
Abstract
Human respiratory syncytial virus (hRSV) is the most common viral pathogen of acute lower respiratory tract infection in infants and young children. The G protein of hRSV is the trans-membrane glycoprotein that is involved in the attachment of virion with the host cell. The nasopharyngeal aspirates were subjected to RT-PCR for the second hypervariable region of the G protein gene in the present investigation. Sequencing and phylogenetic analysis revealed that all the study strains clustered within the BA genotype. The study sequences further clustered in BA-9, BA-7, BA-10 and BA-12 subgroups within the BA genotype. The G proteins of the study sequences were predicted to encode 312 and 319 amino acids. Three different N-linked glycosylation sites were observed in the deduced 93–100 amino acid region. There were 40–43 serine and threonine residues that are the potential O-linked glycosylation sites. The non-synonymous/synonymous (dN/dS) ratio was less than one indicating negative selection pressure for amino acid change in the analyzed region of the G protein. The present investigation provides information on circulating strains of BA genotype from New Delhi, India. Further elaborate investigations of the BA viruses from different regions of the world will establish the basis of the rapid global spread and evolutionary pattern of this expanding genotype.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Human respiratory syncytial virus (hRSV) is one of the most frequently identified pathogen of acute lower respiratory tract infections in children [13]. The G protein of hRSV is the transmembrane glycoprotein that is responsible for attachment of virion to the host cell. The G protein is one of the targets of host immune response and thus a vaccine candidate. hRSV group B strains have been classified into 13 reported genotypes GB1, GB2, GB3, GB4, SAB1, SAB2, SAB3, SAB4, URU1, URU2, JAB1, CB1 and BA [5, 8, 9, 21, 23, 25]. The BA genotype has 60 nucleotide duplication in the second hypervariable region of the G protein gene. This genotype was first detected in Buenos Aires in South America in 1999 [20]. Subsequently, this genotype was reported from various geographical regions such as USA, Japan, Denmark, Canada, United Kingdom, China, Korea, Croatia, Iran, Saudi Arabia, India, New Zealand, South Africa, Brazil [2–4, 6, 9, 12, 15, 16, 19, 20, 22, 26]. Worldwide rapid dissemination of this genotype suggests that it has a selective advantage over other genotypes of hRSV [20].
Relatively few studies have reported genetic diversity of BA viruses from India [3, 7, 15, 16]. In the present investigation, we have carried out a retrospective molecular and phylogenetic analysis of the circulating strain of the BA genotype from New Delhi, India. Information about the circulating strains of RSV will assist in vaccine development efforts. In addition, these epidemiological studies will provide information on the evolution of this emerging viral pathogen.
Materials and methods
Clinical samples
An aliquot of thirteen NPAs (nasopharyngeal aspirates) positive for group B hRSV by N gene PCR was used for the present study. The newborns from four rural villages viz Dayalpur, Dayalpur colony, Khera and Phaphunda in Ballabgarh, Haryana near Delhi were enrolled in the present study from 2007 to 2010. Weekly visits were made by the Medical Officers and field workers to all the villages to identify acute lower respiratory tract infection (ALRI) episodes in enrolled children. The clinical samples were collected from enrolled children <5 years of age with ALRI symptoms and ALRIs were classified according to WHO criteria [24]. The epidemiological details of this study is being reported in another publication. Approval was obtained for the study from the Institutional Ethics Committee, All India Institute of Medical Sciences (AIIMS). Clinical specimens were collected after obtaining written consent from the parents/guardians of the enrolled children.
RNA extraction and cDNA synthesis
RNA from the clinical samples was extracted with RNeasy mini Kit (Qiagen, GmbH, Germany) as per the manufacturer’s instructions. The eluted RNA was incubated at 65 °C for 10–15 min and snap chilled on ice. First strand cDNA was synthesized using 100 U Moloney Murine leukemia virus reverse transcriptase (MuMLV-RT H-) (Sibenzyme, Novosibirsk, Russia), 250 ng random hexamers (Promega, Madison, WI, USA), 1 mM dNTPs (Promega, Madison, WI, USA), four units of RNAsin (Promega, Madison, WI, USA) and 7 μl of RNA in 12.5 μl reaction volume. Reverse transcription reaction was carried out by incubation at 37 °C for 1 h 30 min followed by enzyme inactivation at 65 °C for 10 min and snap chilling on ice.
PCR for G protein gene of hRSV
External and semi-nested rounds of PCR for partial G protein gene amplification were carried out with cDNAs using published protocol from our laboratory [15, 16].
