
Abstract
Dravet syndrome (DS) is a severe developmental and epileptic encephalopathy that is mainly associated with variants in SCN1A. While drug-resistant epilepsy is the most notable feature of this syndrome, numerous symptoms are present that have significant impact on patients’ quality of life. In spite of novel, third-generation anti-seizure treatment options becoming available over the last several years, seizure freedom is often not attained and non-seizure symptoms remain. Precision medicine now offers realistic hope for seizure freedom in DS patients, with several approaches demonstrating preclinical success. Therapeutic approaches such as antisense oligonucleotides (ASO) and adeno-associated virus (AAV)-delivered gene modulation have expanded the potential treatment options for DS, with some of these approaches now transitioning to clinical trials. Several of these treatments may risk the exacerbation of gain-of-function variants and may not be reversible, therefore emphasizing the need for functional testing of new pathogenic variants. The current absence of treatments that address the overall disease, in addition to seizures, exposes the urgent need for reliable, valid measures of the entire complement of symptoms as outcome measures to truly know the impact of treatments on DS. Additionally, with so many treatment options on the horizon, there will be a need to understand how to select appropriate patients for each treatment, whether treatments are complementary or adverse to each other, and long-term risks of the treatment. Nevertheless, precision therapeutics hold tremendous potential to provide long-lasting seizure freedom and even complete cures for this devastating disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
What Is Dravet Syndrome?
Dravet syndrome (DS), a severe developmental and epileptic encephalopathy, is estimated to occur in 1/15,700 live births [1]. It is characterized as a drug-resistant epilepsy presenting in the first year of life with prolonged seizures in the setting of fever or temperature changes, often hemiclonic in nature, followed by unprovoked seizures of varying types. Genetic testing has revealed pathogenic variants in the gene, SCN1A, in 70–80% of children suspected to have DS [1,2,3]. While variants in other genes have been found in children with a clinical diagnosis of DS, it is a largely monogenic disease with a characteristic clinical presentation. Over the past two decades, DS has had the benefit of intense research, resulting in novel therapeutic approaches that target seizure control through genetic modulation.
Characteristics of seizures in DS evolve over the lifespan, starting with prolonged seizures in the first year of life that are often hemiclonic and associated with temperature changes such as fever or ambient temperature changes (e.g., temperature changes associated with bathing). Unprovoked seizures of several types often follow shortly after and can include myoclonic seizures, though myoclonic seizures are not present in all children despite the early name for this syndrome of severe myoclonic epilepsy of infancy. In the toddler years, seizures are often prolonged episodes of status epilepticus, and for many children, frequent admissions to the intensive care unit are the norm. Seizures then begin to evolve into clusters, often during sleep or on the borders of sleep, as children enter into school age years [4,5,6]. While there are only a few reports in the literature of adults with DS, seizures are described as occurring in clusters but becoming less frequent, with individual seizures that are short in nature [4, 7, 8]. Periods of non-convulsive status are often reported in adolescents and young adults. Control with anti-seizure medication is often futile, and sodium channel blockers such as lamotrigine and carbamazepine are reported to exacerbate seizures and should be avoided [6, 9, 10].
While seizures are the most apparent characteristic of DS, several associated symptoms have been reported that impact quality of life as much as, if not more than, seizures. These impact several body systems and include constipation, feeding issues, sleep disruption, lack of perception of pain, behavioral issues, intellectual impairment, short stature, progressive gait abnormalities, balance issues, parkinsonism, and peripheral neuropathy [11,12,13,14,15,16,17,18,19,20]. The severity of many of these symptoms appears to be independent of seizure burden and likely is due to the underlying pathophysiology created by the disruption of SCN1A. Parental/ caregiver questionnaires have identified that these symptoms are as concerning to families as seizures [14, 17]. Additionally, there is a significant risk for sudden unexpected death in epilepsy (SUDEP) in DS patients, up to 20%, which is higher than the general epilepsy population [21, 22].
