
Abstract
Systemic lupus erythematosus (SLE) is considered an autoimmune disease with multiorgan involvement. Many advances have been made during the last decade regarding inflammatory pathways, genetic and epigenetic alterations, adaptive and innate immune system mechanisms specifically involved in SLE pathogenesis. Apoptosis has been proposed as an important player in SLE pathogenesis more than a decade ago. However, only recently new key apoptotic pathways have been investigated and the link between apoptotic debris containing autoantigens, innate immunity and ongoing inflammation has been further elucidated. Better understanding of cellular mechanisms and involved cytokines contributed to the development of new biological drugs specifically addressed for SLE therapy.
Similar content being viewed by others


Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by protean clinical manifestations, multiorgan involvement and unpredictable course [1].
SLE incidence has been estimated ranging 1–10 cases per 100,000 persons for year and the prevalence has been reported to range between 20 and 150 cases every 100,000 persons. Genetic and environmental factors play a key role probably explaining not only different geographical prevalence, but also disease severity and course in different patients. Overall the incidence of SLE is higher in African American, Hispanic and Asian individuals than Caucasians. SLE usually affects women during their reproductive years with a 9:1 female to male ratio [2–5].
SLE has classically been considered an autoimmune disease with a predominant adaptive immune system component, since T cells and recently B cells have been considered the most important pathogenetic player [6]. More recently, many studies also focused on innate immune system, and in particular on dendritic cells and phagocytes, since their ability to interact with autoantigens during the early inflammatory phase. These cells are indeed responsible for antigen processing, presentation and secondarily for activation of the adaptive immune system [7, 8].
SLE genetical consideration
It has been shown that monozygotic twins display a higher rate of concordance, when compared to dizygotic ones (34 vs 3 %), regarding the risk of developing SLE. Several genes seem to increase SLE susceptibility. However, genetic mutations that cause the disease in a Mendelian fashion account only for a small percentage of SLE cases. For the majority of patients, a combination of genes, instead of a single gene, predisposes to the disease, in particular when interaction with environmental factors occurs [9].
For example some class major histocompatibility complex antigens, in particular from class II genes (HLA-DR, DQ and DP), have classically been associated with SLE susceptibility [10]. Homozygous C1q deficiency and genetic mutations determining low levels of C2 and C4 are associated with an increased risk of developing SLE, probably since low complement activity contributes to defective clearance of the apoptotic material and consequently determines accumulation of potential autoantigens [11]. Recently low levels of mannose-binding lectin (MBL), a C1q-related protein with a crucial role in opsonizing mannose-rich microorganisms and activating complement classical pathway, have also been described in SLE patients [12].
Peripheral blood mononuclear cells isolated from SLE patients, particularly when the disease is characterized by severe renal involvement, usually display high IFN-I activity detected using a microarray IFN-genetic signature technique [13, 14]. These patients present indeed an overexpression of the interferon regulatory factor 5, a transcription factor of plasmocytoid dendritic cells (pDCs) [15]. The recent observation that an increased genetic IFN-α activity has been also described as an heritable risk factor for lupus, confirms the importance of IFN-I system for SLE pathogenesis [16].
DNA methylation and histone modification are key mechanisms of human epigenetic control on gene expression. SLE patients with active lupus were found to have a reduced capacity of DNA methylation of several genes, leading to an overexpression of inflammatory proteins such as CD11a, CD70, CD40L. Perforin overexpression, due to gene hypomethylation, is responsible of abnormal CD4+ T lymphocytes killing activity [17, 18]. Some drugs such as hydralazine or procainamide, well known for being associated to drug-induced lupus, could also determine DNA hypomethylation. Furthermore, in SLE patients, DNA hypomethylation increases apoptotic rate on peripheral blood mononuclear cells [19].
MicroRNAs (MiRNAs), non-coding molecules that regulate the expression of target genes in a post-transcriptional manner, could control both DNA methylation and histone modifications. Abnormal patterns of MiRNAs have also been demonstrated in SLE patients [20, 21].
The role of hormonal and environmental factors
Many theories have been postulated in the past to explain why SLE has a so strong female predominance [22, 23]. A key role has been attributed to estrogens’ signal through the receptor α. Previous studies have reported that females with SLE present increased level of estrogens and reduced level of androgens, probably due to increased aromatase activity, an enzyme that converts androgens into estrogens [24, 25]. Very interestingly, the first effective mouse model of renal lupus was described in New Zealand black × white female (NZB/WF) mice. Although there have been subsequent descriptions of lupus in male murine strains, NZB/WF mice model brought the very first scientific evidence for a role of sex hormones in SLE immune responses [26, 27].
The increased risk of SLE flare during pregnancy may be related to the higher amount of sex hormones even if circulating estrogens’ levels are not considered good predictors of disease flares. In vitro studies demonstrated an increased production of inflammatory cytokines, especially IFN-α, from dendritic cells (DCs) exposed to estrogens [28]. An increased cytokine production has also been revealed in T and B lymphocytes exposed to estrogens, probably due to NF-kB activity modulation. By contrast, progesterone could block Toll-like receptor (TLR)-7, inducing a reduction of inflammatory cytokines [29]. During puerperium, the clinical course of several autoimmune diseases can classically get worse. This is probably due to increased levels of circulating prolactin, a hormone well known for its stimulatory effects on the immune system especially on B cells [30].
The role of infections has been also regarded for a long time as an important trigger for both SLE induction during early phases and later for exacerbations during disease course. Complement deficiencies, MBL pathway alteration, FCγRs polymorphism have been already described as susceptibility genes, implicating that abnormalities in defense mechanisms may ultimately predispose individuals to SLE [31, 32].
Cross-reactivity between self and non-self microbial epitopes seems to be a pivotal mechanism in breaking the immune tolerance. Such molecular mimicry has been for example described between EBV nuclear antigen-1 and self-antigens. Chronic active EBV infection could promote the continuous production of IFN-I and up-regulate TLR expression from innate immunity cells [33, 34]. Furthermore, as discussed above, bacterial and viral hypomethylated DNA are very efficient in activating the innate immune system through TLR system.
Ultraviolet (UV) light induces apoptosis in keratinocytes and causes translocation of autoantigens from cellular compartment to membranal surface on apoptotic blebs. UVB exposition seems to specifically promote plasmocytoid DC recruitment into cutaneous lesions of SLE patients. pDC can subsequently present apoptosis-associated autoantigens to lymphocytes and induce specific adaptive humoral and cellular responses. Increased levels of IFN-α have indeed been demonstrated in skin specimens from SLE patients. Moreover, UV light increases the production of other inflammatory cytokines, such as IL-1, IL-6 and TNF-α by keratinocytes and lymphocytes. UV light, apoptotic mechanisms and IFN-α production by pDCs play a crucial role in SLE pathogenesis, in accordance with the well-known described phenomena of cutaneous and not rarely systemic flare after sunlight exposure for SLE patients [35, 36].
Several drugs, such as procainamide, hydralazine and quinidine have been described as trigger factors for SLE. In the majority of cases, however, drug-induced autoimmunity consists only of circulating autoantibodies without the development of overt SLE symptoms. Indeed, true drug-induced lupus usually manifests with skin, joint inflammation or constitutional symptoms (such as fever and asthenia); however, kidney, heart or brain are very rarely involved. As previously stated, reduction in DNA methylation activity can explain for some cases the pathogenesis of drug-induced SLE that usually resolves completely after drug discontinuation [37].
Apoptosis
Apoptosis is a programmed cell death (PCD), that can be triggered by different internal, such as DNA mutation, or external, such as infections and UV exposure, trigger factors. Apoptosis is a fine regulated process, which requires energy with ATP consumption, sequential activation of intracellular enzymes with protease activity (caspases), digestion of chromatin and DNA by DNase enzyme, and finally cytoskeleton modification with formation of microparticles from the membrane. However, unlike necrosis, the integrity of cellular membrane during apoptosis is preserved and the intracellular content is not released into the extracellular compartment. Anyways, a prompt clearance of apoptotic cells is needed under normal circumstances from phagocytes and macrophages, which recognize, adhere, engulf and ultimately digest apoptotic debris. Probably the phosphatidylserine exposed early on the external membrane during apoptosis phases plays a major role for phagocytic recognition; however, the precise involved receptors are not yet completely understood and identified. Notably, the interaction between macrophages and apoptotic cells determines a tolerogenic immunological response, characterized by release into the microenvironment of TGF-beta and IL-10, which ultimately prevents the beginning of inflammation [38, 39].
