 2020 Dec 31];50(5):1457–67. Available from: https://pericles.pericles-prod.literatumonline.com/doi/full/10.1002/art.20219
Kinne RW, Bräuer R, Stuhlmüller B, Palombo-Kinne E, Burmester GR. Macrophages in rheumatoid arthritis [Internet]. Vol. 2, Arthritis Research. BioMed Central; 2000 [cited 2021 Feb 22]. p. 189–202. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC130001/
Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. 2003;423(May):337–42.
Lubberts E. The IL-23-IL-17 axis in inflammatory arthritis. Vol. 11, Nature Reviews Rheumatology. Nature Publishing Group; 2015. p. 415–29.
Yasuda K, Takeuchi Y, Hirota K. The pathogenicity of Th17 cells in autoimmune diseases. 2019;283–97.
Krishnamurthy A, Ytterberg AJ, Sun M, Steen J, Joshua V, Nataliya K, et al. Citrullination controls dendritic cell transdifferentiation into osteoclasts. 2020;
Kocijan R, Harre U, Schett G. ACPA and bone loss in rheumatoid arthritis. 2013;
Rao DA. T cells that help B cells in chronically inflamed tissues. 2018;9(August).
Armas-gonzález E, Domínguez-luis MJ, Díaz-martín A, Arce-franco M, Castro-hernández J, Danelon G, et al. Role of CXCL13 and CCL20 in the recruitment of B cells to inflammatory foci in chronic arthritis. 2018;1–12.
Cascão R, Rosário HS, Souto-Carneiro MM, Fonseca JE. Neutrophils in rheumatoid arthritis: more than simple final effectors. Autoimmun Rev [Internet]. 2010;9(8):531–5. Available from: https://dx.doi.org/10.1016/j.autrev.2009.12.013
Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. 2013;
Wright HL, Moots RJ, Edwards SW. The multifactorial role of neutrophils in rheumatoid arthritis. Nat Rev Rheumatol. 2014;10(10):593–601.
Smolen JS, Landewé RBM, Bijlsma JWJ, Burmester GR, Dougados M, Kerschbaumer A, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis [Internet]. 2020 Jun 1 [cited 2020 Dec 28];79(6):S685–99. Available from: https://ard.bmj.com/
Smolen JS, Landewé RBM, Bijlsma JWJ, Burmester GR, Dougados M, Kerschbaumer A, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis [Internet]. 2020 [cited 2020 Dec 28];79:685–99. Available from: https://ard.bmj.com/
Brown PM, Pratt AG, Isaacs JD. Mechanism of action of methotrexate in rheumatoid arthritis, and the search for biomarkers. Nat Rev Rheumatol. 2016;12(12):731–42.
Friedman B, Cronstein B. Methotrexate mechanism in treatment of rheumatoid arthritis. Jt Bone Spine [Internet]. 2019;86(3):301–7. Available from: https://dx.doi.org/10.1016/j.jbspin.2018.07.004
Malaviya AN. Landmark papers on the discovery of methotrexate for the treatment of rheumatoid arthritis and other systemic inflammatory rheumatic diseases: a fascinating story. Int J Rheum Dis. 2016;19(9):844–51.
Cain DW, Cidlowski JA. Immune regulation by glucocorticoids. Nat Rev Immunol [Internet]. 2017;17(4):233–47. Available from: https://dx.doi.org/10.1038/nri.2017.1
Lane NE, Lukert B. The science and therapy of glucocorticoid-induced bone loss. Endocrinol Metab Clin North Am. 1998;27(2):465–83.
Huscher D, Thiele K, Gromnica-Ihle E, Hein G, Demary W, Dreher R, et al. Dose-related patterns of glucocorticoid-induced side effects. [cited 2021 Jun 22]; Available from: https://ard.bmj.com/
van der Goes MC, Jacobs JW, Bijlsma JW. The value of glucocorticoid co-therapy in different rheumatic diseases - positive and adverse effects [Internet]. Vol. 16, Arthritis Research and Therapy. BioMed Central Ltd.; 2014 [cited 2020 Dec 28]. p. 1–13. Available from: https://arthritis-research.com/content/16/S2/S2
Schett G, Emery P, Tanaka Y, Burmester G, Pisetsky DS, Naredo E, et al. Tapering biologic and conventional DMARD therapy in rheumatoid arthritis: current evidence and future directions. Ann Rheum Dis [Internet]. 2016 Aug 1 [cited 2020 Dec 28];75(8):1428–37. Available from: https://ard.bmj.com/
Abbasi M, Mousavi MJ, Jamalzehi S, Alimohammadi R, Bezvan MH, Mohammadi H, et al. Strategies toward rheumatoid arthritis therapy; the old and the new. J Cell Physiol [Internet]. 2019 Jul 7 [cited 2020 Dec 28];234(7):10018–31. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/jcp.27860
Feldmann M, Brennan FM, Williams RO, Cope AP, Gibbons DL, Katsikis PD, et al. Evaluation of the role of cytokines in autoimmune disease: the importance of TNFα in rheumatoid arthritis. Prog Growth Factor Res. 1992;4(3):247–55.