The most obvious measure of pathology in DS is seizure severity, including frequency and duration; however, it is rare to have good control of seizures, and even in the setting of seizure freedom or near seizure freedom, there are documented ongoing non-seizure symptoms of DS. Cognition has been reported to continue to decline despite control of seizures [23,24,25,26] and may be predictable by variant type, for example, missense or truncating variants that result in haploinsufficiency [27, 28]. Behavior is a major concern for families, which appears to be independent of seizure control [14, 19]. Premature mortality, largely due to SUDEP, occurs at a higher rate than the general epilepsy population [21, 22]. Gait abnormalities emerge at a time period when seizures are slowing [29,30,31]. All of these associated symptoms of DS impact quality of life but are neither well studied nor used as outcome measures for treatment trials. There are new small molecule medications that are leading to improved seizure control in this population, but until people living with DS are seizure-free and have resolution of associated symptoms, the disease is not “cured.” Resolution of seizures, while important, should not be the only measure of “intractable Dravet syndrome”; this definition will need to be expanded to consider all of the associated symptoms of DS that impact quality of life.
Underlying Pathophysiology of DS
SCN1A, encoding the voltage-gated sodium channel Nav1.1 α subunit, was first reported to be associated with DS in 2001 [32], but it is important to note that not all children with SCN1A variants will have DS and not all children with a clinical diagnosis consistent with DS will have an identified variant in SCN1A. Additionally, correlation with variant type and location is not as reliably predictive of disease as would be preferred.
Over 3000 monoallelic pathogenic variants in SCN1A have been reported, including missense and truncation mutations, microdeletions, and gene duplications (ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/?term=SCN1a%5ball). Most pathogenic variants occur de novo, with less than 10% inherited [33]. In most cases, haploinsufficiency of Nav1.1 appears to be sufficient to produce clinical disease. Nav1.1 is expressed most prominently in inhibitory neurons in the brain; therefore, channel dysfunction is postulated to lead to disinhibition, a subsequent increase in network hyperexcitability, and epileptogenesis [34]. Most pathogenic variants in SCN1A result in loss-of-function [34], although some pathogenic variants have been reported to cause gain-of-function and may be associated with a more severe and earlier onset phenotype [35]. As gene modulatory therapies are developed, the need for clarification of variants with functional testing will increase to determine gain-of-function vs. loss-of-function as well as to clarify which variants will lead to disease. Indeed, to avoid further intellectual impairment, theoretically early treatment will lead to improved outcomes; therefore, functional testing to predict disease will be essential to initiating early treatment.
First Precision Therapy for DS to Reach Clinical Trials: STK-001
In spite of the development of many third-generation small molecule anti-seizure drugs [36], the majority of DS patients have drug-resistant epilepsy and associated symptoms that impact quality of life, underscoring the unmet need for precision approaches that directly target the underlying genetic cause, SCN1A haploinsufficiency in the central nervous system (CNS). The first precision therapy for SCN1A-linked DS to reach clinical trials was STK-001, an antisense oligonucleotide (ASO) introduced by Stoke Therapeutics [37]. STK-001 was developed using Targeted Augmentation of Nuclear Gene Output (TANGO) technology [38, 39], which targets a naturally occurring, non-productive alternative splicing event, or poison exon, in SCN1A to specifically reduce levels of non-productive mRNA and increase levels of productive mRNA and Nav1.1 sodium channel protein (Fig. 1). This approach, which prevents expression of exon 20 N in SCN1A [40], upregulates expression of the wild-type allele to compensate for the mutant allele in the context of autosomal dominant SCN1A haploinsufficiency. This therapy is designed to be administered intrathecally to DS patients at regular intervals throughout their lifetime. Preclinical work showed that intracerebroventricular (ICV) administration of STK-001 to wild-type C57BL/6 J mouse brain in vivo increased the expression of productive, full-length Scn1a mRNA and Nav1.1 protein [40, 41]. A single ICV dose of STK-001 at postnatal day 2 (P2) in the Scn1aTmKea (F1:129S-Scn1a+/− × C57BL/6 