
Abstract
IgG4-related disease (IgG4-RD) is relatively a new growing entity of immune-mediated origin, characterized by a mass-forming lesion, the infiltration of IgG4-positive plasma cells and occasionally elevated serum IgG4. It is considered to be both a systemic inflammation and sclerosing disease. The most common manifestations are parotid and lacrimal swelling, lymphadenopathy and autoimmune pancreatitis. Sclerosing cholangitis and retroperitoneal fibrosis are among the other mentioned frequent manifestations. The diagnosis should be approved histo-pathologically but other conditions such as lymphoma should be carefully excluded. Patients with IgG4-RD respond beneficially to glucocorticoid therapy especially when given at early onset stages. In some cases, the combination of immunosuppressive agents is required.
Similar content being viewed by others


Case report
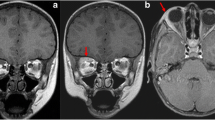
A 68-year-old female was hospitalized due to new onset of abdominal pains, on and off high systemic fever, weight loss and increased inflammation markers (i.e., increased erythrocyte sedimentation rate and C-reactive protein). On abdominal computed tomography (CT), a retroperitoneal mass was noticed, following which a guided biopsy was performed. Histopathology clearly defined the mass to be IgG4-related fibrosis (Figs. 1, 2). The patient was started on oral corticosteroids, which resulted in a slight clinical improvement (i.e., weight gain and improved general status). However, when the abdominal CT was repeated, no improvement in the mass appeared and it was even slightly enlarged. Methotrexate was then added, and after three months when no improvement was seen, it was replaced with rituximab. The addition of rituximab was not beneficial and fever episodes persisted. The mass became bigger and heterogeneous on repeated abdominal CT. In this case, IgG4-related fibrosis was suggested to be a masking finding of underlying malignancy, and abdominal surgery was planned. The mass was surgically removed and the histopathological diagnosis of liposarcoma was established. The patient is currently under follow-up by the Oncology Department.

Immunostaining for IgG and IgG4 is strongly positive in plasma cells. Arrow IgG4 + plasma cells. Anti-IgG4 (MRQ-44) Mouse monoclonal antibody: In formalin fixed, paraffin-embedded tissue optimally diluted for Ventana Roche detection kits in automated slide strainer Benchmark Ultra

Core tissue showing mononuclear inflammation and extensive fibrosis. H&E, F10, X 10
Introduction
IgG4-related disease (IgG4-RD) is increasingly recognized as a serious condition of unknown etiology. It is characterized as having specific locations and a specific histopathology [1]. Previously, this disorder was known as IgG4 multi-organ lymphoproliferative syndrome (IgG4 MOSLP), IgG4 sclerosing disease, or IgG4-related systemic plasmocytic syndrome (SIPS). Recently, it is simply recognized as IgG4-RD, the main features of which are tumor-like swelling of involved organs (mainly lacrimal glands, salivary glands and the pancreas), in association with lymphoplasmacytic infiltrate enriched in IgG4-positive plasma cells and variable degrees of fibrosis with a characteristic “storiform pattern” [2].
Elevated IgG4 serum concentrations are noticed in 60–70 % of patients and responsiveness to glucocorticoids is reported, particularly in early stages of the disease. It is poorly defined, and its incidence in the general population or among different geographic or ethnic groups is indefinite. The estimated incidence in Japan is about 2.6–10.6 per 106. While IgG4-RD occurs mostly in middle-aged persons, the gender distribution differs according to the involved organs. In general, IgG4-RD affects males more than females, yet the group presenting only head and neck symptoms demonstrated an equal sex distribution [2, 3].