MA X, XU S. TNF inhibitor therapy for rheumatoid arthritis. Biomed Reports [Internet]. 2013 Mar 1 [cited 2021 Feb 22];1(2):177–84. Available from: https://www.spandidos-publications.com/10.3892/br.2012.42/abstract
Blair HA, Deeks ED. Abatacept: a review in rheumatoid arthritis. Drugs [Internet]. 2017 Jul 1 [cited 2020 Dec 28];77(11):1221–33. Available from: https://link.springer.com/article/10.1007/s40265-017-0775-4
J.-E. G, J. M, A. C, T. B, A.G. C, B. C, et al. Long-term registry data in 4498 patients with rheumatoid arthritis indicate a similar safety but a different drug retention between abatacept, rituximab and tocilizumab. Arthritis Rheumatol [Internet]. 2016;68:2550–3. Available from: https://www.embase.com/search/results?subaction=viewrecord&from=export&id=L613889014%0Ahttps://dx.doi.org/10.1002/art.39977
Cambridge G, Leandro MJ, Lahey LJ, Fairhead T, Robinson WH, Sokolove J. B cell depletion with rituximab in patients with rheumatoid arthritis: multiplex bead array reveals the kinetics of IgG and IgA antibodies to citrullinated antigens. J Autoimmun [Internet]. 2016;70:22–30. Available from: https://dx.doi.org/10.1016/j.jaut.2016.03.010
Taylor PC. Clinical efficacy of launched JAK inhibitors in rheumatoid arthritis. Rheumatol (United Kingdom). 2019;
Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Vol. 16, Nature Reviews Drug Discovery. Nature Publishing Group; 2017. p. 843–62.
Sokka T, Hetland ML, Mäkinen H, Kautiainen H, Hørslev-Petersen K, Luukkainen RK, et al. Remission and rheumatoid arthritis data on patients receiving usual care in twenty-four countries. Arthritis Rheum. 2008;58(9):2642–51.
Haschka J, Englbrecht M, Hueber AJ, Manger B, Kleyer A, Reiser M, et al. Relapse rates in patients with rheumatoid arthritis in stable remission tapering or stopping antirheumatic therapy: interim results from the prospective randomised controlled RETRO study. Ann Rheum Dis [Internet]. 2016 Jan 1 [cited 2021 Jun 22];75(1):45–51. Available from: https://doi.org/10.1136/annrheumdis-2014-206439
Herman RA, Veng-Pedersen P, Hoffman J, Koehnke R, Furst DE. Pharmacokinetics of low-dose methotrexate in rheumatoid arthritis patients. J Pharm Sci [Internet]. 1989 Feb 1 [cited 2021 Jun 22];78(2):165–71. Available from: https://jpharmsci.org/article/S0022354915478918/fulltext
Langer R, Peppas NA. Advances in biomaterials, drug delivery, and bionanotechnology. AIChE J. 2003;49(12):2990–3006.
VanWijk MJ, VanBavel E, Sturk A, Nieuwland R. Microparticles in cardiovascular diseases [Internet]. Vol. 59, Cardiovascular Research. Oxford Academic; 2003 [cited 2021 Jan 3]. p. 277–87. Available from: https://academic.oup.com/cardiovascres/article-lookup/doi/10.1016/S0008-6363(03)00367-5
Popovic N, Brundin P. Therapeutic potential of controlled drug delivery systems in neurodegenerative diseases. Int J Pharm. 2006;314(2):120–6.
J Rak Microparticles in cancer. [cited, 2021 Jan 3] Available from: https://doi.org/10.1055/s-0030-1267043
Leleux J, Roy K. Micro and nanoparticle-based delivery systems for vaccine immunotherapy: an immunological and materials perspective [Internet]. Vol. 2, Advanced Healthcare Materials. Wiley-VCH Verlag; 2013 [cited 2021 Jan 2]. p. 72–94. Available from: https://www.onlinelibrary.wiley.com/doi/full/10.1002/adhm.201200268
Mitragotri S, Yoo JW. Designing micro- and nano-particles for treating rheumatoid arthritis. Arch Pharm Res. 2011;34(11):1887–97.
Allen R, Chizari S, Ma JA, Raychaudhuri S, Lewis JS. Combinatorial, microparticle-based delivery of immune modulators reprograms the dendritic cell phenotype and promotes remission of collagen-induced arthritis in mice. ACS Appl Bio Mater. 2019;2(6):2388–404.
Lewis JS, Allen RP. An introduction to biomaterial-based strategies for curbing autoimmunity. Exp Biol Med. 2016;241(10):1107–15.
Ben-Akiva E, Est Witte S, Meyer RA, Rhodes KR, Green JJ. Polymeric micro- and nanoparticles for immune modulation. Biomater Sci. 2019;7(1):14–30.