J) mouse model of DS [41], in which exon 1 of Scn1a is deleted, increased productive Scn1a mRNA and Nav1.1 protein expression. Importantly, this single-dose treatment also prevented SUDEP in 97% of DS mice up to 90 days following the single injected dose. Single-dose ASO treatment of DS mice at P14, closer to the time of seizure onset, resulted in a less robust, but still significant, effect on mouse survival. Infrared-video monitoring of 19 DS mice injected with STK-001 at P2 showed a single tonic–clonic seizure followed by SUDEP in only 1 animal, with no behavioral seizures in the other 18. Electroencephalogram (EEG) recording of DS mice injected with STK-001 at P2 showed a reduction in seizure frequency with a prolonged latency to first seizure. While STK-001 administration significantly reduced seizure frequency and latency in mice, the infrequent seizures that did occur were as severe as those observed in untreated animals. Intrathecal lumbar bolus administration of STK-001 was subsequently evaluated at two different dosages in non-human primates (NHPs) for safety, brain biodistribution, target engagement, and pharmacodynamics [42]. The drug was well tolerated at both dosages with no changes noted on physical and neurological examination; no changes in food intake, body weight, hepatic function, or platelet counts; and no abnormal histopathology in all tissues examined. A broad biodistribution was observed in the brain with the highest level found in different regions of the cerebral cortex. Levels of wild-type SCN1A gene expression varied between ASO-treated animals, with trends toward decreased non-productive and increased productive transcript. Nav1.1 protein was increased in the high-dose group on day 29 post-injection in regions of the cerebral cortex. This increase in sodium channel protein varied in different regions of the cortex with as much as a twofold increase observed in some regions. This work showed that a single, intrathecal lumbar bolus injection of STK-001 in NHPs was safe and pharmacologically active. Taken together, this body of preclinical work led to the MONARCH phase 1/2a clinical trial for STK-001, an open-label study of children and adolescents ages 2 to 18 who have an established diagnosis of DS linked to a pathogenic variant in SCN1A [43]. Importantly, the clinical utility of STK-001 is limited to DS patients with SCN1A variants that result in Nav1.1 haploinsufficiency, e.g., truncating, nonsense, or frame shift variants, in which the mutant allele undergoes nonsense-mediated decay, as well as partial or whole-gene SCN1A deletions [44]. In contrast, this therapy is contraindicated for DS patients with missense SCN1A variants that result in the generation of Nav1.1 polypeptides, which may have maladaptive gain-of-function or dominant negative effects [35, 45], as TANGO-mediated increases in protein expression would likely increase disease severity. Nevertheless, introduction of this ASO was a major advance in precision therapeutics for DS patients.

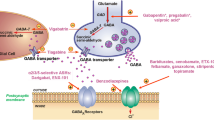
Mechanism of Action of STK-001, a New Application of Steric-Blocking ASOs. A Region of SCN1A wild-type pre-mRNA containing non-productive (non-coding) exon X (yellow rectangle) and flanking coding exons (brown rectangles). SCN1A pre-mRNA is alternatively spliced such that it generates a non-productive mRNA containing the non-productive exon X, which leads to the introduction of a premature truncation codon (PTC), and a productive mRNA lacking exon X. Upon export to the cytoplasm, the non-productive mRNA is degraded by nonsense-mediated mRNA decay and the productive mRNA is translated into wild-type Nav1.1 protein. The pre-mRNA carrying DS mutations undergoes the same alternative splicing processing, but the mutant productive mRNA does not produce a functional protein (not shown in the figure), leading to haploinsufficiency of Nav1.1. B STK-001 (ASO) binding to the non-productive exon X of the SCN1A wild-type and mutant (not shown) pre-mRNA and promotes exon X skipping, which leads to a reduction in non-productive mRNA and increased levels of productive mRNA and wild-type Nav1.1 protein to near normal levels. STK-001 leverages the wild-type gene copy to compensate for the loss-of-function mutant alleles in DS patients. Reproduced with permission from [39]
Other Therapies on the Horizon
Other precision therapeutic strategies for DS are being developed, including other ASOs, viral gene delivery, and virally delivered CRISPR-Cas9-based strategies. This work suggests that genes other than SCN1A, including those encoding other sodium channel α and β subunits, may be effectively targeted to provide benefit to DS patients.