Pathogenesis
Normal sera contain nine immunoglobulin isotypes: IgG1, IgG2, IgG3, IgG4, IgM, IgA1, IgA2, IgD and IgE [4]. IgG4 has a special specificity: (1) The concentration of IgG4 in the serum is very low (less than 5 % of total IgG); in contrast to IgG1-3, however, the variability of IgG4 serum concentrations among healthy people reaches a factor of more than 100 (normal range, 0.01 to 0.4 mg per milliliter) and is stable within individuals [5]. (2) Despite the 95 % homology of the constant domains of IgG4 heavy chains with the other subclasses, there is weak or negligible binding of IgG4 to both C1q and Fcγ receptors, which is attributable to amino acid differences within the second constant domain [6]. IgG4 can bind with a low affinity to Fcγ receptor I (CD64), but not to the other Fcγ receptors (Fcγ-RII-CD32, Fcγ-RIII-CD16) [7]. (3) IgG4 molecules are unable to cross-link antigens due to Fab-arm exchange, and therefore, are unable to form immune complexes [8]. Instead, as shown in vitro analysis, they form ineffective immune complexes with other IgG4 antibodies (by Fc–Fc interaction). Therefore, IgG4 is considered uninvolved in activating classical complement pathways effectively and has traditionally been believed to play only a limited role in immune activation. 4) IgG4 production is controlled by T helper 2 cells, by producing interleukin 4 and 13, leading to the production of both IgG4 and IgE [5].
IgG4 has a unique feature of exchanging Fab arms (“flip-flop” exchange of heavy + light chains), thereby generating a new blocking bivalent IgG4 with a bi-specific reactivity. This characteristic provides a protecting effect [1, 4]. In this respect, the level of IgG4 serum antibodies is increased one to 2 months following immunotherapy and in correlation with the success of this treatment in improving allergic rhinitis [9]. In addition, human anti-acetyl choline receptor IgG4 monoclonal antibodies were experimentally demonstrated to have a degrading effect on AChR when assessed on rhabdomyosarcoma TE671 cells. However, these antibodies failed to demonstrate such a degrading effect following their administration to rhesus monkeys due to Fab-arm exchange and generation of bi-specific IgG4 antibodies in the monkeys’ sera. Bi-specific IgG4 antibodies were capable of blocking the AChR degradation by IgG1 anti-AChR-specific monoclonal antibodies [8].
In contrast to the hypothesis of being protective rather than inflammatory, we know that IgG4 plays a central role in the pathogenesis of some autoimmune diseases. For example, anti-desmoglein-1 IgG4 antibodies are important for the generation of cutaneous blisters in patients with pemphigus vulgaris. Additionally, IgG4 antibodies are probably involved in the pathogenesis of membranous glomerulonephritis and thrombotic thrombocytopenic purpura [1].
The pathogenesis of IgG4-RD is poorly understood, raising the possibility of it being autoimmune or allergic in origin [10]. As in all autoimmune diseases, finding a specific autoantigen or autoantibody in IgG4-RD is necessary to underscore a definitive role in autoimmunity. Aiming to achieve this goal, many researchers have tried to detect autoantigens or pathogens with molecular mimicry. Among the many known autoantibodies identified so far, non-specific antinuclear antibodies are detected in more than half of patients with IgG4-RD. Antibodies against carbonic anhydrase II and lactoferrin are the most frequently detected in autoimmune pancreatitis (AIP) (54 and 73 %, respectively). Immune complex deposition was also reported in the pancreas, kidneys, and other affected organs [11]. Two studies have demonstrated molecular mimicry between Helicobacter pylori (H. pylori) and constituents of pancreatic cells, suggesting that gastric H. pylori infection triggers AIP in genetically predisposed individuals through antibody cross-reactivity. In the first study, significant homology between human CA-II and α-carbonic anhydrase of H. pylori was demonstrated [12]. In the second one, 94 % of patients with AIP had antibodies plasminogen-binding protein of H. pylori, which displays homology with that of ubiquitin-protein ligase E3 component n-recognin 2, an enzyme expressed in pancreatic acinar cells [13]. As yet autoimmunity has not been completely proved. Antibodies directed against potential autoantigens as carbonic anhydrases, lactoferrin, pancreatic secretory trypsin inhibitor, and trypsinogens, some of which are expressed in exocrine glands, may come in association with systemic manifestations of IgG4-RD. However, these autoantibodies are lacking specificity for IgG4-RD and are not known to be of the IgG4 subclass [13].
It also remains unclear whether IgG4 is an autoantibody primarily expressed in IgG4-RD or is overexpressed secondarily in response to an unknown primary inflammatory stimulus. The putative tendency of IgG4 antibodies to fulfill anti-inflammatory functions and the fact that disease-specific IgG4 autoantibodies have not been detected in IgG4-RD support the hypothesis that they are a secondary response to inflammation [13].