During apoptosis, cells undergo profound modification such as nuclear fragmentation, cytoskeleton rearrangement and membrane blebbing. As a consequence, apoptosis-modified or cryptic autoantigens, which are normally not exposed to the immune system, are transitory expressed on apoptotic cell membranes or apoptotic debris, such as microparticles, generating during the process of cell death. A normal and effective function of phagocytic cells is physiologically fundamental for maintaining a rapid and complete clearance of apoptotic cells, which would otherwise evolve into secondary necrosis. This last circumstance would determine the release of “danger signals” molecules, normally enclosed inside the cytoplasm, into the extracellular environment, and would begin an inflammatory response characterized by cytokine release and production of autoantibodies [40, 41]. For example, high mobility group box protein 1 (HMGB1), produced during cell activation and during apoptosis in attempt to stabilize the nucleosome structure, shows strong proinflammatory activity when released into extracellular compartment. HMGB1 has indeed been identified as “alarmin” able to amplify inflammation and enhance immune responses by interacting with the receptor for Advanced Glycation End Products (RAGE) and TLRs 2,4 and 9 [42, 43].
As a consequence of defective apoptosis clearance in human SLE, patients present circulating self-DNA or self-RNA complex that becomes antigenic targets for both humoral and cell-mediated autoimmune responses.
In a murine model of SLE, an increased apoptosis rate usually results in lupus-like features. On the other hand, such an increase has not been shown in human SLE that has been predominantly associated with a reduction of apoptotic bodies’ clearance from phagocytic/macrophage system. In both cases, the final result is always an increased apoptotic burden that determines the recognition of apoptotic-derived autoantigens and hyperactivation of innate and adaptive immune system cells.
Dendritic cells and innate immunity activation
DCs can recognize damage-associated molecular pattern, as not only microbial but also as endogenous ligands, via pathogen recognition receptors and, particularly, through TLRs. TLRs are a family of transmembrane proteins, expressed by not only innate and adaptive immune system cells, but also by some epithelial cells, which recognize various pathogen molecular patterns from bacteria, viruses, fungi or protozoan parasites [44]. TLRs are located both on the surface and in the cytosol of different cells; anyways, these receptors are particularly expressed in antigen-presenting cells. For SLE pathogenesis, nucleic acid-sensing TLRs (TLR3, TLR7, TLR8 and TLR9) are of particular interest since they recognize and bind DNA- or RNA-containing antigens. In particular, TLR7 recognizes single-stranded RNA, while TLR9 is considered very efficient in binding unmethylated CpG DNA. The strategic intracellular location of these two TLRs minimizes accidental exposure to self-nucleic material, since their activation is under normal circumstance triggered by nuclear material from viral or microbiological origin. The inflammatory cascade from TLRs is particularly effective when the nucleic acid material is delivered in the form of immunocomplex, since a second signal from Fcγ receptors is usually needed to amplify the immunological response. Both TLR7 and TLR9 stimulate the production of type I interferon through key adaptor molecules such as MyD88, a critical protein in IFN alfa production. During SLE pathogenesis, the activation of intracellular TLRs is not driven by microbiological nuclear material, since the inflammatory response is maintained by material of apoptotic origin [45, 46]. Actually in SLE predisposed subjects, endogenous TLRs ligands, such as DNA- or RNA-containing particles generated from apoptosis, can activate the innate immune system. An increased level of circulating DNA, RNA and nuclear proteins has been shown in blood from SLE patients [47].
The most significant amount of evidence in the context of SLE involves the above-mentioned endosomal receptors TLR7 and TLR9. For example, an increased expression of TLR9 in B cells and monocytes has been described in active SLE patients [48]. Moreover, in this context, environmental factors, such as viral infections, could also contribute to disease flares determining a further activation of TLRs and IFN-I systems.
Under normal circumstances, immature DCs present self-antigens without costimulatory signals, inducing a tolerogenic effect on autoreactive lymphocytes. However, under certain proinflammatory conditions, which are able to induce DC maturation, self-antigens are presented to T lymphocytes in the presence of costimulatory signals, promoting an autoimmune response [49, 50]. While myeloid DCs (mDCs) are able to recognize, phagocyte and present uncomplexed apoptotic material, pDCs usually recognize immune complex (IC) containing apoptotic material and are very efficient in producing large amounts of IFN-α. Although any cell can virtually produce type I IFN, pDCs are undoubtedly considered the major producers of this family of cytokines, which comprises 13 different IFN-α isoforms as well as IFN-β, IFN-ε, IFN-κ, and IFN-ω. Many different ICs can activate pDCs, but as discussed above RNA-containing ICs seems to be the best IFN-α inducing stimulus, owing to the simultaneous binding of Fcγ and intracytosolic TLRs. Type I IFN family presents many immunological functions, such as promotion of B cells’ differentiation, immunoglobulin class switch, production of autoantibodies and increase of activated B and T cells survival.
In healthy individuals, IFN-I released from pDCs is usually triggered by viral infection, but under such circumstance IFN-I production is only temporary, strictly regulated and limited in time with resolution of the viral infection. This is not the case of SLE, since IFN release under such condition is independent from the infectious stimulus and driven mainly by nuclear debris recognized as autoantigens. Direct estimate of circulating IFN level is usually very complicated and not completely reliable; however, recent studies have shown a good correlation between expression of IFN-inducible genes in peripheral blood mononuclear cells, using a microarray device, and SLE activity [51]. Such a hyperactivation of IFN-genes, more commonly known as “interferon signature”, in SLE patients has not only been demonstrated on the circulating mononuclear cells, but also locally on the glomerular, synovial and cutaneous tissues, suggesting a key pathogenetic role of IFN type I family in such disease [52]. Interestingly, interferon pathway has been shown to induce and drive SLE inflammation in several, but not in all, murine experimental models, suggesting different SLE pathogenetic subsets [53–56]. Nevertheless, lupus-like syndrome is a very well-known complication of recombinant IFN-α therapy administered for chronic hepatitis or cancer treatment [57, 58].
Although neutrophils have long been considered to be associated with lupus, their potential role in disease pathogenesis has only been recently more deeply investigated [59]. Neutrophils, the most abundant leukocyte in humans, are typically recruited to sites of infection during the early phases of inflammatory responses and are considered the primary cellular defense against bacterial and fungal infections. They are able to kill pathogens using phagocytosis, producing reactive oxygen species or releasing preformed cytotoxic molecules from cytoplasmatic granules into the extracellular compartment. Very recently, another killing mechanism has been discovered, during which neutrophil extracellular traps (NETs), meshworks structures consisting of chromatin and peptides with antimicrobial activity, are released from dying cells [60]. This specific form of neutrophils PCD has been so-called “NETosis” and has been involved in autoimmune diseases pathogenesis [61].
Neutrophil-derived structures, containing a significant amount of DNA and ribonucleoproteins, are potentially able to stimulate pDC to produce interferon alfa, principally through interaction with TLR9 [62]. In about one-third of SLE patients, an abnormal NET accumulation has recently been demonstrated, due to low DNase (deoxyribonuclease) I activity, the main enzyme responsible of NET clearance in humans, in these “non degraders” patients. This is particularly important since the non-degrader phenotype carries a more than 70 % risk of lupus nephritis compared to just 25 % of normal degrader SLE patients [63].
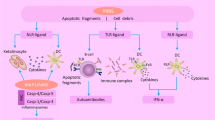
Further studies will be required for a better comprehension of IFN-I and DC role in SLE pathogenesis but, to date, they seem to play a key role at the interface between innate and adaptive immunity, as reported in Fig. 1.