DNA Sequencing
The PCR products were purified using mini-elute gel extraction kit (Qiagen, GmbH, Germany). The purified amplicons were cycle sequenced in forward and reverse directions using Big Dye Terminator kit (v 3.1, ABI, USA). The nested set of primers BG517 and F164 were used as forward and reverse primers respectively, for the sequence determination of partial G protein genes. Sequencing reaction products were cleaned up with 3 M sodium acetate (pH4.8) and 125 mM EDTA as per the manufacturer’s instructions and loaded to ABI genetic analyzer 310 (Applied Bio systems Inc., Foster City, CA, USA).
Sequence editing and alignment
Sequence electropherograms were analyzed using Chromas software (version 1.5) and BioEdit version 7.0.0 was used to resolve nucleotide ambiguities. Sequences were handled, edited and formatted using Genedoc version 2.6.002. Sequence similarities of study sequences were achieved by NCBI-BLAST tool (http://www.ncbi.nlm.nih.gov/BLAST/).
Phylogenetic analysis
The nucleotide sequences spanned 655–900 bp of prototype BA strain (BA/4128/99B; GenBank accession number AY333364) [20]. The study sequences were aligned with the sequences downloaded from GenBank (Supplementary Table 1) using CLUSTAL X (version 1.83) software. Phylogenetic and molecular evolutionary analysis was conducted using MEGA version 6.0. The evolutionary history was inferred by using the Maximum Likelihood method based on the Kimura-2 parameter model. The statistical significance of tree topology was tested with bootstrapping (1000 replicas).
Synonymous and non-synonymous mutations
Synonymous and non-synonymous mutations were analyzed by Nei and Gojobori method [14]. The program, SNAP (Synonymous/Non-synonymous Analysis Program) provided by the HIV database web site (http://www.hiv.lanl.gov/content/hiv-db/SNAP/WEBSNAP/SNAP.html) was used for analysis of synonymous verses nonsynonymous mutations.
Nucleotide sequence accession numbers
The sequences from this study were deposited in the GenBank and there accession numbers are KJ690591 to KJ690602 and KJ690604.
Results
Sequence and phylogenetic analysis
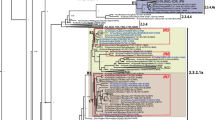
RT-PCR for the second hypervariable region of the G protein gene was done on the clinical samples. Thirteen study sequences were characterized by DNA sequencing and phylogenetic analysis. A total of 49 unique group B partial G protein gene sequences were used to construct the phylogenetic tree including 19 sequences from India (Supplementary Table 1). The alignment was 288 bp (96 amino acid) in length spanning 658–948 bp of G protein gene with reference to prototype group B reference strain. The study sequences were genotyped by phylogenetic comparisons with other sequences reported from different parts of the world [4, 6, 10] (Fig. 1). All the 13 study strains clustered in the BA genotype with 60 bp duplication in the second hypervariable region of the G protein gene. Different subgroups have been described within the BA genotype BA-1 to BA-12 [7, 10]. Six of the study sequences clustered with BA-9 and two sequences with the BA-4 subgroup. One study sequence was grouped in each of the following subgroups, BA-7, BA-10 and BA-12. Two of the study sequences (DEL/S191P/10 and DEL/S192P/10) were not assigned to any specific subgroup of the BA genotype. The study sequences had a 94.1–97.8 % similarity at the nucleotide level and 86.8–95.6 % similarity at the amino acid level as compared to BA prototype strain. On the other hand a similarity of 92.7–100 % at the nucleotide level and 85.7–100 % at the amino acid level was identified among the study sequences.

Phylogenetic tree for hRSV group B nucleotide sequences from the second hypervariable region of the G protein gene. The details of the GenBank sequences that were used for the analysis are given in Supplementary Table 1. The nucleotide sequences were aligned with CLUSTAL X program and the phylogenetic tree was generated by Maximum Likelihood method using MEGA6. The bootstrap values more than 60 % are shown at the nodes. The study sequences are denoted by solid diamonds
Amino acid analysis
The predicted amino acid sequence of the study strains were compared with the BA prototype strain (Fig. 2). The study strains were predicted to encode G proteins of 312 and 319 amino acids with reference to the prototype BA strain (315 amino acids). The stop codon, TAA was utilized by all the study sequences except one sequence that had TAG as the stop codon. A total of 31 amino acid mutations were identified in the 93–100 amino acid region that was analyzed for the study strains as compared to the prototype BA strain. The strain DEL/S154P/09 displayed the highest number of amino acid mutations (ten). The three mutations (L223P, 247P, T270I) were identified in all the study sequences. Two different amino acid substitutions (V271A and H287Y) were observed in most of the study sequences. Certain amino acid substitutions were subgroup specific. The BA-9 subgroup showed five common mutations. Similarly, six mutations were identified in BA-10, ten mutations in BA-12, seven mutations in BA-4 and 4 mutations in the BA-7 subgroup. Some amino acid substitutions were identified in a few sequences of the BA subgroup. Two different amino acid substitutions (R262G and S267P) were observed in two study strains of the BA-9 subgroup (DEL/158P/09 and DEL/159P/09). The two study strains which did not cluster in any subgroup (DEL/191P/10 and DEL/192P/10) showed nine and eight mutations respectively. Four out of these mutations were common among the two sequences (Fig. 3).