IgG4-RD is also characterized by a dominance of Th2 immune response, where IL-4 and IL-13 cytokines were proven to increase the production of IgG4 and IgE [14]. In parallel with this, IgG4-RD is characterized by increased numbers of T regulatory (Treg) cells and levels of cytokines produced by them such as IL-10 and TGF-β in both peripheral blood and affected tissues. In this respect, IL-10 interferes with the Ig class switch contributing to increase both IgE and IgG4 production. These findings are consistent with the theory that the production of IgG4 in vivo is induced mainly by a Th2-cell-dominant immune reaction, in the presence of increased IL-10 producing Treg cells [4, 5, 14, 15].
Clinical manifestation
Many studies have reported that 60–90 % of patients with IgG4-RD have multiple organ involvements [16]. IgG4-RD is usually found incidentally when imaging or histopathology procedures are performed. Asymptomatic lymphadenopathy is common, and is present in 80 % of patients with AIP; however, constitutional symptoms such as fever and fatigue—though reported—are generally absent at diagnosis [17]. In a cross-sectional study involving 114 cases with multiple organ involvement, lymphadenopathy was found in 41 % of patients [2]. Symptomatic lymphadenopathy, when present, was due to mass effect, which may range up to 5 cms [18]. About 40 % of patients suffer usually from allergic diseases, mainly allergic rhinitis or asthma (consistent with the above-mentioned dominance of Th2 immune responses) [14]. Yet, IgG4 Mikulicz disease and type 1 AIP are considered the two most common IgG4-related diseases. Initial observations were made in patients with type 1 AIP, also called lymphocytic sclerosing pancreatitis, which normally affects elderly males. This condition often presents as pancreatic mass or as painless obstructive jaundice, and can be misdiagnosed as pancreatic cancer [19]. IgG4-related cholangitis is the most common extra pancreatic manifestation of type 1 AIP and may affect over 70 % of such patients [17]. Normally, IgG4 Mikulicz disease presents as a bilateral swelling of lacrimal and salivary glands but without signs of dryness; unilateral involvement has also been reported [19]. About 40 % of patients with IgG4-related pancreatitis have, in addition, salivary or lacrimal gland involvement, while AIP may be detected in only 17 % of patients presenting with sialadenitis (inflammation of salivary glands) [20]. Sclerosing sialadenitis (Kuttner’s tumor) often precedes AIP in patients who develop both conditions. IgGRD is likely to be responsible for 25–50 % of orbital pseudotumors and can cause orbital myositis (IgG4-related orbital myositis) [21]. In some cases, patients with idiopathic retroperitoneal fibrosis have histologic and serologic changes consistent with IgG4-RD [22].
In a review in which 14 published cases of IgG4-related retroperitoneal fibrosis were summarized, the involvement of other organs included the pancreas (11 patients), salivary glands (3 patients), lymph nodes (2 patients), one affected the pituitary gland, and one the mediastinal periaortitis. On the other hand, isolated IgG4-related retroperitoneal fibrosis was also reported. IgG4-RD is considered to be a cause of noninfectious aortitis. Data have been derived from retrospective pathologic studies of patients who had undergone aortic resections [23, 24]. In a study of 638 patients, who underwent thoracic aortic resections, three patients exhibited histology characteristics of IgG4-RD, representing 9 % of the 33 cases with noninfectious aortitis or 0.5 percent of the entire series of thoracic resections [25]. Thyroid involvement in IgG4-RD takes two forms: Riedel’s thyroiditis and the fibrous variant of Hashimoto’s thyroiditis [26]. IgG4-related pulmonary disease is mostly asymptomatic but may present with cough, hemoptysis, and dyspnea or chest pain. Manifestations as pseudotumors and interstitial pneumonia have been associated with AIP [27]. Visceral or parietal pleural thickening may occur. The most common finding of renal involvement in patients with IgG4-RD is tubulointerstitial nephritis [28]. In a Japanese retrospective study of 153 patients with suspected IgG4-RD, 23 patients (15 %) were identified with tubulointerstitial nephritis (TIN) secondary to IgG4-RD, and 96 % of TIN patients exhibited extrarenal manifestations, which included sialadenitis (83 %), lymphadenopathy (44 %), AIP (39 %), dacryoadenitis (30 %), and lung lesions (26 %). Symptoms—when present—were usually secondary to extrarenal manifestations. The involvement of other organs or tissues in IgG4-RD includes skin, liver (resembling autoimmune hepatitis), stomach (lymphoplasmacytic associated with AIP), breast (sclerosing mastitis and inflammatory pseudotumors of the breast), hypopituitarism with IgG4-related hypophysitis, pachymeningitis, prostatitis and constrictive pericarditis [29–33].