Tran S, DeGiovanni P, Piel B, Rai P. Cancer nanomedicine: a review of recent success in drug delivery. Clin Transl Med [Internet]. 2017;6(1). Available from: https://doi.org/10.1186/s40169-017-0175-0
Pacifici N, Bolandparvaz A, Lewis JS. Stimuli-responsive biomaterials for vaccines and immunotherapeutic applications. Adv Ther [Internet]. 2020 Nov [cited 2021 Feb 22];3(11):2000129. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7435355/pmc/articles/PMC7435355/
Rahman M, Beg S, Anwar F, Kumar V, Ubale R, Addo RT, et al. Liposome-based nanomedicine therapeutics for rheumatoid arthritis. Crit Rev Ther Drug Carrier Syst. 2017;34(4):283–316.
Kapoor B, Singh SK, Gulati M, Gupta R, Vaidya Y. Application of liposomes in treatment of rheumatoid arthritis: quo vadis. Sci World J. 2014;2014.
Lian T, Ho RJY. Trends and developments in liposome drug delivery systems. J Pharm Sci [Internet]. 2001;90(6):667–80. Available from: https://doi.org/10.1002/jps.1023
Sercombe L, Veerati T, Moheimani F, Wu SY, Sood AK, Hua S. Advances and challenges of liposome assisted drug delivery. Front Pharmacol. 2015;6(DEC):1–13.
Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Advanced Drug Delivery Reviews. 2013.
Ren H, He Y, Liang J, Cheng Z, Zhang M, Zhu Y, et al. Role of liposome size, surface charge, and PEGylation on rheumatoid arthritis targeting therapy. ACS Appl Mater Interfaces. 2019;11(22):20304–15.
Jia M, Deng C, Luo J, Zhang P, Sun X, Zhang Z, et al. A novel dexamethasone-loaded liposome alleviates rheumatoid arthritis in rats. Int J Pharm. 2018;
Wang Q, He L, Fan D, Liang W, Fang J. Improving the anti-inflammatory efficacy of dexamethasone in the treatment of rheumatoid arthritis with polymerized stealth liposomes as a delivery vehicle. J Mater Chem B [Internet]. 2020 Mar 7 [cited 2021 Jan 4];8(9):1841–51. Available from: https://pubmed.ncbi.nlm.nih.gov/32016224/
Kadry MO. Liposomal glutathione as a promising candidate for immunological rheumatoid arthritis therapy. Heliyon. 2019;
Shen Q, Shu H, Xu X, Shu G, Du Y, Ying X. Tofacitinib citrate-based liposomes for effective treatment of rheumatoid arthritis. Pharmazie [Internet]. 2020 Apr 1 [cited 2021 Jan 4];75(4):131–5. Available from: https://pubmed.ncbi.nlm.nih.gov/32295688/
Wang P, Li A, Yu L, Chen Y, Xu D. Energy conversion-based nanotherapy for rheumatoid arthritis treatment [Internet]. Vol. 8, Frontiers in Bioengineering and Biotechnology. Frontiers Media S.A.; 2020 [cited 2020 Dec 28]. p. 652. Available from: /pmc/articles/PMC7366901/?report=abstract
Panyam J, Dali MM, Sahoo SK, Ma W, Chakravarthi SS, Amidon GL, et al. Polymer degradation and in vitro release of a model protein from poly(D,L-lactide-co-glycolide) nano- and microparticles. J Control Release [Internet]. 2003 Sep 19 [cited 2021 Jun 22];92(1–2):173–87. Available from: https://pubmed.ncbi.nlm.nih.gov/14499195/
Brannon-Peppas L. Recent advances on the use of biodegradable microparticles and nanoparticles in controlled drug delivery. Int J Pharm. 1995;116(1):1–9.
Shen H, Ackerman AL, Cody V, Giodini A, Hinson ER, Cresswell P, et al. Enhanced and prolonged cross-presentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles. Immunology. 2006;117(1):78–88.
Fernando LP, Lewis JS, Evans BC, Duvall CL, Keselowsky BG. Formulation and characterization of poly(propylacrylic acid)/poly(lactic-co-glycolic acid) blend microparticles for pH-dependent membrane disruption and cytosolic delivery. J Biomed Mater Res - Part A [Internet]. 2018 Apr 1 [cited 2021 Feb 27];106(4):1022–33. Available from: https://pubmed.ncbi.nlm.nih.gov/29164777/
Harriman R, Lewis JS. Bioderived materials that disarm the gut mucosal immune system: potential lessons from commensal microbiota. Acta Biomater [Internet]. 2021 Jun 5 [cited 2021 Jun 21]; Available from: https://linkinghub.elsevier.com/retrieve/pii/S174270612100355X
Lee J, Jeong L, Jung E, Ko C, Seon S, Noh J, et al. Thrombus targeting aspirin particles for near infrared imaging and on-demand therapy of thrombotic vascular diseases. J Control Release. 2019;28(304):164–72.
Kohane DS. Microparticles and nanoparticles for drug delivery [Internet]. Vol. 96, Biotechnology and Bioengineering. John Wiley & Sons, Ltd; 2007 [cited 2021 Jan 11]. p. 203–9. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/bit.21301
Elshabrawy HA, Chen ∙ Zhenlong, Volin M V, Ravella S, Virupannavar S, Shahrara S. The pathogenic role of angiogenesis in rheumatoid arthritis.