Both myeloid DCs (mDCs) and plasmocytoid DCs (pDCs) produce IFN-α in response to self-nucleic acids antigens and self-nucleic acids in the form of immune complexes, respectively. IFN-α presents many immunological functions, including promotion of B cells differentiation, immunoglobulin switch, autoantibodies’ production and increased survival of activated B and T lymphocytes. IFN-α also activates mDCs and contribute to direct endothelial cell damage and promotes accelerated atherosclerosis (mDC myeloid dendritic cells, pDC plasmacytoid dendritic cells, Lymph B B lymphocytes, CTL cytotoxic T lymphocytes, IFN interferon
Adaptive immunity
Under physiological conditions, T cells undergo activation only when mature DCs present self-antigen in MHC-restricted conditions. In SLE patients, T cells hyperactivity has been described, since the engagement of T cell receptor (TCR) with MHC, in conjunction with costimulatory signals, leads to a vigorous intracytoplasmic calcium flux and produce an aberrant downstream cellular activation. CD3ζ chain is a crucial component of the TCR complex, playing an important role in determining the intracellular signal transduction pathway [64, 65].
CD4+ T helper cells are classically distinct in Th1 and Th2, according to cytokine production and to their functions (allergic reaction for Th2, defense against infection for Th1). IL-12 stimulates the differentiation of naïve CD4+ T cells into Th1 cells, which in turn produce IFN-γ. On the other hand, IL-4, IL-5 and IL-13, cytokines mainly produced by Th2 lymphocytes, are involved in many functions of T and B cells including proliferation, activation and isotype switching.
Th17 cells, involved in pathogenesis of several autoimmune diseases such as rheumatoid arthritis or inflammatory bowel diseases, are a subset of T helper lymphocytes producing the cytokines of IL-17 family. Recently, a crucial role has also been demonstrated for Th17 lymphocytes in SLE [66, 67]. Th17 cells indeed have been found in glomerular tissue from patient with active lupus nephritis. Increased levels of IL-6 and IL-21, both influencing Th-17 differentiation and response, have also been demonstrated in SLE [68, 69]. Recently, Savino et al. have shown, both in mice and humans, a possible role of the SHC adapter family member, Rai. Indeed Rai(−/−) mice develop a lupus-like phenotype with a spontaneous activation of self-reactive lymphocytes; moreover, it has been demonstrated that Rai(−/−) mice present Th1 and Th17 cell infiltrates in the kidneys, suggesting that Rai knockout mice (−/−) is more susceptible to lupus nephritis. Finally, T cells derived from SLE patients demonstrated a defect in Rai expression, suggesting a possible role of this adapter protein as an immunomodulator/immunosoppressor in SLE pathogenesis [70].
Several studies focused on regulatory T cells (Treg) as potential players of the break down of immune tolerance, since both quantitative and qualitative abnormalities of peripheral regulatory T lymphocytes (CD4+ CD25+ high) were demonstrated in SLE patients [71, 72]. The deficiency of regulatory activity in SLE can be explained partially both by decreased production of IL-2, the main cytokine driving development and survival of T reg, and by the contemporary increased levels of IL-6, a circulating factor profoundly influencing the differentiation of Th17, whose functions usually counteract T reg functions [73–75]. Further studies, however, are needed to better understand the role of Treg in SLE pathogenesis [76, 77].
B lymphocytes are new player of adaptive immunity in SLE pathogenesis. They were classically considered fundamental for the production of a broad array of autoantibodies against soluble and cellular component, such as nuclear antigens, typical of SLE patients. Moreover, B lymphocytes present efficiently autoantigens and activate T cells, so their action is not only limited to production of antibodies [78, 79]. Similar to T cells, hyperactivation has recently also been described for B lymphocytes, since increased phosphorylation of several signaling molecules and abnormal calcium flux have been reported in B cells from SLE patients [80, 81].
B lymphocyte can be classified into at least two lineages: B1 and B2 cells. B1 lymphocytes, which have been mainly studied in mice, are considered self-renewing and long-lived cells and are specialized in producing polyreactive ‘natural’ IgM class antibodies for immediate defense; they are also thought to clear apoptotic material and debris, linking innate and adaptive immunity together. B2 cells are generated in the bone marrow, where autoreactive cells are first removed (central tolerance), and then undergo further selection in the spleen microenvironment (peripheral tolerance). After this selection phase, B2 cells either become mature follicular cells, that migrate throughout the secondary lymphoid organs waiting for T cell-dependent activation, or marginal zone (MZ) B2 cells, which, like B1 cells, are able to respond immediately to pathogens independently of T cell help.
Although the role of MZ B cells in autoimmunity and in lupus is still debating, they are probably involved in some autoimmune diseases, such as autoimmune thrombocytopenia, for which splenectomy is beneficial [82–84].
The number of MZB cells has been demonstrated to be increased in the NZB/WF1 mice model of SLE; moreover, IFN is a potent driver of their activation and an efficient enhancer of costimulatory molecules’ expression, making MZB cells an important player in SLE in particular for autoantibody production [85, 86]. Finally IgD+ CD27+ memory B cells, which include a particular transient differentiation stage of memory B cell and circulating MZ B cells, are fundamental for determining clinical outcome in RA and SLE after B depletive therapy, since a delayed repopulation after such a treatment is associated with a better clinical response [87, 88].
Very recently, a new subset of B lymphocytes has been described, endowed with immunosuppressive activity and so referred as regulatory B cells or simply as “B reg”. The main function of regulatory B cells is the production of IL-10, so that the intracellular staining for this cytokine is currently the main method for identifying these cells, even if the recently described B population CD24highCD27++ probably include the large proportion of human Breg [89, 90]. The immunosuppressive properties of IL-10 are well established in animal models of collagen-induced arthritis and experimental autoimmune encephalitis; however, the function of IL-10 in SLE has been very controversial [91, 92]. Recent data seem to indicate a role of IL-10 producing B reg in SLE too, particularly during active disease to control the inflammatory response and restore immune tolerance [93]. Interestingly, after depletion of B cells using anti-CD20 treatment (Rituximab), the following repopulation phase is probably constituted mainly of regulatory B cells and this is particularly true for patients who achieved a good clinical response. However, further data are necessary to better clarify the role of Breg cells and IL-10 in SLE pathogenesis [94, 95].
As previously stated, an elevated level of cytokines that affect B cell activation and proliferation has been described in SLE [96]. Increased levels of B lymphocyte stimulator (BAFF/BLyS) and A proliferation-inducing ligand (APRIL), promoting autoreactive lymphocytes survival and autoantibody responses, were also abundantly described in human and mice SLE models, indeed both these factors are target of recently developed drugs for lupus [97, 98]. Interestingly, memory B lymphocytes are independent from BLyS; survival of mature B cells is promoted both by BLyS and APRIL; plasma cells survival is essentially stimulated by BLyS. SLE activity correlates with BLyS mRNA expression in leukocytes and recently BLyS has emerged as a new therapeutic target in SLE treatment [99, 100].
Figures 2 and 3 illustrate the complex interplay between T and B lymphocytes, underlining the role of different pathogenetic pathways. The deregulation of the adaptive immunity results in a wide broad of effects. Autoantibody production, as well known, represents one of the most important mechanisms contributing to tissue damage.