Association with malignancy
Pancreato-biliary cancers and other malignancies were reported to have increased IgG4 infiltration. The finding of elevated numbers of IgG4 + plasma cells within a tumor is misleading and may mistakenly be considered as IgG4-RD. In such cases, a needle biopsy sampling from the periphery of a malignant neoplasm may lead to misdiagnoses of IgG4-RD. Some types of lymphoma have been reported in patients with IgG4-RD. In one study involving 111 patients with IgG4-RD (91 % with AIP), three cases of non-Hodgkin lymphoma were found within three to five years after diagnosing IgG4-RD. The standardized incidence ratio was 16.0 (95 % CI 3.3–45.5), indicating an increased risk of non-Hodgkin lymphoma [34]. In another series of 108 patients with IgG4-related pancreatitis, 18 cancers were diagnosed in 15 patients within a median follow-up of 3.3 years. The highest risk was in the first year following the diagnosis, and gastric cancer was the most common involved, with the other organs being lung, prostate, colon, non-Hodgkin lymphoma, bile duct, and thyroid [35]. In six patients suffering from IgG4-related pancreatitis and cancer, the diagnosis of cancer was histologically approved before being treated with corticosteroids. Interestingly, IgG4-related pancreatitis remained in full remission after successful treatment of the cancer. According to this observation, the finding of IgG4-RD is considered a para-neoplastic syndrome in some cases. The possibility that patients with IgG4-RD have increased prevalence of malignancy requires further study.
Diagnosis
IgG4RD should be suspected if any of the following is present: Pancreatitis of unknown origin, sclerosing cholangitis or bilateral salivary and/or lacrimal gland enlargement. Sah et al. reported that normal IgG4 serum levels are present in 20–30 % of patients with typical IgG4 histopathological infiltrates [36]. Thus, elevated serum levels of IgG4 are considered characteristic of, but not specific to, IgG4-RD. A multinational, multidisciplinary group of experts defined criteria for the diagnosis of IgG4-RD [37]. The three major histopathological features associated with IgG4-related disease are as follows:
-
1.
Dense lymphoplasmacytic infiltrate
-
2.
Focally arranged fibrosis in a storiform pattern
-
3.
Obliterative phlebitis.
The other histopathological features associated with IgG4-RD are as follows: 1. Phlebitis without obliteration of the lumen; and 2. Increased numbers of eosinophils. These two features alone are neither sensitive nor specific for the diagnosis of IgG4-RD. A confident pathological diagnosis of IgG4-RD necessitates the presence of two of the three major histological features. The usually detected features are a dense lymphoplasmacytic infiltrate and storiform-type fibrosis. The group of experts mentioned above proposed a scheme and three options for the diagnosis of IgG4-related disease that is based primarily on the morphological appearance on biopsy (Table 1). Three clinical options were suggested:
-
1.
Histology highly suggestive of IgG4-RD: This category requires at least two of the major histological features (a dense lymphoplasmacytic infiltrate, storiform-type fibrosis and obliterative phlebitis). Exceptions are the lymph nodes, lung, minor salivary glands and the lacrimal gland, where both storiform fibrosis and obliterative phlebitis may be absent (Table 2). In this case, one major histological feature compatible with IgG4-RD might suffice for dacryoadenitis (37). The required number of IgG4 + plasma cells (cell/hpf) for establishing the diagnosis of IgG4-RD is broad and in accordance with the affected organs, ranging from 10 to 200 cells/hpf. The IgG4 +/IgG + cell ratio of > 40 % was also suggested as a requirement for a full diagnosis. In the case of aortic specimens, a cell ratio of >50 % is considered a minimal requirement for diagnosis due to the fact that atherosclerosis giant cell arteritis and infectious aortitis may have a IgG4 +/IgG + ratio close to 40 % [23].
-
2.
Probable histological features of IgG4-RD: These cases lack the full histopathological features associated with IgG4-RD, and therefore, require additional clinical, serological, or radiological findings to confirm a full diagnosis. Of these, IgG4 serum >135 mg/dl and another organ involvement based on radiological or pathological examination are the most common.
-
3.