The deduced amino acid alignment of the second hypervariable region of the G protein of Indian strains. The alignment is shown relative to the prototype BA strain (BA/4128/99b) (AY333364) corresponding to 220–315 amino acids. The two copies of the 20-amino acid duplicated region are marked by rectangles. Asterisks indicate the stop codons. The potential N-linked glycosylation sites are underlined

The amino acid sequence of the second hypervariable region of the G protein of prototype BA strain (BA/4128/99b) (AY333364). The sequence corresponds to the 220–315 amino acids of the BA prototype strain. The amino acid changes in the Indian strains are indicated by arrows
Glycosylation sites
The N-linked glycosylation sites were predicted for the study sequences with reference to the BA prototype strain. One site at 296 amino acids was conserved in all the study strains. An additional N-glycosylation site at 310 amino acid was observed in one of the strain (DEL/M85P/07). Two study strains (DEL/M258P/08 and DEL/M281P/08) showed the N-linked glycosylation sites at 273 amino acid. This site was located in the 20 amino acid duplicated region. The O-linked glycosylation sites in the G protein are the serine and threonine residues. The program NetOGlyc predicted 40–43 serine and threonine residues that had potential for O-linked glycosylation with G score of 0.5–0.8 in the 93–100 amino acid region. Thirty four of these sites were conserved among all the study strains. Further, the 20 amino acid duplication region had a total of ten O-linked glycosylation sites, including six threonine and four serine residues. Eight of these ten glycosylation sites were conserved among all the study strains.
Analysis of synonymous and non-synonymous mutations
On an average, the nonsynonymous/synonymous (dN/dS) ratio for the partial G protein gene sequences used in this study was 0.47 while the average synonymous/non-synonymous (dS/dN) ratio was 2.13. These values suggest lower dN/dS ratios (<1) and lower distances suggesting negative selection pressure for amino acid change in the analyzed region of the G protein.
Discussion
hRSV is major viral pathogen of acute respiratory infection (ARI) in India [3, 15, 16]. The rapid global spread of the BA genotype suggests that these viruses may have a selective advantage over other hRSV genotypes [21]. In addition, the BA viruses have also replaced the existing group B genotypes in different geographical regions [1]. Limited data is available on molecular characterization of BA viruses from India [3, 7, 15, 16]. In the present investigation, we carried out a retrospective molecular and phylogenetic analysis of circulating strains of BA genotype from New Delhi, India during 2007–1010.
The G protein of hRSV exhibits high genetic and antigenic variability. The mechanism for this high genetic variability includes amino acid substitutions, deletions, insertions, duplications and changes in stop codon usage. An important source of variation in the G protein of BA genotype is protein length polymorphism [21]. The G protein of the study sequences were of 312 and 319 amino acids in length. However, it has been described that changes in stop codon usage have been associated with important antigenic variations found in hRSV escape mutants that were selected with monoclonal antibodies recognizing strain specific epitopes [11, 17, 18]. The BA genotype has been divided into subgroups (BA-I to BA-XII) based on the genetic variability in the 60 bp duplicated region [7]. Our earlier study from New Delhi, India [16] reported two mutations in the 20 amino acid duplicated region. However the Indian strains of the present study had eight amino acid substitutions in the duplicated region. A recent study from Saudi Arabia reported nine amino acid mutations in this region [4]. Thus, it is clear that there is a gradual accumulation of mutations over time in the duplicated region of BA genotype suggesting rapid evolution of these viruses. It is thus evident by these investigations that the duplicated region of G protein is under evolutionary pressure. Therefore, in future we can expect more mutations in this region, which may evolve into new subgroups within this rapidly expanding genotype.