A As known a breakdown in the mechanisms that control the central and/or peripheral tolerance may lead to an expansion and differentiation of autoreactive T cells in turn able to activate B autoreactive lymphocytes. B B cells may be activated classically by T lymphocytes or alternatively by direct link between immunocomplexes containing self-DNA/RNA and TLRs. Moreover, BAFF/Blys has broad potential implications in SLE pathogenesis, since it influences peripheral B cell survival, maturation and immunoglobulin class switch. Moreover, BAFF/Blys is upregulated by IFN-γ, IL-10 and CD40 ligand produced during inflammatory conditions (TLRs Toll-like receptors, BAFF B cell activating factor, Blys B lymphocyte stimulator)

A defective ICs’ clearance due to complement alteration and increased apoptosis (i.e., UV light induced) lead to the formation of IC consisting of self-DNA/RNA rich in CpG motifs, hypomethylated, oxidized. These ICs may bind other molecules such as HMGB1 (released from dead cells) or the antimicrobial peptide LL37-forming structures capable of inducing pDCs’ activation through different pathways (i.e., RAGE, FcγRIIA and/or TLRs). Of note in SLE patients, IFN-α activity is increased, since IFN-alfa genetic signature can be detecting in circulating monocytes using a microarray technique. (ICs immune complexes, FcγRIIA low affinity receptor for IgG, BCR B cell receptor, HMGB1 high mobility group box 1, RAGE receptor for advanced glycation end products, LL37 antimicrobial peptide or cathelicidin)
New therapeutic perspectives: biological agents for SLE
Until recently, few biological agents have been used on a limited number of SLE patients often with no large controlled-randomized controlled trials supporting efficacy for such therapies. The lack of new drugs or biological agents, if compared to other autoimmune disease such as rheumatoid arthritis or inflammatory bowel diseases, accounts for the current broad use of traditional immunosoppressor drugs, in particular of cyclophosphamide and mycophenolate, for severe SLE cases and for the broad number of drug side effects experienced by SLE patients during their lifetime. New data coming from research studies on different inflammatory pathways and cellular interplay recently produced essential information on new targets for developing biological drugs in SLE. As summarized in Fig. 3, the main targets of new SLE therapies are represented by neutralization of autoreactive B cells, induction of tolerance, inhibition of costimulatory signals and modulation of cytokines pathways. Due to the recent light on SLE pathogenesis, B cells in particular represent a major therapeutic target of new biological agents. Current and ongoing therapeutic approaches targeting B cells include direct depletion of B cells and inhibition of specific B cell-stimulating cytokines [101, 102].
Rituximab is a chimeric monoclonal antibody (mAb) against B lymphocytes CD20 antigen, already approved for lymphoma and rheumatoid arthritis that induces profoundly circulating B cells’ depletion soon after administration. Rituximab has been regarded as a promising agent for active SLE refractory to traditional immunosuppressive drugs. In murine models, rituximab worked depleting B cells from the pre-B stage to the mature lymphocyte and memory B cells. Unlike mice, in humans, rituximab targets a more narrow spectrum of B lymphocytes, ranging pre-B–mature B cells. Plasma cells or memory B cells are indeed not affected by rituximab therapy in humans, since CD20 is not anymore expressed on membrane surface of these cells. This also explains why circulating antibodies’ level is usually preserved during rituximab treatment, unless repeated cycles of the drug are used [103, 104]. Clinical experience and several open case series suggested a clinical improvement and a consistent reduction of SLE activity disease index (SLEDAI, BILAG) when this biological agent was used for SLE patients [105]. However, two double-blind placebo-controlled trials (EXPLORER and LUNAR) subsequently failed to meet the primary endpoints in systemic and renal SLE, respectively [106, 107]. Despite the normalization of anti-dsDNA and complement levels in the treated group, clinical outcomes did not differ significantly between rituximab and placebo group. Further investigation using different study designs could probably be useful to clarify the discrepancy between the reported effectiveness of rituximab for SLE, both in several open case series and generally in clinical practice, with the failure to demonstrate any efficacy of the drug in larger randomized placebo-controlled trials. Important reasons limiting the potential rituximab benefit in the trials setting could be represented by the aggressive background immunosuppressive therapy, the influence of corticosteroids used during the study and the too much limited sample size of enrolled patients. Such considerations should be kept in mind for further clinical trials with any investigating agents for SLE [108, 109].
Epratuzumab is a humanized monoclonal antibody against CD22 antigen, a surface molecule associated to B cell receptor and endowed with downregulation function. It also induces reduction of circulating B cells, even if with a less profound extent comparing to rituximab, since only the latter is able to induce antibody-dependent cellular cytotoxicity on target cells. Unlike rituximab, a slight reduction of immunoglobulin levels is usually observed early during treatment. A significant benefit for SLE was observed during a phase-IIb trial, which employed a new combined clinical index for evaluating responses. A phase-III trial on epratuzumab is currently ongoing to confirm these initial positive results [110].
Belimumab, a fully human IgG1λ monoclonal antibody against BLyS, decreases B cells and level of autoantibodies. BAFF/BLyS pathway plays a key role in the survival and proliferation of autoreactive B cells [111]. Even if a phase-II trial was initially not able to reach its primary endpoints, a post hoc analysis of the data was able to demonstrate a significant greater response to belimumab in a subgroup of patients with low complement levels and anti-DNA positivity [112]. Subsequently, two randomized, double-blind, placebo-controlled phase-III trials (BLISS-52 and BLISS-76) were performed, using a new index of clinical response (SLE responder index or SRI). The BLISS-52 trial, largely conducted in Asia, South America and Eastern Europe, demonstrated a good response rate at 52 weeks of treatment. SRI rate of responders was 51 % with 1 mg/kg belimumab dose and 58 % with 10 mg/kg, in comparison with 44 % in the placebo group [113]. BLISS-76 trial, which conversely involved US, Western Europe and Canada, demonstrated a 41 % SRI rate of responders at 1 mg/kg belimumab dose and 43 % at 10 mg/kg, in comparison with 34 % in the placebo group [114]. In brief, phase-III belimumab trials demonstrated the efficacy of belimumab in addition to standard therapy compared to placebo. Belimumab is the only drug currently approved by the US Food and Drug Administration and recently also by the European Medicines Agency as the first biologic agent for SLE patients. Several questions, such as the usefulness of a long-term treatment and the effectiveness of belimumab in any SLE subset, remain still open. Recent evidences, however, confirmed the safety of belimumab treatment over 7 years [115].
In addition to belimumab, other agents target the BLyS/BAFF pathway. Atacicept, a fusion protein between TACI and the Fc portion of IgG, binds both BLyS and APRIL. Despite the favorable safety profile demonstrated in preclinical and phase-I studies, a clinical phase-II/III trial in lupus nephritis was early interrupted because of severe reduction of B cell number and serum immunoglobulin levels, which considerably increased severe infections risk. A new trial, however, is actually ongoing to confirm the safety and efficacy profile of atacicept [116].
Costimulatory signals, in particular CD40–CD40L interaction, are crucial for cognate interaction since T cells are able to stimulate B cells and innate immunity cells when CD40L (also called CD154) is expressed on their surface [117, 118]. Despite the effectiveness in a mouse model, the anti-CD40L monoclonal antibody trials in humans were early interrupted because of lack of efficacy compared to placebo and more importantly because of unexpected elevated incidence of thrombosis in the treated arm [119, 120]. Despite the negative results, an increasing amount of data supports the importance of CD40–CD40L interaction in SLE and prompts the interest in further exploratory trials with alternative biological agents targeting this pathway [121].
Abatacept, a fusion protein between the extracellular domain of CTLA-4 and the Fc of IgG1, binds to B7-1 and B7-2 receptors expressed on antigen-presenting cells such as B lymphocytes and “professional” APC. A distinguish feature of CTLA4 is its higher affinity for B7 receptors, if compared to CD28 expressed on T cells, since its main function is to downregulate costimulatory signals and limit immune response. Positive results were initially obtained from mouse models of SLE and other autoimmune diseases [122]. For this reasons abatacept was recently evaluated in SLE with a phase-II randomized, placebo-controlled trial [123]. Despite a reduction in disease flares, particularly in patient with articular involvement, no differences were observed between abatacept and control group regarding the main endpoints. As mentioned for rituximab, the use of different response indexes may probably reveal a usefulness of abatacept in SLE [124, 125].