Insufficient histopathological evidence of IgG4-RD: Patients in this category are not entirely excluded from a diagnosis of IgG4-RD. Sampling artifact, the effects of previous therapy, and progression to a fibrotic stage may constitute potential reasons for making the diagnosis.
To establish the extent of disease, because of the frequency of subclinical features and the involvement of two organs, a CT scan of chest, abdomen, and pelvis should be performed. Organ infiltration in IgG4RD demonstrates high uptake on gallium scan and positron emission tomography-computed tomography (PET-CT) [38]. When available, PET-CT is considered at baseline.
Several diseases, being outside the range of IgG4-RD, are associated with increased numbers of IgG4 + plasma cells in tissue. These include: (1) Inflammatory diseases: primary sclerosing cholangitis, ANCA-associated vasculitis, rheumatoid arthritis, inflammatory bowel disease, cutaneous plasmocytosis, and autoimmune atrophic gastritis (pernicious anemia) [39, 40]. None of these conditions, however, consistently show IgG4-rich inflammation and all lack the characteristic histopathological features of IgG4-RD. (2) Lymphoma: both extranodal marginal zone lymphomas and sometimes follicular lymphomas and angioimmunoblastic lymphomas can mimic IgG4-RD. In cases of possible IgG4-RD with plasma cells exhibiting atypical features, low-grade B-cell lymphomas must be excluded. The majority of infiltrating lymphocytes in IgG4RD are T-and B lymphocytes, and are mostly found in nodular aggregates, often within reactive germinal centers [37].
Differential diagnosis
The differential diagnosis of IgG4-RD depends upon the involved organ and clinical presentation. Differential diagnosis of AIP type 1 includes other types of pancreatitis (e.g., biliary, alcoholic, drug induced), type 2 AIP (young adults with inflammatory bowel disease) and adenocarcinoma of pancreas [38]. Differentiation is usually difficult when based on clinical and radiological findings. For example, painless jaundice is common to both AIP type 1 and pancreatic adenocarcinoma. In some cases, patients have mistakenly undergone Whipple procedures with the intention of treating pancreatic cancer. IgG4-RD of salivary glands has many common features with Sjögren’s syndrome (SS). Clinical features that are helpful in distinguishing between SS and IgG4-RD include relatively mild dryness of the eyes and of the mouth, a higher frequency of allergic rhinitis and bronchial asthma, a higher frequency of AIP and interstitial nephritis and low frequencies of autoantibodies (including RF, ANA, anti-SSA, and anti-SSB) [41, 42].
The clinical differentiation between primary sclerosing cholangitis, cholangiocarcinoma and IgG4-related sclerosing cholangitis is difficult, but distinguishing between the diseases is critical because of the drastically different therapies and prognoses. Differentiation could be done using the classification of Nakazawa et al., where IgG4-related sclerosing cholangitis is classified into four types according to findings in the cholangiograms [43, 44]. IgG4-RD can also mimic sarcoidosis. In one study observing patients with sarcoidosis due to bilateral hilar adenopathy and/or lung nodules on a chest CT, patients with IgG4-RD were found among a subset with elevated IgG4 serum, which also displays significantly higher levels of IgG4, IgG4/IgG and IgG4/IgG3 in bronchoalveolar lavage compared with those with normal IgG4 serum levels [45].
Treatment
The issue of how to treat IgG4-RD is based on the experience from observational data, case reports and small series, most of which came from patients with AIP [17]. There are no randomized trials that have compared and evaluated different approaches to the treatment of IgG4-RD. IgG4-RD is responsive to steroids, which remain the first line of therapy. Responsiveness is expected to be within weeks, including symptomatic improvement, reductions in the size of masses or organ enlargement, improvement in organ function, and usually a decrease in IgG4 serum levels. However, some patients respond only within a few months, some relapse, and others respond partially or not at all. Less responsive patients are those with more advanced fibrotic changes, but such cases have not been well defined. In a few case reports, spontaneous remission with regression of organ infiltrates was reported.
The measurement of IgG4 serum concentration as an indicator of disease activity remains controversial [30]. In patients with elevated IgG4 serum concentrations at baseline, such concentrations may decrease with glucocorticoid treatment, but they remain above the normal values in most cases [36]. The value of IgG4 serum concentrations in predicting disease relapse is highly questionable, mostly due to limited follow-up periods. In some patients, the monitoring of IgG4 concentrations may indicate early relapse; however, disease relapses were recorded in 10 % of patients having normal IgG4 serum concentrations [46]. Spontaneous remission with regression of organ infiltrates is very rare.