SCN8A ASO
Sodium channel Nav1.6, encoded by SCN8A, is critical for neuronal firing in the CNS. Gain-of-function variants in this gene are linked to developmental and epileptic encephalopathies with similar presentation to DS [46]. Previous work in a mouse model showed that Scn8a is a genetic modifier of Scn1a-linked DS. Reduction of Scn8a expression by intercrossing Scn8a+/med−jo mice with Scn1a+/− DS mice to produce double heterozygous animals rescued premature lethality and extended lifespan compared to Scn1a+/− animals [47]. Meisler and colleagues expanded on this work using the ASO technology [48]. They first developed a gapmer ASO [39] that reduced Scn8a expression, delayed seizure onset, and increased survival in the Scn8aR1872W/+ gain-of-function mouse model of developmental and epileptic encephalopathy (Fig. 2). They then tested this ASO in the Scn1aTmKea (F1:129S-Scn1a+/− × C57BL/6 J) mouse model of DS [41]. Remarkably, DS mice treated with a single ICV dose of Scn8a ASO at P2 survived beyond 5 months of age without behavioral seizures or SUDEP. Scn8a ASO administration resulted in 50% reduction of Scn8a transcript in brain and spinal cord of DS mice with, remarkably, no effect on the level of Scn1a transcript. While ASO treatment of the Scn8aR1872W/+ mice required repeated drug administration to be effective in the long term, 100% of the DS mice survived for 5 months following a single Scn8a ASO dose at P2. This result, combined with the observation that a single dose of STK-001 at P2 prevented SUDEP in 97% of DS mice during a 90-day observation period [37], has led to the hypothesis that single-dose ASO administration during the early critical period of postnatal brain development may provide long-term seizure control in DS patients. It will be interesting to determine whether reduction of Scn8a expression via ASO administration is effective in DS mouse models expressing missense Scn1a variants that result in the generation of Nav1.1 polypeptides. If so, then SCN8A ASO therapy may provide seizure relief for a wider range of DS patient variants than STK-001.

Mechanism of Action of SCN8A Gapmer ASO. A Expression of the gene target, SCN8A, from transcription through translation of a wild-type or mutant Nav1.6 protein. B Gapmer ASO with the typical structure of modified chemistry in the wings to protect the ends from nucleases and an internal stretch of DNA that leads to the formation of the RNA–DNA hybrid when bound to the target transcript. The gapmer SCN8A ASO binds to an exon in the pre-mRNA and mRNA and recruits RNase H that recognizes the RNA/DNA hybrid and cleaves the RNA. The cleavage triggers RNA degradation, leading to reduction of wild-type or mutant Nav1.6 protein levels. Gapmer ASOs can be designed to target other transcript regions, e.g., introns. Even though RNase H is more abundant in the nucleus, RNase H–mediated cleavage of RNA–DNA hybrids can also occur in the cytoplasm. Reproduced and adapted with permission from [39]
Viral Approaches
Because standard AAV vectors used in gene therapy cannot accommodate large payloads like sodium channel α subunit encoding genes [49], investigators are developing novel strategies to modulate SCN1A gene expression through the activities of smaller gene products like transcription factors and accessory subunits. Although high-capacity adenoviral vectors, with expanded cloning capacities up to 37 KB, are in development [50], their utility for SCN1A expression has not yet been reported.
ETX101
While a number of neuronal cell types are affected in DS [41, 51, 52], GABAergic neurons, especially parvalbumin-positive, fast-spiking interneurons, are particularly vulnerable, and thus, disinhibition is proposed to be a primary contributor to DS mechanisms [34]. Encoded Therapeutics has taken a gene therapy approach in the development of ETX101, an adeno-associated virus serotype 9 (AAV9) vector-based, GABAergic neuron-selective, therapeutic agent expressing an engineered transcription factor that upregulates endogenous SCN1A gene expression. Unlike ASO therapies, which must be readministered to patients throughout their lifetime, AAV9-based approaches are intended for one-time administration to permanently alter gene expression in the brain. Preclinical work published in abstract form showed that ETX101 administration, similar to STK-001, increased Nav1.1 protein expression, prolonged survival, and decreased the occurrence of spontaneous and hyperthermia-induced seizures in a DS mouse model [53]. A single, unilateral ICV injection of ETX101 in NHPs resulted in wide vector biodistribution and transgene expression throughout the brain, including in the cerebral cortex and hippocampus [54]. Transgene mRNA expression in response to ICV injection was limited to the CNS, reaching similar levels within various brain structures after 28 days. ETX101 was reported to be well tolerated in NHPs with no detectable changes in clinical findings, including body weight and body temperature. No macroscopic or microscopic findings were observed in NHP brain, liver, dorsal root ganglion, or spinal cord. All animals survived until necropsy. Serum titers of AAV9 neutralizing antibodies increased 28 days post-ETX101 administration while cerebrospinal fluid neutralizing antibody titers were not different from pre-treatment levels. The authors concluded that ETX101 was well tolerated and that unilateral ICV injection is the appropriate delivery route for this AAV-mediated gene therapy. Importantly, and similar to STK-001, the clinical utility of ETX101 is limited to DS patients with SCN1A variants that result in Nav1.1 haploinsufficiency, and is contraindicated for DS patients with missense SCN1A variants that result in the generation of Nav1.1 polypeptides, which may have maladaptive gain-of-function or dominant negative effects [35, 45], as increases in mutant channel expression may increase disease severity.