TNF-α is a pleiotropic cytokine, produced mainly by macrophages, and involved in many inflammatory pathways, with broad stimulatory effects on B and T cells. TNF-α levels are increased in SLE patients, often in association with disease activity parameters [126, 127]. Anti-TNF-α agents are the most prescribed biological drugs used to treat many autoimmune disorders, particularly rheumatoid arthritis, spondyloarthritis and inflammatory bowel diseases. Despite the presumed TNF-α involvement in SLE pathogenesis, monoclonal antibodies against TNF-α have not generally used for SLE therapy, except in limited case series [128, 129]. It is well known from the literature that TNF blockage can induce autoantibody production and, more rarely, overt drug-induced lupus-like syndromes, so the use of anti-TNF agents is not generally accepted or recommended for treating lupus patients [130, 131].
Tocilizumab, a humanized IgG1 monoclonal antibody against IL-6 receptor, inhibits the IL-6 pathway that, as mentioned above, is involved in the development of inflammation and in B cell activation [132]. The experience with tocilizumab for SLE is nowadays limited to sporadic but significant case reports from refractory patients. Based on the growing evidence suggesting a pathogenetic role of IL-6, a key cytokine stimulating Th17 differentiation and downregulating T reg cells, the blockage of IL6 receptor using tocilizumab is considered a very promising therapeutic option also in SLE, in particular for patients refractory to conventional therapy [133–135]. Interestingly, a lupus nephritis double-blind phase-II RCT with sirukumab, an antibody targeting directly IL-6 rather than IL-6R, is currently on investigation after the drug showed no toxicity in a previous phase-I study [136].
As described above, IFN-α production is mainly the result of pDCs’ activation by intracytosolic TLRs binding with IC containing nuclear material. The subsequent inflammatory response is therefore responsible of several steps of SLE pathogenesis, such as T and B lymphocytes’ proliferation. Sifalimumab and rontalizumab, monoclonal antibodies against IFN-α, are currently evaluated in phase-II clinical trials [137, 138]. Probably the routine evaluation of IFN-genes’ overexpression in peripheral monocytes, using the so-called “IFN-gene signature”, will be useful in the future to identify the subgroup of SLE patients who can benefit the most from anti-IFN agents. However, it is not still clear if blocking selectively IFN-alfa will be sufficient to reverse the inflammatory response and the IFN signature, considering the many and redundant cytokines belonging to IFN type I family [139, 140].
Conclusions
Recent advances in our understanding of SLE pathogenesis have pointed out new targets for treatment but several fundamental questions remain unsolved. As for pathogenesis, clinical aspects and therapy, SLE remains a very complex disease, that requires very skilled and highly trained physicians for a correct clinical evaluation and diagnosis, for deciding the most suitable therapy in every single patients and for conducting properly clinical trials. Indeed different SLE subsets of patients present with different pathogenetic and clinical profiles, requiring diverse and rather individualized therapeutic approaches, to obtain the best clinical outcome. Belimumab is the only biological drug approved for SLE; however, other promising agents are currently under evaluation with clinical phase-II/III trials.
References
Rahman A, Isemberg DA (2008) Systemic lupus erythematosus. N Engl J Med 358:929–939
Jimenez S, Cervera R, Font J, Ingelmo M (2003) The epidemiology of systemic lupus erythematosus. Clin Rev Allergy Immunol 25:3–12
Borchers AT, Naguwa SM, Shoenfeld Y, Gershwin ME (2010) The geoepidemiology of systemic lupus erythematosus. Autoimmun Rev 9:277–287
Pons-Estel GJ, Alarcón GS, Sconfield L, Reinlib L, Cooper GS (2010) Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum 39(4):257–268
Cervera R, Khamashta MA, Font J, Sebastiani GD (1999) Morbidity and mortality in systemic lupus erythematosus during a 5-year period. A multicenter prospective study of 1,000 patients. European Working Party on Systemic Lupus Erythematosus. Medicine (Baltimore) 78:167–175
Gualtierotti R, Biggioggero M, Penatti AE, Meroni PL (2010) Updating on the pathogenesis of systemic lupus erythematosus. Autoimmun Rev 10:3–7
Janko C, Schorn C, Grossmayer GE, Frey B, Herrmann M, Gaipl US, Munoz LE (2008) Inflammatory clearance of apoptotic remnants in systemic lupus erythematosus (SLE). Autoimmun Rev 8(1):9–12
Shrivastav M, Niewold TB (2013) Nucleic acid sensors and type I interferon production in systemic lupus erythematosus. Front Immunol 7(4):319
Flesher DL, Sun X, Behrens TW, Graham RR, Criswell LA (2010) Recent advances in the genetics of systemic lupus erythematosus. Expert Rev Clin Immunol 6:461–479
Crow MK (2008) Collaboration, genetic associations and lupus erythematosus. N Eng J Med 358:956–961
Bowness P, Davies KA, Norsworthy PJ, Athanassiou P, Taylor-Wiedeman J, Borysiewicz LK, Meyer PA, Walport MJ (1994) Hereditary C1q deficiency and systemic lupus erythematosus. QJM 87(8):455–464
Monticielo OA, Mucenic T, Machado Xavier R, Tavares Brenol JC, Bogo Chies JA (2008) The role of mannose-binding lectin in systemic lupus erythematosus. Clin Rheumatol 27:413–419
Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ et al (2003) Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA 100(5):2610–2615
Kirou KA, Lee C, George S, Louca K, Papagiannis IG, Peterson MG et al (2004) Coordinate overexpression of interferon-alpha-induced genes in systemic lupus erythematosus. Arthritis Rheum 50(12):3958–3967
Rönnblom L, Alm GV, Eloranta ML (2011) The type I interferon system in the development of lupus. Semin Immunol 23:113–121
Deng Y, Tsao BP (2010) Genetic susceptibility to systemic lupus erythematosus in the genomic era. Nat Rev Rheumatol 6(12):683–92.10
Zhao S, Long H, Qiaanjin L (2010) Epigenetic perspectives in systemic lupus erythematosus: pathogenesis, biomarkers and therapeutic potentials. Clin Rev Allergy Immunol 39:3–9
Wen ZK, Xu W, Xu L, Cao QH, Wang Y, Chu YW, Xiong SD (2007) DNA hypomethylation is crucial for apoptotic DNA to induce systemic lupus erythematosus-like autoimmune disease in SLE-non-susceptible mice. Rheumatology (Oxford) 46:1796–1803
Ballestar E, Esteller M, Richardson BC (2006) The epigenetic face of systemic lupus erythematosus. J Immunol 176:7143–7147
Rigby RJ, Vinuesa CG (2008) Silencing SLE: the power and promise of small noncoding RNAs. Curr Opin Rheumatol 20:526–531
Stagakis E, Bertsias G, Verginis P, Nakou M, Hatziapostolou M, Kritikos H, Iliopoulos D, Boumpas DT (2011) Identification of novel microRNA signatures linked to human lupus disease activity and pathogenesis: mir-21 regulates aberrant T-cells response trough the regulation of PDCD4 expression. Ann Rheum Dis 70:1496–1506
Weckerle CE, Niewold TB (201) The unexplained female predominance of systemic lupus erythematosus: clues from genetic and cytokine studies. Clin Rev Allergy Immunol 40:42–49