N- and O-linked glycosylation pattern of hRSV has been associated with variable host immune response. The duplication of 20 amino acids resulted in the addition of a few more O-linked glycosylation sites in the G protein. It is worth mentioning that the study strains had ten additional O-linked glycosylation sites in the 20 amino acid duplication region. Thus, it is postulated that the additional amino acids as well as enhanced glycosylation of the G protein of BA viruses may result in antigenically different virus. This may influence the expression of some antigenic epitopes by augmentation of antibody recognition or by camouflaging the antigenic sites [15, 21]. This may further enhance the rapid global spread and survival of the BA genotype among the immunological naïve population. Alternately, the duplication may modify G protein mediated attachment of virion with the host cell which may further result in enhanced fitness of BA viruses.
Estimation of the rates of synonymous and nonsynonymous substitutions is important to understand the selective pressure operating on a gene fragment or gene. The lower dN/dS ratio (<1) of the study sequences indicates shorter distances thus suggesting negative selection pressure and therefore fewer amino acid changes in the G protein. These results indicate that in a closely related virus population, such as in a genotype, neutral or negative selection pressure occurs even in the variable region of the G protein. Other investigations have similarly reported a negative selection pressure in the C-terminal region of the G protein [4, 16].
In conclusion, molecular and phylogenetic characterization of BA genotype from India revealed genetic variation in these viruses occurs probably due to the accumulation of mutations in the duplication region and changes in stop codon usage. The frequency and pattern of the N- and O-linked glycosylation sites of the G protein may alter the antigenic structure of the BA viruses resulting in antigenic variation. This antigenic change in BA viruses may assist in avoidance of host immune response and thus may provide a selective advantage to them. The BA genotype is therefore continuously expanding and spreading to different geographical regions. Further comprehensive studies of the BA viruses from different parts of the world will determine the epidemiological and evolutionary pattern of this emerging genotype.
References
Agoti CN, Mwihuri AG, Sande CJ, Onyango CO, Medley GF, Cane PA, et al. Genetic relatedness of infecting and reinfecting respiratory syncytial virus strains identified in a birth cohort from rural Kenya. J Infect Dis. 2012;206(10):1532–41. doi:10.1093/infdis/jis570.
Agoti CN, Gitahi CW, Medley GF, Cane PA, Nokes DJ. Identification of group B respiratory syncytial viruses that lackthe 60-nucleotide duplication after six consecutive epidemics of total BA dominance at coastal Kenya. Influenza Other Respir Viruses. 2013;7(6):1008–12. doi:10.1111/irv.12131.
Agrawal AS, Sarkar M, Ghosh S, Chawla-Sarkar M, Chakraborty N, Basak M, et al. Prevalence of respiratory syncytial virus group B genotype BA-IV strains among children with acute respiratory tract infection in Kolkata, Eastern India. J Clin Virol. 2009;45(4):358–61. doi:10.1016/j.jcv.2009.05.013.
Almajhdi FN, Farrag MA, Amer HM. Group B strains of human respiratory syncytial virus in Saudi Arabia: molecular and phylogenetic analysis. Virus Genes. 2014;48(2):252–9. doi:10.1007/s11262-013-1030-z.
Arnott A, Vong S, Mardy S, Chu S, Naughtin M, Sovann L, et al. A study of the genetic variability of human respiratory syncytial virus (HRSV) in Cambodia reveals the existence of a new HRSV group B genotype. J Clin Microbiol. 2011;49(10):3504–13. doi:10.1128/JCM.01131-11.
Bose ME, He J, Shrivastava S, Nelson MI, Bera J, Halpin RA, et al. Sequencing and analysis of globally obtained human respiratory syncytial virus a and B genomes. PLoS One. 2015;10(3):e0120098. doi:10.1371/journal.pone.0120098.
Choudhary ML, Anand SP, Wadhwa BS, Chadha MS. Genetic variability of human respiratory syncytial virus in Pune, Western India. Infect Genet Evol. 2013;20:369–77. doi:10.1016/j.meegid.2013.09.025.
Cui G, Zhu R, Qian Y, Deng J, Zhao L, Sun Y, et al. Genetic variation in attachment glycoprotein genes of human respiratory syncytial virus subgroups A and B in children in recent five consecutive years. PLoS One. 2013;8(9):e75020. doi:10.1371/journal.pone.0075020.
Dapat IC, Shobugawa Y, Sano Y, Saito R, Sasaki A, Suzuki Y, et al. New genotypes within respiratory syncytial virus group B genotype BA in Niigata, Japan. J Clin Microbiol. 2010;48(9):3423–7. doi:10.1128/JCM.00646-10.
Khor CS, Sam IC, Hooi PS, Chan YF. Displacement of predominant respiratory syncytial virus genotypes in Malaysia between 1989 and 2011. Infect Genet Evol. 2013;14:357–60. doi:10.1016/j.meegid.2012.12.017.