AAV-Navβ1
Sodium channel β1 subunits, encoded by SCN1B, are multi-functional [55, 56]. β1 subunits associate with all known sodium channel α subunits, including Nav1.1, as well as with some potassium channel α subunits, modulate sodium, potassium, and calcium currents; function as molecular chaperones to increase sodium channel and potassium channel α subunit expression at the plasma membrane; participate in cell–cell and cell–matrix adhesion as immunoglobulin-superfamily cell adhesion molecules; and modulate gene transcription through regulated intramembrane proteolysis-excitation coupling. In response to this body of literature, Hampson and colleagues hypothesized that viral-driven overexpression of sodium channel β1 subunits in CNS GABAergic neurons may enhance Nav1.1 plasma membrane expression and thus excitability, countering the effects of SCN1A haploinsufficiency [57]. They developed an AAV9-based vector driving β1 cDNA expression via the Gad1 promoter (AAV-Navβ1) and injected this agent into the cerebral spinal fluid of Scn1aTmKea (F1:129S-Scn1a+/− × C57BL/6 J) WT and DS mice [41] at P2. Interestingly, they found that untreated female DS mice showed a higher degree of SUDEP than males, uncovering a previously unknown sexual dimorphism in DS mice. AAV-Navβ1-treated DS mice displayed modest, but significant, increases in survival compared to untreated mice, and this effect was more pronounced in females over males. Male, but not female, DS mice treated with AAV-Navβ1 showed significantly reduced spontaneous seizures. However, AAV-Navβ1 treatment had no effect on febrile seizure susceptibility in either sex. Behavioral analyses using the open field, elevated plus maze, rotarod test, and passive avoidance tests showed that male, but not female, DS mice displayed motor hyperactivity and performed abnormally on tests of fear, anxiety, and learning and memory. AAV-Navβ1 treatment of male, but not female, DS mice normalized their motor activity and performance on the elevated plus maze test. The limited therapeutic efficacy of AAV-Navβ1 in a mouse model of DS suggests a potentially new therapeutic avenue for the treatment of this developmental and epileptic encephalopathy; however, the broad multi-functionality of sodium channel β1 subunits and limited effects in DS mice must be thoroughly considered before this agent could be tested in humans.
CRISPR-Cas9/AAV Strategies
CRISPR-Cas9 (clustered, regularly interspaced, short palindromic repeats, and CRISPR-associated protein) genome editing technology, originally discovered in prokaryotes, has transformed all of biology. Dr. Emmanuelle Charpentier and Dr. Jennifer Doudna were awarded the 2020 Nobel Prize in Chemistry for this remarkable work. CRISPR-Cas9 has provided robust tools for the generation of new animal models and offers powerful new precision gene therapy approaches for the treatment of genetic diseases in humans, including neurodevelopmental disorders [58,59,60]. Reports from two groups, each using a modified version of the CRISPR-Cas9 system, CRISPRa, also called CRISPR-ON, that features nuclease deficient Cas9 (dCas9) and guide RNAs (gRNAs) complementary to promoter regions of a target gene to activate its transcription, have suggested that CRISPR-Cas9 technology may be useful in developing gene therapies to treat DS [61, 62].
Broccoli and co-workers screened single-guide RNAs (sgRNAs) for their ability to stimulate Scn1a transcription in association with the dCas9 activation system. They identified a specific sgRNA, sg1P, targeting a sequence close to the Scn1a proximal promoter, that selectively increased Scn1a mRNA and Nav1.1 protein expression in cell lines and primary mouse neurons, including neurons isolated from Scn1a+/R1407X DS pups [62, 63]. They then delivered this Scn1a-dCas9 activation system ICV to P0 Scn1a+/R1407X pups using a dual AAV9-based system that included the mDlx5/6 promoter to drive expression in forebrain GABAergic neurons. In response to treatment, DS mouse interneuron firing rates, measured in brain slices, were increased and hyperthermia-induced seizures, recorded from 1-month old pups using EEG, were attenuated but not completely suppressed, with increased threshold for seizure induction. Spontaneous seizures and SUDEP rates were not reported. The authors commented that the relatively low co-infection efficiency of the two separate AAVs in the interneuron population (approximately 20%) along with the considerable size of the SpCas9 requiring the use of two independent AAVs, significantly limited the utility of their system. They proposed future work using smaller Cas9 orthologs to improve delivery and efficacy. Nevertheless, this work holds great promise for future gene therapy. Notably, this transcriptional activation strategy does not distinguish between wild-type and mutant Scn1a alleles, and expression of the R1407X truncated allele was stimulated along with wild type. Thus, similar to the STK-001 and ETX101 approaches described above, this approach may be contraindicated for DS patients with gain-of-function or dominant negative SCN1A variants.