González DA, Díaz BB, Rodríguez Pérez Mdel C, Hernández AG, Chico BN, De León AC (2010) Sex hormones and autoimmunity. Immunol Lett 133:6–13
Kassi E, Moutsatsou P (2010) Estrogen receptor signaling and its relationship to cytokines in systemic lupus erythematosus. J Biomed Biotechnol 2010:317–452
Wang J, Nuite M, McAlindon TE (2010) Association of estrogen and aromatase gene polymorphisms with systemic lupus erythematosus. Lupus 19(6):734–740
Shoenfeld Y, Tincani A, Gershwin ME (2012) Sex gender and autoimmunity. J Autoimmun 38(2–3):J71–J73
Hudson CA, Cao L, Kasten-Jolly J, Kirkwood JN, Lawrence DA (2003) Susceptibility of lupus-prone NZM mouse strains to lead exacerbation of systemic lupus erythematosus symptoms. J Toxicol Environ Health A 66(10):895–918
Hughes GC, Clark EA (2007) Regulation of dendritic cells by female sex steroids: relevance to immunity and autoimmunity. Autoimmunity 40(6):470–481
Tayel SS, Helmy AA, Ahmed R, Esmat G, Hamdi N, Abdelaziz AI (2013) Progesterone suppresses interferon signaling by repressing TLR-7 and MxA expression in peripheral blood mononuclear cells of patients infected with hepatitis C virus. Arch Virol 158(8):1755–1764
Shelly S, Boaz M, Orbach H (2012) Prolactin and autoimmunity. Autoimmun Rev 11(6–7):A465–A467
Doria A, Canova M, Tonon M, Zen M, Rampudda E, Bassi N, Atzeni F, Zampieri S, Ghirardello A (2008) Infections as trigger and complications of systemic lupus erythematosus. Autoimmun Rev 8:24–28
Zandman-Goddard G, Schoenfeld Y (2005) Infections and SLE. Autoimmunity 38:473–485
Poole BD, Scofield RH, Harley JB, James JA (2006) Epstein–Barr virus and molecular mimicry in systemic lupus erythematosus. Autoimmunity 39:63–70
James J, Harley JB, Scofield RH (2006) Role of Epstein–Barr virus in systemic lupus erythematosus. Curr Opin Rheumatol 18:462–467
Werth VP (2007) Cutaneous lupus: insight into pathogenesis and disease classification. Bull NYU Hosp Jt Dis 65:200–204
Kuhn A, Wenzel J, Weyd H (2014) Photosensitivity, apoptosis, and cytokines in the pathogenesis of lupus erythematosus: a critical review. Clin Rev Allergy Immunol. (Epub ahead of print)
Chang C, Gershwin ME (2010) Drugs and autoimmunity—a contemporary review and mechanistic approach. J Autoimmun 34(3):J266–J275
Muñoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M (2010) The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol 6:280–289
Ravichandran KS, Lorenz U (2007) Engulfment of apoptotic cells: signals for a good meal. Nat Rev Immunol 7:964–974
Mevorach D, Trahtemberg U, Krispin A, Attalah M, Zazoun J, Tabib A, Grau A, Verbovetski-Reiner I (2010) What do we mean when we write “senescence,” “apoptosis,” “necrosis,” or “clearance of dying cells”? Ann N Y Acad Sci 1209:1–9
Poon IK, Lucas CD, Rossi AG, Ravichandran KS (2014) Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol 14(3):166–180
Pisetsky DS, Erlandsson-Harris H, Andersson U (2008) High-mobility group box protein 1 (HMGB1): an alarmin mediating the pathogenesis of rheumatic disease. Arthritis Res Ther 10:209
Ma CY, Jiao YL, Zhang J, Yang QR, Zhang ZF, Shen YJ, Chen ZJ, Zhao YR (2012) Elevated plasma level of HMGB1 is associated with disease activity and combined alterations with IFN-alpha and TNF-alpha in systemic lupus erythematosus. Rheumatol Int 32:395–402
Hurst J, Von Landenberg P (2008) Toll-like receptors and autoimmunity. Autoimmun Rev 7:204–208
Marshak-Rothstein A (2006) Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol 6:823–835
Theofilopoulos AN (2012) TLRs and IFNs: critical pieces of the autoimmunity puzzle. J Clin Invest 122(10):3464–3466
Pisetsky DS, Ullal AJ (2010) The blood nucleome in the pathogenesis of SLE. Autoimmun Rev 10:35–37
Kontaki E, Boumpas DT (2010) Innate immunity in systemic lupus erythematosus: sensing endogenous nucleic acids. J Autoimmun 35:206–211
Fransen JH, van der Vlag J, Ruben J, Adema GJ, Berden JH, Hilbrands LB (2010) The role of dendritic cells in the pathogenesis of systemic lupus erythematosus. Arthritis Res Ther 12:207
Crispín JC, Liossis SN, Kis-Toth K, Lieberman LA, Kyttaris VC, Juang YT, Tsokos GC (2010) Pathogenesis of human systemic lupus erythematosus: recent advances. Trends Mol Med 16:47–57
Kirou KA, Lee C, George S, Louca K, Peterson MG, Crow MK (2005) Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum 52(5):1491–1503
Nzeusseu Toukap A, Galant C, Theate I, Maudoux AL, Lories RJ, Houssiau FA, Lauwerys BR (2007) Identification of distinct gene expression profiles in the synovium of patients with systemic lupus erythematosus. Arthritis Rheum 56(5):1579–1588
Liu Z, Davidson A (2013) IFNα inducible models of murine SLE. Front Immunol 2(4):306
Koss MN, Stohl W, Kovats S, Jacob CO (2009) Deficiency of type I IFN receptor in lupus-prone New Zealand mixed 2328 mice decreases dendritic cell numbers and activation and protects from disease. J Immunol 183(9):6021–6029
Agrawal H, Jacob N, Carreras E, Bajana S, Putterman C, Turner S, Neas B, Mathian A, Weinberg A, Gallegos M, Banchereau J, Koutouzov S (2005) IFN-α induces early lethal lupus in preautoimmune (New Zealand Black × New Zealand White) F1 but not in BALB/c mice. J. Immunol 174:2499–2506
Fairhurst AM et al (2008) Systemic IFN-α drives kidney nephritis in B6.SLE123 mice. Eur J Immunol 38:1948–1960
Wilson LE, Widman D, Dikman SH, Gorevic PD (2002) Autoimmune disease complicating antiviral therapy for hepatitis C virus infection. Semin Arthritis Rheum 32(3):163–173
Rönnblom LE, Alm GV, Öberg KE (1990) Possible induction of systemic lupus erythematosus by interferon-α treatment in a patient with a malignant carcinoid tumor. J Intern Med 227:207–210
Hargraves MM, Richmond H, Morton R (1948) Presentation of two bone marrow elements; the tart cell and the LE cell. Proc Staff Meet Mayo Clin 23:25–28
Branzk N, Papayannopoulos V (2013) Molecular mechanisms regulating NETosis in infection and disease. Semin Immunopathol 35(4):513–530
Knight JS, Kaplan MJ (2012) Lupus neutrophils: ‘NET’ gain in understanding lupus pathogenesis. Curr Opin Rheumatol 24:441–450
Garcia-Romo GS et al (2011) Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med 3:73ra20
Hakkim A, Furnrohr BG, Amann K et al (2010) Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA 107:9813–9818
Scheinecker C, Bonelli M, Smolen JS (2010) Pathogenetic aspects of systemic lupus erythematosus with an emphasis on regulatory T cells. J Autoimmun 35:269–275
Peng SL (2009) Altered T and B lymphocyte signaling pathways in lupus. Autoimmun Rev 8:179–183
Truchetet ME, Mossalayi MD, Boniface K (2013) IL-17 in the rheumatologist’s line of sight. Biomed Res Int 2013:295132
Nalbandian A, Crispín JC, Tsokos GC (2009) Interleukin-17 and systemic lupus erythematosus: current concepts. Clin Exp Immunol 157(2):209–215
Chen DY, Chen YM, Wen MC, Hsieh TY, Hung WT, Lan JL (2012) The potential role of Th17 cells and Th17-related cytokines in the pathogenesis of lupus nephritis. Lupus 21(13):1385–1396
Ohl K, Tenbrock K (2011) Inflammatory cytokines in systemic lupus erythematosus. J Biomed Biotechnol 2011, Art id 432595