McDonald DM. Angiogenesis and remodeling of airway vasculature in chronic inflammation. Am J Respir Crit Care Med [Internet]. 2001 Nov 15 [cited 2021 Jun 25];164(10 Pt 2):S39–45. Available from: https://www.atsjournals.org/doi/abs/10.1164/ajrccm.164.supplement_2.2106065
Yang M, Feng X, Ding J, Chang F, Chen X. Nanotherapeutics relieve rheumatoid arthritis. Vol. 252, Journal of Controlled Release. Elsevier B.V.; 2017. p. 108–24.
Catrina AI, Svensson CI, Malmström V, Schett G, Klareskog L. Mechanisms leading from systemic autoimmunity to joint-specific disease in rheumatoid arthritis. Nat Rev Rheumatol. 2017;13(2):79–86.
Zhang CY, Dong X, Gao J, Lin W, Liu Z, Wang Z. Nanoparticle-induced neutrophil apoptosis increases survival in sepsis and alleviates neurological damage in stroke. Sci Adv. 2019;5(11):1–14.
Rosales C. Neutrophil: a cell with many roles in inflammation or several cell types? [Internet]. Vol. 9, Frontiers in Physiology. Frontiers Media S.A.; 2018 [cited 2021 Jan 17]. p. 113. Available from: /pmc/articles/PMC5826082/?report=abstract
Lyu J, Wang L, Bai X, Du X, Wei J, Wang J, et al. Treatment of rheumatoid arthritis by serum albumin nanoparticles coated with mannose to target neutrophils. ACS Appl Mater Interfaces [Internet]. 2020 Dec 30 [cited 2021 Jan 11];acsami.0c19468. Available from: https://pubs.acs.org/doi/10.1021/acsami.0c19468
Hu L, Luo X, Zhou S, Zhu J, Xiao M, Li C, et al. Neutrophil-mediated delivery of dexamethasone palmitate-loaded liposomes decorated with a sialic acid conjugate for rheumatoid arthritis treatment. Pharm Res [Internet]. 2019 Jul 1 [cited 2021 Jan 4];36(7). Available from: https://pubmed.ncbi.nlm.nih.gov/31076925/
Ivetic A, Green HLH, Hart SJ. L-Selectin: a major regulator of leukocyte adhesion, migration and signaling [Internet]. Vol. 10, Frontiers in Immunology. Frontiers Media S.A.; 2019 [cited 2021 Feb 22]. p. 1068. Available from: https://www.frontiersin.org
Zhang Q, Dehaini D, Zhang Y, Zhou J, Chen X, Zhang L, et al. Neutrophil membrane-coated nanoparticles inhibit synovial inflammation and alleviate joint damage in inflammatory arthritis. Nat Nanotechnol [Internet]. 2018 Dec 1 [cited 2021 Jan 11];13(12):1182–90. Available from: https://pubmed.ncbi.nlm.nih.gov/30177807/
Chandrupatla DMSH, Molthoff CFM, Lammertsma AA, van der Laken CJ, Jansen G. The folate receptor β as a macrophage-mediated imaging and therapeutic target in rheumatoid arthritis. Drug Delivery and Translational Research. 2019.
Xia W, Hilgenbrink AR, Matteson EL, Lockwood MB, Cheng JX, Low PS. A functional folate receptor is induced during macrophage activation and can be used to target drugs to activated macrophages. Blood. 2009;
Verma A, Jain A, Tiwari A, Saraf S, Panda PK, Agrawal GP, et al. Folate conjugated double liposomes bearing prednisolone and methotrexate for targeting rheumatoid arthritis. Pharm Res [Internet]. 2019 Aug 1 [cited 2021 Jan 4];36(8). Available from: https://pubmed.ncbi.nlm.nih.gov/31218557/
Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduction and Targeted Therapy. 2017.