The involvement of vital organs in IgG4-RD should be treated aggressively to avoid serious organ dysfunction and failure. Untreated IgG4-RD often progresses to extensive fibrosis, but some manifestations of the disease remain stable over time and do not require immediate treatment. For example, for IgG4-related lymphadenopathy or incidentally-detected lung nodules, watchful waiting is considered to be appropriate. Reports of indolent cases of lymphadenopathy persisting for decades have been described [18]. Prednisolone at a dose of 0.6 mg per kilo of body weight per day for 2–4 weeks is recommended by a consensus statement from 17 referral centers in Japan. They also recommended tapering down the level of prednisolone over a period of 3–6 months to 5 mg per day, and continuing at a maintenance dose between 2.5 and 5.0 mg per day for up to 3 years. Discontinuing glucocorticoids entirely within 3 months is considered another approach [46, 47]. Azathioprine, mycophenolate mofetil, and methotrexate are offered as glucocorticoid-sparing agents or remission-maintenance drugs, although their efficacy has never been tested in clinical trials. A disease responder index is a tool suggested by the Organizing Committee of the International IgG4-related Disease Symposium to detect changes in disease activity and to quantify objectively the treatment response by providing standardized outcome measures [48].
By recurrence or refractoriness, B-cell depletion with rituximab can be attempted and is considered a useful approach. In two studies (consisting altogether of 14 patients), the clinical and serologic responses to B lymphocyte depletion therapy in IgG4-RD patients who remained refractory to steroids and DMARDs were assessed. Treatment with rituximab (2 infusions of 1000 mg, 15 days apart) led to prompt clinical and serologic improvement in these patients with refractory IgG4-RD within one month of starting rituximab. One patient with advanced thyroid fibrosis did not show improvement in the thyroid gland, but disease did not progress to involve new organs. Four patients were re-treated with RTX after 6 months, two patients because of symptom recurrence and increasing IgG4 concentration, and the other two because of physician discretion. Repeated courses of RTX maintained their effectiveness and further decreased IgG4 concentrations. The reduction in IgG-subclass levels appeared to be specific for IgG4. Rituximab achieves its effects in IgG4-RD by depleting B lymphocytes that replenish short-lived IgG4-secreting plasma cells [49, 50]. Patients with significant fibrosis in biopsy are less likely to have a response to glucocorticoids and rituximab and have a poor prognosis.
The natural history of IgG4-RD is not well defined. Various complications influencing morbidity and mortality in untreated patients were observed. These complications include cirrhosis and portal hypertension; retroperitoneal fibrosis; complications from aortic aneurysms, including dissection; biliary obstruction; diabetes mellitus; and others [17, 22]. Sustained benefit may be observed during therapy, but relapses are common after discontinuation of therapy.
In conclusion, IgG4-RD remains a field not yet fully explored or understood. Early and efficient therapies aiming to achieve a beneficial outcome, and to improve prognosis are still required. Malignancy should be considered in each case of IgG4-RD to remove as early as possible.