Savino MT, Ulivieri C, Emmi G, Prisco D, De Falco G, Ortensi B, Beccastrini E, Emmi L, Pelicci G, D’Elios MM, Baldari CT (2013) The Shc family protein adaptor, Rai, acts as a negative regulator of Th17 and Th1 cell development. J Leukoc Biol 93(4):549–559
Valencia X, Yarboro C, Illei G, Lipsky PE (2007) Deficient CD4+ CD25high T regulatory cell function in patients with active systemic lupus erythematosus. J. Immunol. 178:2579–2588
Bonelli M et al (2008) Quantitative and qualitative deficiencies of regulatory T cells in patients with systemic lupus erythematosus (SLE). Int Immunol 20:861–868
Yu A, Zhu L, Altman NH, Malek TR (2009) A low interleukin-2 receptor signaling threshold supports the development and homeostasis of T regulatory cells. Immunity 30:204–217
Wan S, Xia C, Morel L (2007) IL-6 produced by dendritic cells from lupus-prone mice inhibits CD4+ CD25+ T cell regulatory functions. J. Immunol 178:271–279
Chen X, Oppenheim JJ (2014) Th17 cells and Tregs: unlikely allies. J Leukoc Biol
Gerli R, Nocentini G, Alunno A, Bocci EB, Bianchini R, Bistoni O, Riccardi C (2009) Identification of regulatory T cells in systemic lupus erythematosus. Autoimmun Rev 8:426–430
Kuhn A, Beissert S, Krammer PH (2009) CD4(+)CD25 (+) regulatory T cells in human lupus erythematosus. Arch Dermatol Res 301:71–81
Jenks SA, Sanz I (2009) Altered B cell receptor signaling in human systemic lupus erythematosus. Autoimmun Rev 8(3):209–213
Grammer AC, Lipsky PE (2003) B cell abnormalities in systemic lupus erythematosus. Arthritis Res Ther 5:S22–S27
Liossis SN, Kovacs B, Dennis G, Kammer GM, Tsokos GC (1996) B cells from patients with systemic lupus erythematosus display abnormal antigen receptor-mediated early signal transduction events. J Clin Invest 98(11):2549–2557
Liu K, Mohan C (2009) Altered B-cell signaling in lupus. Autoimmun Rev 8:214–218
Lopes-Carvalho T, Kearney JF (2005) Marginal zone B cell physiology and disease. Curr Dir Autoimmun 8:91–123
Martinez-Gamboa L, Mei H, Loddenkemper C, Ballmer B, Hansen A, Lipsky PE, Emmerich F, Radbruch A, Salama A, Dörner T (2009) Role of the spleen in peripheral memory B-cell homeostasis in patients with autoimmune thrombocytopenia purpura. Clin Immunol 130(2):199–212
Sang A, Zheng YY, Morel L (2013) Contributions of B cells to lupus pathogenesis. Mol Immunol [Epub ahead of print]
Wither JE, Roy V, Brennan LA (2000) Activated B cells express increased levels of costimulatory molecules in young autoimmune NZB and (NZB × NZW)F(1) mice. Clin Immunol 94(1):51–63
Wang JH, Wu Q, Yang P, Li H, Li J, Mountz JD, Hsu HC (2011) Type I interferon-dependent CD86(high) marginal zone precursor B cells are potent T cell costimulators in mice. Arthritis Rheum 63(4):1054–1064
Weller S, Braun MC, Tan BK, Rosenwald A, Cordier C, Conley ME, Plebani A, Kumararatne DS, Bonnet D, Tournilhac O, Tchernia G, Steiniger B, Staudt LM, Casanova JL, Reynaud CA, Weill JC (2004) Human blood IgM “memory” B cells are circulating splenic marginal zone B cells harboring a prediversified immunoglobulin repertoire. Blood 104(12):3647–3654
Anolik JH et al (2007) Delayed memory B cell recovery in peripheral blood and lymphoid tissue in systemic lupus erythematosus after B cell depletion therapy. Arthritis Rheum 56:3044–3056
Goode I, Xu H, Ildstad ST (2014) Regulatory B cells: the new “it” cell. Transplant Proc 46(1):3–8
Iwata Y, Matsushita T, Horikawa M et al (2011) Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood 117:530–541
Matsushita T, Horikawa M, Iwata Y, Tedder TF (2010) Regulatory B cells (B10 cells) and regulatory T cells have independent roles in controlling experimental autoimmune encephalomyelitis initiation and late-phase immunopathogenesis. J Immunol 185:2240–2252
Carter NA, Rosser EC, Mauri C (2012) Interleukin-10 produced by B cells is crucial for the suppression of Th17/Th1 responses, induction of T regulatory type 1 cells and reduction of collagen-induced arthritis. Arthritis Res Ther 14(1):R32
Yang X, Yang J, Chu Y, Xue Y, Xuan D, Zheng S, Zou H (2014) T follicular helper cells and regulatory B cells dynamics in systemic lupus erythematosus. PLoS One 9(2):e88441
Sanz I, Lee FE (2010) B cells as therapeutic targets in SLE. Nat Rev Rheumatol 6(6):326–337
Manjarrez-Orduño N, Quách TD, Sanz I (2009) B cells and immunological tolerance. J Invest Dermatol 129(2):278–288
Jacob N, Stohl W (2011) Cytokine disturbances in systemic lupus erythematosus. Arthritis Res Ther 13:228
Ding H, Wang L, Wu X, Yan J, He Y, Ni B et al (2010) Blockade of B-cell-activating factor suppresses lupus-like syndrome in autoimmune BXSB mice. J Cell Mol Med 14:1717–1725
Chu VT, Enghard P, Schürer S, Steinhauser G, Rudolph B, Riemekasten G (2009) Berek C Systemic activation of the immune system induces aberrant BAFF and APRIL expression in B cells in patients with systemic lupus erythematosus. Arthritis Rheum 60:2083–2093
Vincent FB, Saulep-Easton D, Figgett WA, Fairfax KA, Mackay F (2013) The BAFF/APRIL system: emerging functions beyond B cell biology and autoimmunity. Cytokine Growth Factor Rev 24(3):203–215
Yildirim-Toruner C, Diamond B (2011) Current and novel therapeutics in the treatment of systemic lupus erythematosus. J Allergy Clin Immunol 127:303–312
Vincent FB, Morand EF, Schneider P, Mackay F (2014) The BAFF/APRIL system in SLE pathogenesis. Nat Rev Rheumatol 10(6):365–373
Dörner T, Radbruch A, Burmester GR (2009) B-cell-directed therapies for autoimmune disease. Nat Rev Rheumatol 5(8):433–441
Mok MY (2010) The immunological basis of B-cell therapy in systemic lupus erythematosus. Int J Rheum Dis 13:3–11
Silverman GJ (2006) Therapeutic B cell depletion and regeneration in rheumatoid arthritis: emerging patterns and paradigms. Arthritis Rheum 54(8):2356–2367
García-Carrasco M, Jiménez-Hernández M, Escárcega RO, Mendoza-Pinto C, Galarza-Maldonado C, Sandoval-Cruz M, Zamudio-Huerta L, López-Colombo A, Cervera R (2009) Use of rituximab in patients with systemic lupus erythematosus. Autoimmun Rev 8:343–348
Merrill JT, Neuwelt CM, Wallace DJ, Shanahan JC, Latinis KM, Oates JC, Utset TO, Gordon C, Isenberg DA, Hsieh HJ, Zhang D, Brunetta PG (2010) Efficacy and safety of Rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum 62:222–233
Rovin BH, Furie R, Latinis K, Looney RJ, Fervenza FC, Sanchez-Guerrero J, Maciuca R, Zhang D, Garg JP, Brunetta P, Appel G, LUNAR Investigator Group (2012) Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum 64:1215–1226
Isenberg D (2012) Rituximab-it was the best of times, it was the worst of times. Autoimmun Rev 11(11):790–791
Reddy V, Jayne D, Close D, Isenberg D (2013) B-cell depletion in SLE: clinical and trial experience with rituximab and ocrelizumab and implications for study design. Arthritis Res Ther 15(Suppl 1):S2
Wallace DJ, Kalunian K, Petri MA, Strand V, Houssiau FA, Pike M, Kilgallen B, Bongardt S, Barry A, Kelley L, Gordon C (2014) Efficacy and safety of epratuzumab in patients with moderate/severe active systemic lupus erythematosus: results from EMBLEM, a phase IIb, randomised, double-blind, placebo-controlled, multicentre study. Ann Rheum Dis 73(1):183–190