Duan W, Li H. Combination of NF-kB targeted siRNA and methotrexate in a hybrid nanocarrier towards the effective treatment in rheumatoid arthritis. J Nanobiotechnology. 2018;
Chen M, Kambere Amerigos Daddy JC, Su Z, El Islem Guissi N, Xiao Y, Zong L, et al. Folate receptor-targeting and reactive oxygen species-responsive liposomal formulation of methotrexate for treatment of rheumatoid arthritis. Pharmaceutics. 2019;
Ni R, Song G, Fu X, Song R, Li L, Pu W, et al. Reactive oxygen species-responsive dexamethasone-loaded nanoparticles for targeted treatment of rheumatoid arthritis via suppressing the iRhom2/TNF-α/BAFF signaling pathway. Biomaterials [Internet]. 2020 Feb 1 [cited 2021 Jan 11];232. Available from: https://pubmed.ncbi.nlm.nih.gov/31918224/
Vellozo NS, Pereira-Marques ST, Cabral-Piccin MP, Filardy AA, Ribeiro-Gomes FL, Rigoni TS, et al. All-trans retinoic acid promotes an M1-to M2-phenotype shift and inhibits macrophage-mediated immunity to Leishmania major. Front Immunol [Internet]. 2017 Nov 17 [cited 2021 Jan 20];8(NOV):1560. Available from: /pmc/articles/PMC5698282/?report=abstract
Li P, Yang X, Yang Y, He H, Chou CK, Chen F, et al. Synergistic effect of all-trans-retinal and triptolide encapsulated in an inflammation-targeted nanoparticle on collagen-induced arthritis in mice. J Control Release [Internet]. 2020 Mar 10 [cited 2021 Jan 20];319:87–103. Available from: https://pubmed.ncbi.nlm.nih.gov/31862360/
Chen Y, Guan M, Ren R, Gao C, Cheng H, Li Y, et al. Improved immunoregulation of ultra-low-dose silver nanoparticle-loaded TiO2 nanotubes via M2 macrophage polarization by regulating GLUT1 and autophagy. Int J Nanomedicine [Internet]. 2020 Mar 24 [cited 2021 Jun 22];Volume 15:2011–26. Available from: https://www.dovepress.com/improved-immunoregulation-of-ultra-low-dose-silver-nanoparticle-loaded-peer-reviewed-article-IJN
Yang Y, Guo L, Wang Z, Liu P, Liu X, Ding J, et al. Targeted silver nanoparticles for rheumatoid arthritis therapy via macrophage apoptosis and re-polarization. Biomaterials [Internet]. 2021 Jan 1 [cited 2021 Jan 11];264. Available from: https://pubmed.ncbi.nlm.nih.gov/32980634/
Bluestone JA. Mechanisms of tolerance. Immunol Rev. 2011;241(1):5–19.
Audiger C, Rahman MJ, Yun TJ, Tarbell K V., Lesage S. The importance of dendritic cells in maintaining immune tolerance. J Immunol [Internet]. 2017 Mar 15 [cited 2020 Aug 29];198(6):2223–31. Available from: /pmc/articles/PMC5343761/?report=abstract
Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711.
Švajger U, Rožman P. Induction of tolerogenic dendritic cells by endogenous biomolecules: an update. Front Immunol. 2018;9(OCT):1–17.
Adorini L. Tolerogenic dendritic cells induced by vitamin D receptor ligands enhance regulatory T cells inhibiting autoimmune diabetes. Major targets of VDR ligands in the immune system: antigen-presenting cells and T cells. Vol. 987, Ann. N.Y. Acad. Sci. 2003.
Zhao Y, Zhang A, Du H, Guo S, Ning B, Yang S. Tolerogenic dendritic cells and rheumatoid arthritis: current status and perspectives. Rheumatol Int. 2012;32(4):837–44.
Harry RA, Anderson AE, Isaacs JD, Hilkens CMU. Generation and characterisation of therapeutic tolerogenic dendritic cells for rheumatoid arthritis. Ann Rheum Dis. 2010;69(11):2042–50.
Morelli AE, Thomson AW. Tolerogenic dendritic cells and the quest for transplant tolerance. Nat Rev Immunol. 2007;7(8):610–21.
Lewis JS, Stewart JM, Marshall GP, Carstens MR, Zhang Y, Dolgova NV, et al. Dual-sized microparticle system for generating suppressive dendritic cells prevents and reverses type 1 diabetes in the nonobese diabetic mouse model. ACS Biomater Sci Eng. 2019;5(5):2631–46.
Cho JJ, Stewart JM, Drashansky TT, Brusko MA, Zuniga AN, Lorentsen KJ, et al. An antigen-specific semi-therapeutic treatment with local delivery of tolerogenic factors through a dual-sized microparticle system blocks experimental autoimmune encephalomyelitis. Biomaterials [Internet]. 2017 Oct 1 [cited 2021 Jan 18];143:79–92. Available from: https://pubmed.ncbi.nlm.nih.gov/28772190/
Adorini L, Giarratana N, Penna G. Pharmacological induction of tolerogenic dendritic cells and regulatory T cells. Semin Immunol. 2004;
Bell GM, Anderson AE, Diboll J, Reece R, Eltherington O, Harry RA, et al. Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Ann Rheum Dis. 2017;76(1):227–34.
Galea R, Nel HJ, Talekar M, Liu X, Ooi JD, Huynh M, et al. PD-L1– and calcitriol-dependent liposomal antigen-specific regulation of systemic inflammatory autoimmune disease. JCI Insight. 2019;
O’Sullivan BJ, Thomas R. CD40 ligation conditions dendritic cell antigen-presenting function through sustained activation of NF-κB. J Immunol [Internet]. 2002 Jun 1 [cited 2021 Jun 24];168(11):5491–8. Available from: https://www.jimmunol.org/content/168/11/5491 and http://www.jimmunol.org/content/168/11/5491.full#ref-list-1
Capini C, Jaturanpinyo M, Chang H-I, Mutalik S, McNally A, Street S, et al. Antigen-specific suppression of inflammatory arthritis using liposomes. J Immunol [Internet]. 2009 Mar 15 [cited 2021 Jun 24];182(6):3556–65. Available from: https://www.jimmunol.org/content/182/6/3556
Benham H, Nel HJ, Law SC, Mehdi AM, Street S, Ramnoruth N, et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients. Sci Transl Med [Internet]. 2015 Jun 3 [cited 2021 Jan 18];7(290):290ra87–290ra87. Available from: https://www.ScienceTranslationalMedicine.org
Hilliard BA, Mason N, Xu L, Sun J, Lamhamedi-Cherradi S-E, Liou H-C, et al. Critical roles of c-Rel in autoimmune inflammation and helper T cell differentiation. J Clin Invest. 2002;110(6):843–50.