Abbreviations
- AIP:
-
Autoimmune pancreatitis
- IgG4-RD:
-
IgG4-related disease
- IgG4 SIPS:
-
IgG4-related systemic plasmocytic syndrome
- IgG4 MOSLP:
-
IgG4 multi-organ lymphoproliferative syndrome
- RTX:
-
Rituximab
References
Stone JH, Zen Y, Deshpande V et al (2012) IgG4-related disease. N Engl J Med 366:539
Zen Y, Nakanuma Y (2010) IgG4-related disease: a cross-sectional study of 114 cases. Am J Surg Pathol 34:1812–1819
Nishimori I, Tamakoshi A, Otsuki M et al (2007) Prevalence of autoimmune pancreatitis in Japan from a nationwide survey in 2002. J Gastroenterol 42(Suppl 18):6–8
Aalberse RC, Stapel SO, Schuurman J et al (2009) Immunoglobulin G4: an odd antibody. Clin Exp Allergy 39:469–477
Nirula A, Glaser SM, Kalled SL et al (2011) What is IgG4? A review of the biology of a unique immunoglobulin subtype. Curr Opin Rheumatol 23:119–124
Canfield SM, Morrison SL (1991) The binding affinity of human IgG for its high affinity Fc receptor is determined by multiple amino acids in the CH2 domain and is modulated by the hinge region. J Exp Med 173:1483–1491
Huizinga TW, Roos D, von dem Borne AE et al (1990) Neutrophil Fc-gamma receptors: a two-way bridge in the immune system. Blood 75(6):1211–1214
Van der Neut Kolfschoten M, Schuurman J, Losen M et al (2007) Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science 317:1554–1557
Nouri-Aria Kayhan T, Petra A et al (2004) Grass pollen immunotherapy induces mucosal and peripheral IL-10 responses and blocking IgG activity. J Immunol 172(5):3252–3259
Cheuk W, Chan JK (2010) IgG4-related sclerosing disease: a critical appraisal of an evolving clinicopathologic entity. Adv Anat Pathol 17:303
Zen Y, Nakanuma Y (2011) Pathogenesis of IgG4-related disease. Curr Opin Rheumatol 23:114
Guarneri F, Guarneri C, Benvenga S et al (2005) Helicobacter pylori and autoimmune pancreatitis: role of carbonic anhydrase via molecular mimicry? J Cell Mol Med 9:741–744
Frulloni L, Lunardi C, Simone R et al (2009) Identification of a novel antibody associated with autoimmune pancreatitis. N Engl J Med 361:2135–2142
Kamisawa T, Anjiki H, Egawa N et al (2009) Allergic manifestations in autoimmune pancreatitis. Eur J Gastroenterol Hepatol 21:1136–1139
Miyoshi H, Uchida K, Taniguchi T et al (2008) Circulating naïve and CD4 + CD25high regulatory T cells in patients with autoimmune pancreatitis. Pancreas 36:133
Okazaki K, Uchida K, Koyabu M et al (2011) Recent advances in the concept and diagnosis of autoimmune pancreatitis and IgG4-related disease. J Gastroenterol 46:277
Khosroshahi A, Stone JH (2011) A clinical overview of IgG4-related systemic disease. Curr Opin Rheumatol 23:57
Cheuk W, Yuen HK, Chu SY et al (2008) Lymphadenopathy of IgG4-related sclerosing disease. Am J Surg Pathol 32:671
Divatia M, Kim SA, Ro JY et al (2012) IgG4-related sclerosing disease, an emerging entity: a review of a multi-system disease. Yonsei Med J 53(1):15–34
Hamano H, Arakura N, Muraki T et al (2006) Prevalence and distribution of extrapancreatic lesions complicating autoimmune pancreatitis. J Gastroenterol 41:1197
Mehta M, Jakobiec F, Fay A et al (2009) Idiopathic fibroinflammatory disease of the face, eyelids, and periorbital membrane with immunoglobulin G4-positive plasma cells. Arch Pathol Lab Med 133:1251
Zen Y, Onodera M, Inoue D et al (2009) Retroperitoneal fibrosis: a clinicopathologic study with respect to immunoglobulin G4. Am J Surg Pathol 33:1833
Stone JR (2011) Aortitis, periaortitis, and retroperitoneal fibrosis, as manifestations of IgG4-related systemic disease. Curr Opin Rheumatol 23:88
Zen Y, Sawazaki A, Miyayama S et al (2006) A case of retroperitoneal and mediastinal fibrosis exhibiting elevated levels of IgG4 in the absence of sclerosing pancreatitis (autoimmune pancreatitis). Hum Pathol 37:239
Stone JH, Khosroshahi A, Deshpande V et al (2010) IgG4-related systemic disease accounts for a significant proportion of thoracic lymphoplasmacytic aortitis cases. Arthritis Care Res (Hoboken) 62:316
Li Y, Nishihara E, Kakudo K et al (2011) Hashimoto’s thyroiditis: old concepts and new insights. Curr Opin Rheumatol 23:102
Zen Y, Kitagawa S, Minato H et al (2005) IgG4-positive plasma cells in inflammatory pseudotumor (plasma cell granuloma) of the lung. Hum Pathol 36:710