Yamagata and colleagues used a similar CRISPRa strategy to develop an inhibitory neuron-selective Scn1a gene activation approach [61]. A complex experimental system was developed using triple mutant mice (floxed-dCas9-VPRVPR/+/Vgat-CreCre/+/Scn1aR1407X/+) injected with AAV harboring four synergistically effective mouse gRNAs. A high level of lethality in the triple mutant mouse model due to toxicity of the dCas-9-VPR confounded the experiments. Nevertheless, intravenous delivery by tail injection of the AAV particles containing the gRNAs into surviving pups at 4 weeks of age, following seizure onset at ~ P18 and a period of high lethality, resulted in a significant increase in temperature threshold for hyperthermia-induced seizures measured at 12 weeks of age. In addition, there was a significant improvement in latency to clonic seizures, wild-jumps, and generalized tonic–clonic seizures when the temperature of the animals was held at 43 °C. Electrocorticography recorded in freely moving animals showed a significant decrease in the frequency of abnormal spike discharges in CRISPRa-treated mice. Behavioral analyses showed a partial phenotypic rescue. Hyperactivity and thigmotaxis observed in the open field test, as well as decreased anxiety in the elevated plus maze test, observed in DS mice were improved to a state intermediate between DS and wild-type animals. Interestingly, while Scn1a mRNA and Nav1.1 protein levels were significantly increased in total brain lysates prepared from CRISPRa-treated mice, the amount of membrane-associated Nav1.1 in CRISPRa-treated brains was not different from that measured in untreated Scn1aR1407X/+ brains, suggesting that, even though overall levels of Nav1.1 protein increased, the excess channels may not have been functional. Consistent with this result, immunofluorescence imaging of brain slices with anti-Nav1.1 antibodies revealed excess cytoplasmic staining in the CRISPRa brains rather than in the axon initial segment, as normally seen in wild-type mice. Taken together, while the significance of this work was limited, and more efficient delivery systems need to be developed, the results suggest that gene therapy treatment to increase SCN1A expression in DS patients after seizure onset may effectively treat some aspects of their seizures and behavioral deficits.
Pros and Cons of Precision Therapy
There are several aspects of precision therapy that must be considered regardless of the mechanism: viral vector delivery or ASO. Timing of treatment for DS seems particularly important and related to outcomes, as there may be aspects of the disease that are not reversible. Significant SCN1A overexpression, or having “too much of a good thing,” must not result in adverse effects, as there is currently little ability to regulate the amount of response in a single patient. Little is known about long-term side effects and stability of response, which will not be able to be answered in short-term clinical trials. Finally, CRISPR approaches can result in significant off-target effects and viral treatments in general are not reversible. Theoretically, there is the potential for gene modification of germ cell lines, though none have yet been reported. Current treatments target symptoms of DS, predominantly seizures, though the syndrome has an impact on many other organ systems and functions. The promise of precision therapy targeting SCN1A expression has the potential to improve all of these aspects of DS. Early resolution of SCN1A expression may lead to improvements in overall function, including cognition and behavior.
Concerns with Viral Vectors
There are several challenges associated with viral vector therapies, including impact of the immune response and gene size limitations. AAV administration induces an immune response from the body leading to many negative issues that must be considered. The immune response can lead to destruction of the AAV vector prior to reaching its target, as well as a systemic inflammatory response. Repeated exposures are expected to demonstrate a greater response; therefore, the vector can, in practice, only be used once. If antibodies to the viral vector are already present, this may prohibit treatment. This single treatment requirement means that there must be a clinically meaningful response with the first delivery. As therapeutic strategies are improved, the presence of an immune response to AAV may preclude patients who have received one treatment from receiving subsequently new treatments. When treating very young infants, the presence of maternal antibodies may lead to a delay in treatment. Dosing must also be considered, since there is only one opportunity; the dose must be large enough to have an effect but cannot be so large that it induces a strong inflammatory response [64]. Viral vectors, even when administered directly in the CNS, can have a broad distribution, leading to increased uptake in the liver [65, 66].