Melero JA, Garcia-Barreno B, Martinez I, Pringle CR, Cane PA. Antigenic structure, evolution and immunobiology of human respiratory syncytial virus attachment (G) protein. J Gen Virol. 1997;78(Pt 10):2411–8.
Nagai K, Kamasaki H, Kuroiwa Y, Okita L, Tsutsumi H. Nosocomial outbreak of respiratory syncytial virus subgroup B variants with the 60 nucleotides-duplicated G protein gene. J Med Virol. 2004;74(1):161–5. doi:10.1002/jmv.20160.
Nair H, Nokes DJ, Gessner BD, Dherani M, Madhi SA, Singleton RJ, et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet. 2010;375(9725):1545–55.
Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3(5):418–26.
Parveen S, Broor S, Kapoor SK, Fowler K, Sullender WM. Genetic diversity among respiratory syncytial viruses that have caused repeated infections in children from rural India. J Med Virol. 2006;78(5):659–65. doi:10.1002/jmv.20590.
Parveen S, Sullender WM, Fowler K, Lefkowitz EJ, Kapoor SK, Broor S. Genetic variability in the G protein gene of group A and B respiratory syncytial viruses from India. J Clin Microbiol. 2006;44(9):3055–64. doi:10.1128/JCM.00187-06.
Rueda P, Delgado T, Portela A, Melero JA, Garcia-Barreno B. Premature stop codons in the G glycoprotein of human respiratory syncytial viruses resistant to neutralization by monoclonal antibodies. J Virol. 1991;65(6):3374–8.
Rueda P, Palomo C, Garcia-Barreno B, Melero JA. The three C-terminal residues of human respiratory syncytial virus G glycoprotein (Long strain) are essential for integrity of multiple epitopes distinguishable by antiidiotypic antibodies. Viral Immunol. 1995;8(1):37–46.
Scott PD, Ochola R, Ngama M, Okiro EA, Nokes DJ, Medley GF, et al. Molecular epidemiology of respiratory syncytial virus in Kilifi district, Kenya. J Med Virol. 2004;74(2):344–54. doi:10.1002/jmv.20183.
Trento A, Galiano M, Videla C, Carballal G, Garcia-Barreno B, Melero JA, et al. Major changes in the G protein of human respiratory syncytial virus isolates introduced by a duplication of 60 nucleotides. J Gen Virol. 2003;84(Pt 11):3115–20.
Trento A, Viegas M, Galiano M, Videla C, Carballal G, Mistchenko AS, et al. Natural history of human respiratory syncytial virus inferred from phylogenetic analysis of the attachment (G) glycoprotein with a 60-nucleotide duplication. J Virol. 2006;80(2):975–84. doi:10.1128/JVI.80.2.975-984.2006.
Trento A, Casas I, Calderon A, Garcia-Garcia ML, Calvo C, Perez-Brena P, et al. Ten years of global evolution of the human respiratory syncytial virus BA genotype with a 60-nucleotide duplication in the G protein gene. J Virol. 2010;84(15):7500–12. doi:10.1128/JVI.00345-10.
Venter M, Madhi SA, Tiemessen CT, Schoub BD. Genetic diversity and molecular epidemiology of respiratory syncytial virus over four consecutive seasons in South Africa: identification of new subgroup A and B genotypes. J Gen Virol. 2001;82(Pt 9):2117–24.
WHO 2005, Integrated management of childhood illness handbook. In: World Health Organization: WHO/FCH/CAH/0012.
Zhang ZY, Du LN, Chen X, Zhao Y, Liu EM, Yang XQ, et al. Genetic variability of respiratory syncytial viruses (RSV) prevalent in Southwestern China from 2006 to 2009: emergence of subgroup B and A RSV as dominant strains. J Clin Microbiol. 2010;48(4):1201–7. doi:10.1128/JCM.02258-09.
Zlateva KT, Lemey P, Moes E, Vandamme AM, Van Ranst M. Genetic variability and molecular evolution of the human respiratory syncytial virus subgroup B attachment G protein. J Virol. 2005;79(14):9157–67. doi:10.1128/JVI.79.14.9157-9167.2005.
Acknowledgments
We would like to thank Department of Biotechnology and Indian Council of Medical Research, Government of India for their financial assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
VLN Raghuram S and Wajihul Hasan Khan have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Raghuram S, V., Khan, W.H., Deeba, F. et al. Retrospective phylogenetic analysis of circulating BA genotype of human respiratory syncytial virus with 60 bp duplication from New Delhi, India during 2007–2010. VirusDis. 26, 276–281 (2015). https://doi.org/10.1007/s13337-015-0283-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13337-015-0283-7