Fan T, Zhong F, Liu R, Chen YH, Wang T, Ruan Q. siRNA-mediated c-Rel knockdown ameliorates collagen-induced arthritis in mice. Int Immunopharmacol [Internet]. 2018 Mar 1 [cited 2021 Jan 20];56:9–17. Available from: https://pubmed.ncbi.nlm.nih.gov/29324391/
Lee S-Y, Lee SH, Yang E-J, Kim J-K, Kim E-K, Jung K, et al. Coenzyme Q10 inhibits Th17 and STAT3 signaling pathways to ameliorate colitis in mice. J Med Food [Internet]. 2017 Sep 1 [cited 2021 Jan 21];20(9):821–9. Available from: https://www.liebertpub.com/doi/10.1089/jmf.2016.3859
Jhun JY, Lee SH, Byun JK, Jeong JH, Kim EK, Lee J, et al. Coenzyme Q10 suppresses Th17 cells and osteoclast differentiation and ameliorates experimental autoimmune arthritis mice. Immunol Lett. 2015;166(2):92–102.
Jhun J, Moon J, Ryu J, Shin Y, Lee S, Cho K-H, et al. Liposome/gold hybrid nanoparticle encoded with CoQ10 (LGNP-CoQ10) suppressed rheumatoid arthritis via STAT3/Th17 targeting. Chung Y, editor. PLoS One [Internet]. 2020 Nov 6 [cited 2021 Jan 20];15(11):e0241080. Available from: https://doi.org/10.1371/journal.pone.0241080
Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;
Fisher JD, Zhang W, Balmert SC, Aral AM, Acharya AP, Kulahci Y, et al. In situ recruitment of regulatory T cells promotes donor-specific tolerance in vascularized composite allotransplantation. Sci Adv. 2020;6(11).
Lima AC, Campos CF, Cunha C, Carvalho A, Reis RL, Ferreira H, et al. Biofunctionalized liposomes to monitor rheumatoid arthritis regression stimulated by interleukin-23 neutralization. Adv Healthc Mater [Internet]. 2020 [cited 2021 Jan 4]; Available from: https://pubmed.ncbi.nlm.nih.gov/33103383/
Bolandparvaz A, Harriman R, Alvarez K, Lilova K, Zang Z, Lam A, et al. Towards a nanoparticle-based prophylactic for maternal autoantibody-related autism. Nanomedicine Nanotechnology, Biol Med [Internet]. 2019 Oct 1 [cited 2021 Feb 22];21. Available from: https://pubmed.ncbi.nlm.nih.gov/31349087/
Rubin SJS, Bloom MS, Robinson WH. B cell checkpoints in autoimmune rheumatic diseases. Nat Rev Rheumatol [Internet]. 2019;15(5):303–15. Available from: https://dx.doi.org/10.1038/s41584-019-0211-0
SE Elliott S Kongpachith N Lingampalli JZ Adamska BJ Cannon LK Blum et al 2019 B cells in rheumatoid arthritis synovial tissues encode focused antibody repertoires that include antibodies that stimulate macrophage TNF-α production Clin Immunol [Internet]. 2020(212), 108360 Available from: https://doi.org/10.1016/j.clim.2020.108360
Hofmann K, Clauder A, Manz RA, Howie D, Steptoe RJ, Hofmann K. Targeting B cells and plasma cells in autoimmune diseases. 2018;9(April).
Ng LG, Sutherland APR, Newton R, Qian F, Cachero TG, Scott ML, et al. B cell-activating factor belonging to the TNF family (BAFF)-R is the principal BAFF receptor facilitating BAFF costimulation of circulating T and B cells. J Immunol. 2004;
Huard B, Arlettaz L, Ambrose C, Kindler V, Mauri D, Roosnek E, et al. BAFF production by antigen-presenting cells provides T cell co-stimulation. Int Immunol [Internet]. 2004 Mar [cited 2021 Feb 23];16(3):467–75. Available from: https://pubmed.ncbi.nlm.nih.gov/14978020/
Wu H, Su S, Wu Y, Wu Y, Zhang Z, Chen Q. Nanoparticle-facilitated delivery of BAFF-R siRNA for B cell intervention and rheumatoid arthritis therapy. Int Immunopharmacol [Internet]. 2020 Nov 1 [cited 2021 Jan 11];88. Available from: https://pubmed.ncbi.nlm.nih.gov/32866781/
Pozsgay J, Babos F, Uray K, Magyar A, Gyulai G, Kiss É, et al. In vitro eradication of citrullinated protein specific B-lymphocytes of rheumatoid arthritis patients by targeted bifunctional nanoparticles. Arthritis Res Ther [Internet]. 2016 Jan 16 [cited 2021 Jan 11];18(1). Available from: https://pubmed.ncbi.nlm.nih.gov/26780830/
Bednar KJ, Nycholat CM, Rao TS, Paulson JC, Fung-Leung WP, MacAuley MS. Exploiting CD22 to selectively tolerize autoantibody producing B-cells in rheumatoid arthritis. ACS Chem Biol. 2019;
Steffen U, Schett G, Bozec A. How autoantibodies regulate osteoclast induced bone loss in rheumatoid arthritis. 2019;10(July):1–9.