Stohl W, Scholz JL, Cancro MP (2011) Targeting BLyS in rheumatic disease: the sometimes-bumpy road from bench to bedside. Curr Opin Rheumatol 23:305–310
Wallace DJ, Stohl W, Furie RA, Lisse JR, McKay JD, Merrill JT, Petri MA, Ginzler EM, Chatham WW, McCune WJ, Fernandez V, Chevrier MR, Zhong ZJ, Freimuth WW (2009) A phase II, randomized, double-blind, placebo-controlled, dose-ranging study of belimumab in patients with active systemic lupus erythematosus. Arthritis Rheum 61(9):1168–1178
Navarra SV, Guzmán RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, Li EK, Thomas M, Kim HY, León MG, Tanasescu C, Nasonov E, Lan JL, Pineda L, Zhong ZJ, Freimuth W, Petri MA, BLISS-52 Study Group (2011) Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet 377(9767):721–731
Furie R, Petri M, Zamani O, Cervera R, Wallace DJ, Tegzová D, Sanchez-Guerrero J, Schwarting A, Merrill JT, Chatham WW, Stohl W, Ginzler EM, Hough DR, Zhong ZJ, Freimuth W, van Vollenhoven RF, BLISS-76 Study Group (2011) A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum 63(12):3918–3930
Ginzler EM et al (2014) Disease control and safety of belimumab plus standard therapy over 7 years in patients with systemic lupus erythematosus. J Rheumatol 41(2):300–309
Ginzler EM, Wax S, Rajeswaran A, Copt S, Hillson J, Ramos E, Singer NG (2012) Atacicept in combination with MMF and corticosteroids in lupus nephritis: results of a prematurely terminated trial. Arthritis Res Ther 14:R33
Wang X, Huang W, Mihara M, Sinha J, Davidson A (2002) Mechanism of action of combined short-term CTLA4Ig and anti-CD40 ligand in murine systemic lupus erythematosus. J Immunol 168:2046–2053
Merrill JT (2013) Co-stimulatory molecules as targets for treatment of lupus. Clin Immunol 148(3):369–375
Kalunian KC, Davis JC Jr, Merrill JT, Totoritis MC, Wofsy D, IDEC-131 Lupus Study Group (2002) Treatment of systemic lupus erythematosus by inhibition of T cell costimulation with anti-CD154: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum 46(12):3251–3258
Boumpas DT, Furie R, Manzi S, Illei GG, Wallace DJ, Balow JE, Vaishnaw A, BG9588 Lupus Nephritis Trial Group (2003) A short course of BG9588 (anti-CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum 48(3):719–727
Sidiropoulos PI, Boumpas DT (2004) Lessons learned from anti-CD40L treatment in systemic lupus erythematosus patients. Lupus 13(5):391–397
Wang X, Huang W, Mihara M, Sinha J, Davidson A (2002) Mechanism of action of combined short-term CTLA4Ig and anti-CD40 ligand in murine systemic lupus erythematosus. J Immunol 168(4):2046–2053
Merrill JT, Burgos-Vargas R, Westhovens R, Chalmers A, D’Cruz D, Wallace DJ, Bae SC, Sigal L, Becker JC, Kelly S, Raghupathi K, Li T, Peng Y, Kinaszczuk M, Nash P (2010) The efficacy and safety of abatacept in patients with non-life-threatening manifestations of systemic lupus erythematosus: results of a twelve-month, multicenter, exploratory, phase IIb, randomized, double-blind, placebo-controlled trial. Arthritis Rheum 62(10):3077–3087
Mok CC (2012) Abatacept for systemic lupus erythematosus: the outlook. Expert Opin Biol Ther 12(12):1559–1561
Van Vollenhoven RF, Parodis I, Levitsky A (2013) Biologics in SLE: towards new approaches. Best Pract Res Clin Rheumatol 27(3):341–349
Aderka D, Wysenbeek A, Engelmann H, Cope AP, Brennan F, Molad Y, Hornik V, Levo Y, Maini RN, Feldmann M et al (1993) Correlation between serum levels of soluble tumor necrosis factor receptor and disease activity in systemic lupuserythematosus. Arthritis Rheum 36(8):1111–1120
Postal M, Appenzeller S (2011) The role of tumor necrosis factor-alpha (TNF-α) in the pathogenesis of systemic lupus erythematosus. Cytokine 56(3):537–543
Aringer M, Smolen JS (2012) Therapeutic blockade of TNF in patients with SLE-promising or crazy? Autoimmun Rev 11(5):321–325
Aringer M, Graninger WB, Steiner G, Smolen JS (2004) Safety and efficacy of tumor necrosis factor alpha blockade in systemic lupus erythematosus: an open-label study. Arthritis Rheum 50(10):3161–3169
Ramos-Casals M, Brito-Zerón P, Soto MJ, Cuadrado MJ, Khamashta MA (2008) Autoimmune diseases induced by TNF-targeted therapies. Best Pract Res Clin Rheumatol 22(5):847–861
Soforo E, Baumgartner M, Francis L, Allam F, Phillips PE, Perl A (2010) Induction of systemic lupus erythematosus with tumor necrosis factor blockers. J Rheumatol 37(1):204–205
Linker-Israeli M, Deans RJ, Wallace DJ, Prehn J, Ozeri-Chen T, Klinenberg JR (1991) Elevated levels of endogenous IL-6 in systemic lupus erythematosus. A putative role in pathogenesis. J Immunol 147(1):117–123
Woodrick RS, Ruderman EM (2011) Interleukin 6 inhibition—RA and beyond. Bull NYU Hosp Jt Dis 69(3):225–229
Illei GG, Shirota Y, Yarboro CH, Daruwalla J, Tackey E, Takada K, Fleisher T, Balow JE, Lipsky PE (2010) Tocilizumab in systemic lupus erythematosus: data on safety, preliminary efficacy, and impact on circulating plasma cells from an open-label phase I dosage-escalation study. Arthritis Rheum 62(2):542–552
Kamata Y, Minota S (2012) Successful treatment of massive intractable pericardial effusion in a patient with systemic lupus erythematosus with tocilizumab. BMJ Case Rep 21:2012
Szepietowski JC, Nilganuwong S, Wozniacka A, Kuhn A, Nyberg F, van Vollenhoven RF, Bengtsson AA, Reich A, de Vries DE, van Hartingsveldt B, Robinson DW Jr, Gordon R, Hsu B (2013) Phase I, randomized, double-blind, placebo-controlled, multiple intravenous, dose-ascending study of sirukumab in cutaneous or systemic lupus erythematosus. Arthritis Rheum 65(10):2661–2671
Yao Y, Richman L, Higgs BW, Morehouse CA, de los Reyes M, Brohawn P, Zhang J, White B, Coyle AJ, Kiener PA, Jallal B (2009) Neutralization of interferon-alpha/beta-inducible genes and downstream effect in a phase I trial of an anti-interferon-alpha monoclonal antibody in systemic lupus erythematosus. Arthritis Rheum 60(6):1785–1796
McBride JM, Jiang J, Abbas AR, Morimoto A, Li J, Maciuca R, Townsend M, Wallace DJ, Kennedy WP, Drappa J (2012) Safety and pharmacodynamics of rontalizumab in patients with systemic lupus erythematosus: results of a phase I, placebo-controlled, double-blind, dose-escalation study. Arthritis Rheum 64(11):3666–3676
Lichtman AI, Helfgott SM, Kriegel MA (2012) Emerging therapies for systemic lupus erythematosus—focus on targeting interferon-alpha. Clin Immunol 143:210–221
Lauwerys BR, Ducreux J, Houssiau FA (2013) Type I interferon blockade in systemic lupus erythematosus: where do we stand? Rheumatology (Oxford) 53(8):1369–1376
Conflict of interest
None.
Informed consent
I declare and confirm that our study was conducted according to the declaration of Helsinki and to the italian legislation.
Human and Animal rights
No animal or human studies were carried out by the authors for this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Squatrito, D., Emmi, G., Silvestri, E. et al. Pathogenesis and potential therapeutic targets in systemic lupus erythematosus: from bench to bedside. Autoimmun Highlights 5, 33–45 (2014). https://doi.org/10.1007/s13317-014-0058-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13317-014-0058-y