Murashima M, Tomaszewski J, Glickman JD et al (2007) Chronic tubulointerstitial nephritis presenting as multiple renal nodules and pancreatic insufficiency. Am J Kidney Dis 49:e7
Ikeda T, Oka M, Shimizu H et al (2013) IgG4-related skin manifestations in patients with IgG4-related disease. Eur J Dermatol 23:241
Umemura T, Zen Y, Hamano H et al (2007) IgG4 associated autoimmune hepatitis: a differential diagnosis for classical autoimmune hepatitis. Gut 56:1471
Uehara T, Hamano H, Kawa S et al (2010) Chronic gastritis in the setting of autoimmune pancreatitis. Am J Surg Pathol 34:1241
Cheuk W, Chan AC, Lam WL et al (2009) IgG4-related sclerosing mastitis: description of a new member of the IgG4-related sclerosing diseases. Am J Surg Pathol 33:1058
Sugimoto T, Morita Y, Isshiki K et al (2008) Constrictive pericarditis as an emerging manifestation of hyper-IgG4 disease. Int J Cardiol 130:e100
Takahashi N, Ghazale AH, Smyrk TC et al (2009) Possible association between IgG4-associated systemic disease with or without autoimmune pancreatitis and non-Hodgkin lymphoma. Pancreas 38:523
Shiokawa M, Kodama Y, Yoshimura K et al (2013) Risk of cancer in patients with autoimmune pancreatitis. Am J Gastroenterol 108:610
Sah RP, Chari ST (2011) Serologic issues in IgG4-related systemic disease and autoimmune pancreatitis. Curr Opin Rheumatol 23:108–113
Deshpande V, Zen Y, Chan JK et al (2012) Consensus statement on the pathology of IgG4-related disease. Mod Pathol 25:1181
Sato Y, Notohara K, Kojima M et al (2010) IgG4-related disease: historical overview and pathology of hematological disorders. Pathol Int 60:247–258
Zhang L, Smyrk TC (2010) Autoimmune pancreatitis and IgG4-related systemic diseases. Int J Clin Exp Pathol 3:491–504
Kuo TT, Chen TC, Lee LY et al (2009) Sclerosing angiomatoid nodular transformation of the spleen (SANT): clinicopathological study of 10 cases with or without abdominal disseminated calcifying fibrous tumors, and the presence of a significant number of IgG4 + plasma cells. Pathol Int 59:844–850
Masaki Y, Dong L, Kurose N et al (2009) Proposal for a new clinical entity, IgG4-positive multiorgan lymphoproliferative syndrome: analysis of 64 cases of IgG4-related disorders. Ann Rheum Dis 68:1310
Masaki Y, Sugai S, Umehara H et al (2010) IgG4-related diseases including Mikulicz’s disease and sclerosing pancreatitis: diagnostic insights. J Rheumatol 37:1380
Webster GJ, Pereira SP, Chapman RW et al (2009) Autoimmune pancreatitis/IgG4-associated cholangitis and primary sclerosing cholangitis–overlapping or separate diseases? J Hepatol 51:398
Nakazawa T, Ohara H, Sano H et al (2006) Schematic classification of sclerosing cholangitis with autoimmune pancreatitis by cholangiography. Pancreas 32:229
Tsushima K, Yokoyama T, Kawa S et al (2011) Elevated IgG4 levels in patients demonstrating sarcoidosis-like radiologic findings. Medicine (Baltimore) 90:194
Kamisawa T, Shimosegawa T, Okazaki K et al (2009) Standard steroid treatment for autoimmune pancreatitis. Gut 58:1504–1507
Kamisawa T, Okazaki K, Kawa S et al (2010) Japanese consensus guidelines for management of autoimmune pancreatitis. III. Treatment and prognosis of AIP. J Gastroenterol 45:471–477
Mollie N. Carruthers, John H. Stone, Vikram Deshpande et al (2012) Development of an IgG4-RD Responder Index. Inter J Rheumatol, Article ID 259408
Khosroshahi A, Carruthers M, Deshpande V et al (2012) Rituximab for the treatment of IgG4-related disease: lessons from ten consecutive patients. Medicine (Baltimore) 91:57–66
Khosroshahi A, Bloch DB, Deshpande V et al (2010) Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum 62:1755–1762
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent and animal rights
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Informed consent
Informed consent was obtained from all individual participants included in the study. For this type of study formal consent is not required.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Nizar, AH., Toubi, E. IgG4-related disease: case report and literature review. Autoimmun Highlights 6, 7–15 (2015). https://doi.org/10.1007/s13317-015-0069-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13317-015-0069-3