Yavropoulou MP, Yovos JG. Osteoclastogenesis - current knowledge and future perspectives. Journal of Musculoskeletal Neuronal Interactions. 2008.
Morishima S, Morita I, Tokushima T, Kawashima H, Miyasaka M, Omura K, et al. Expression and role of mannose receptor/terminal high-mannose type oligosaccharide on osteoclast precursors during osteoclast formation. Journal of Endocrinology. 2003.
Neog MK, Joshua Pragasam S, Krishnan M, Rasool M. p-Coumaric acid, a dietary polyphenol ameliorates inflammation and curtails cartilage and bone erosion in the rheumatoid arthritis rat model. BioFactors. 2017;
Neog MK, Rasool M. Targeted delivery of p-coumaric acid encapsulated mannosylated liposomes to the synovial macrophages inhibits osteoclast formation and bone resorption in the rheumatoid arthritis animal model. Eur J Pharm Biopharm. 2018;
Sujitha S, Rasool M. Berberine coated mannosylated liposomes curtail RANKL stimulated osteoclastogenesis through the modulation of GSK3β pathway via upregulating miR-23a. Int Immunopharmacol. 2019;
Sujitha S, Dinesh P, Rasool M. Berberine modulates ASK1 signaling mediated through TLR4/TRAF2 via upregulation of miR-23a. Toxicol Appl Pharmacol. 2018;
Sul OJ, Kim JC, Kyung TW, Kim HJ, Kim YY, Kim SH, et al. Gold nanoparticles inhibited the receptor activator of nuclear factor-κB ligand (RANKL)-induced osteoclast formation by acting as an antioxidant. Biosci Biotechnol Biochem [Internet]. 2010 [cited 2021 Jan 20];74(11):2209–13. Available from: https://www.tandfonline.com/action/journalInformation?journalCode=tbbb20
Lee D, Heo DN, Kim HJ, Ko WK, Lee SJ, Heo M, et al. Inhibition of osteoclast differentiation and bone resorption by bisphosphonate-conjugated gold nanoparticles. Sci Rep [Internet]. 2016 Jun 2 [cited 2021 Jan 20];6(1):1–11. Available from: https://www.nature.com/scientificreports
Heo DN, Ko WK, Moon HJ, Kim HJ, Lee SJ, Lee JB, et al. Inhibition of osteoclast differentiation by gold nanoparticles functionalized with cyclodextrin curcumin complexes. ACS Nano [Internet]. 2014 Dec 23 [cited 2021 Jan 20];8(12):12049–62. Available from: https://www.acsnano.org12049
L Z, H G, W G, S M, Y Q, S X, et al. Au nanoparticles attenuate RANKL-induced osteoclastogenesis by suppressing pre-osteoclast fusion. J Nanosci Nanotechnol [Internet]. 2019 [cited 2021 Jan 20];19(4). Available from: https://pubmed.ncbi.nlm.nih.gov/30486961/
Tu J, Hong W, Zhang P, Wang X, Körner H, Wei W. Ontology and function of fibroblast-like and macrophage-like synoviocytes: how do they talk to each other and can they be targeted for rheumatoid arthritis therapy? [Internet]. Vol. 9, Frontiers in Immunology. Frontiers Media S.A.; 2018 [cited 2021 Feb 25]. p. 1. Available from: www.frontiersin.org
Tang Q, Cui J, Tian Z, Sun J, Wang Z, Chang S, et al. Oxygen and indocyanine green loaded phase-transition nanoparticle-mediated photo-sonodynamic cytotoxic effects on rheumatoid arthritis fibroblast-like synoviocytes. Int J Nanomedicine [Internet]. 2017 [cited 2021 Feb 24];12:381–93. Available from: https://www.dovepress.com/oxygen-and-indocyanine-green-loaded-phase-transition-nanoparticle-medi-peer-reviewed-fulltext-article-IJN
Caló E, Khutoryanskiy V V. Biomedical applications of hydrogels: a review of patents and commercial products [Internet]. Vol. 65, European Polymer Journal. Elsevier Ltd; 2015 [cited 2021 Feb 24]. p. 252–67. Available from: https://doi.org/10.1016/j.eurpolymj.2014.11.024
Norouzi M, Nazari B, Miller DW. Injectable hydrogel-based drug delivery systems for local cancer therapy. Vol. 21, Drug Discovery Today. Elsevier Ltd; 2016. p. 1835–49.
Singh A, Peppas NA. Hydrogels and scaffolds for immunomodulation. Adv Mater. 2014;26(38):6530–41.
Lee KY, Mooney DJ. Hydrogels for tissue engineering [Internet]. Vol. 101, Chemical Reviews. American Chemical Society ; 2001 [cited 2021 Feb 24]. p. 1869–79. Available from: https://pubs.acs.org/sharingguidelines
Dhandayuthapani B, Yoshida Y, Maekawa T, Kumar DS. Polymeric scaffolds in tissue engineering application: a review. Vol. 2011, International Journal of Polymer Science. Hindawi Publishing Corporation; 2011.
Masuko K, Murata M, Yudoh K, Kato T, Nakamura H. Anti-inflammatory effects of hyaluronan in arthritis therapy: not just for viscosity [Internet]. Vol. 2, International Journal of General Medicine. Dove Medical Press Ltd.; 2009 [cited 2021 Feb 25]. p. 77–81. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2840553/
Seo J, Park SH, Kim MJ, Ju HJ, Yin XY, Min BH, et al. Injectable click-crosslinked hyaluronic acid depot to prolong therapeutic activity in articular joints affected by rheumatoid arthritis. ACS Appl Mater Interfaces [Internet]. 2019 Jul 2 [cited 2021 Feb 24];11(28):24984–98. Available from: https://pubmed.ncbi.nlm.nih.gov/31264830/
Wu H, Wang K, Wang H, Chen F, Huang W, Chen Y, et al. Novel self-assembled tacrolimus nanoparticles cross-linking thermosensitive hydrogels for local rheumatoid arthritis therapy. Colloids Surfaces B Biointerfaces. 2017;1(149):97–104.
Chen W, Li Z, Wang Z, Gao H, Ding J, He Z. Intraarticular injection of infliximab-loaded thermosensitive hydrogel alleviates pain and protects cartilage in rheumatoid arthritis. J Pain Res [Internet]. 2020 [cited 2021 Feb 24];13:3315–29. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7733037/
Zhao Y, Gao C, Liu H, Liu H, Feng Y, Li Z, et al. Infliximab-based self-healing hydrogel composite scaffold enhances stem cell survival, engraftment, and function in rheumatoid arthritis treatment. Acta Biomater. 2021;1(121):653–64.
Yin N, Tan X, Liu H, He F, Ding N, Gou J, et al. A novel indomethacin/methotrexate/MMP-9 siRNA: in situ hydrogel with dual effects of anti-inflammatory activity and reversal of cartilage disruption for the synergistic treatment of rheumatoid arthritis. Nanoscale [Internet]. 2020 Apr 21 [cited 2021 Feb 24];12(15):8546–62. Available from: https://pubs.rsc.org/en/content/articlehtml/2020/nr/d0nr00454e
Akalin E, Azzi Y, Bartash R, Seethamraju H, Parides M, Hemmige V, et al. Covid-19 and kidney transplantation. N Engl J Med [Internet]. 2020 Jun 18 [cited 2021 Feb 27];382(25):2475–7. Available from: https://www.nejm.org/doi/10.1056/NEJMc2011117
Saigal S, Gupta S, Sudhindran S, Goyal N, Rastogi A, Jacob M, et al. Liver transplantation and COVID-19 (Coronavirus) infection: guidelines of the liver transplant Society of India (LTSI). Hepatol Int [Internet]. 2020 Jul 1 [cited 2021 Feb 27];14(4):429–31. Available from: https://doi.org/10.1007/s12072-020-10041-1
Defilippis EM, Farr MA, Givertz MM. Challenges in heart transplantation in the era of COVID-19 [Internet]. Vol. 141, Circulation. Lippincott Williams and Wilkins; 2020 [cited 2021 Feb 27]. p. 2048–51. Available from: http://ahajournals.org
Grabbe S, Beissert S, Enk A. Systemic immunosuppression in times of COVID-19: do we need to rethink our standards? JDDG J der Dtsch Dermatologischen Gesellschaft [Internet]. 2020 Aug 2 [cited 2021 Feb 27];18(8):810–3. Available from: https://onlinelibrary.wiley.com/doi/10.1111/ddg.14194
Medeiros M, Lumini J, Stern N, Castañeda-Hernández G, Filler G. Generic immunosuppressants. Pediatr Nephrol. 2018;33(7):1123–31.
Funding
ABT and JSL were supported by the following NIH grants: T32GM099608 and R01AI139399.
Author information
Authors and Affiliations
Contributions
ABT and JSL conceived and co-wrote this manuscript together.
Corresponding author
Ethics declarations
Consent for publication
The authors give consent for publication.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Tu, A.B., Lewis, J.S. Biomaterial-based immunotherapeutic strategies for rheumatoid arthritis. Drug Deliv. and Transl. Res. 11, 2371–2393 (2021). https://doi.org/10.1007/s13346-021-01038-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13346-021-01038-w