There are several reports of adverse reactions to existing AAV vector treatments. Some examples are from the recently approved onasemnogene for use in children with spinal muscular atrophy (SMA). These include elevated transaminases in 90 of 100 patients [67], liver-associated adverse events were reported for 34 of 100 [67], and transient liver failure has been reported in 2 patients [68]. Similar events were also reported in animal models [69]. The medication has a black box warning for acute hepatoxicity, although there is some evidence that steroids may help [70]. Decreased platelets have been reported, though this effect was transient and did not require intervention [71]. Thrombotic microangiopathy (hemolytic anemia, thrombocytopenia, and acute kidney injury), a very rare entity, has been reported in 3 of 500 children treated with onasemnogene [67]. Virus is known to be shed in the stool; therefore, caution will need to be taken by caretakers that may be immunocompromised. Families that have more than one child with DS will also need to be cautious about exposure to the rest of the family, so that antibodies to the vector do not develop in the other affected family member and preclude treatment.
Concerns with ASO Treatment
ASO treatment has reported side effects, although milder than with AAV. In patients treated with nusinerson for SMA, the largest population treated with ASOs to date, almost all patients in the trials reported adverse events, although only one was thought to be treatment-related, post-procedural nausea [72]. Elevation of hepatic enzymes, proteinuria, and thrombocytopenia have been reported across several ASO treatment trials [73, 74]; however, these were often mild and transient in nature.
Administration of Treatment
In selecting gene-modifying treatments, several critical factors should be considered. The duration of action of these treatments is not yet known, but as mentioned above, treatments that are administered via AAV will only be able to be administered once, whereas treatment with ASO allows and may require multiple doses. ASO requires administration within the CNS, as ASOs do not cross the blood–brain barrier. AAV therapy has been administered intravenously, but current animal models have used ICV administration. This may be required for AAV treatment in DS to ensure that the majority of the treatment reaches the target cell population.
The cost of current gene therapy products on the market is very high, approaching millions of dollars per patient, and while uncomfortable to consider, cannot be ignored. Clinically, cost is considered in the treatment of SMA where ethical discussions are ongoing regarding if treatments are cost-effective [75] and, if so, who should carry the burden of the cost of care. Regardless of treatment choice, the cost of care for a child with a life-long disability like SMA [76] or DS [77,78,79] is very expensive, and these discussions will certainly continue. As more long-term data are obtained, we will gain a better understanding of the true reduction in cost of care created by precision therapeutics.
Study Planning
Clinical trials are just now starting for STK-001 in patients with DS, and there are several key aspects that need to be considered in the study design. Timing of treatment is likely important. As observed in mouse models [37], earlier treatment leads to better outcomes. However, treatment prior to clinical presentation is not as easy to replicate in the real-world setting as it is in the laboratory environment. Genetic testing is currently not performed until after clinical presentation in infants, and pathogenic variants do not correlate as closely to disease to allow differentiation between DS and generalized epilepsy with febrile seizures (GEFS +). Additionally, whether a particular variant is loss-of-function or gain-of-function is difficult to discern unless previously reported in the literature. Importantly, STK-001, as well as other treatments under development, may not be beneficial for patients with gain-of-function variants. All of these factors create barriers to early treatment.
Primary outcome measures in clinical trials for DS have only thus far included seizure occurrence. This is an obvious outcome that is measurable both accurately and in a short period of time; however, very young children may not yet have seizures frequent enough to be measurable in a time-limited trial and, with the recent addition of newer anti-seizure medications, a larger percentage of children have had a reduction of seizures such that they may not qualify for a trial. There are several other possible outcomes that are concerning to families but may be challenging to measure consistently and in a short time period, such as behavior, cognition, attention, and gait with changes noticed over months to years rather than days to weeks. Additionally, outcome measures will need to be considered for long-term evaluation and safety that are applicable over the lifespan. Furthermore, there is lack of clarity as to which aspects of DS are reversible with precision therapy and which are not. Older children and adults may benefit from treatment in ways that are not yet clearly measurable, such as improvement in behavior, reduction in rigidity, or improvement in mobility.
There is great excitement regarding the several developing options to treat DS that will require AAV administration, but due to the immune response, unless unique vectors are used, a single child will only be able to receive one of these therapies. Development of these therapies will require consideration of how to use unique vectors that will allow for several different approaches to be used in a single person and how to select patients who are most likely to benefit from an individual therapy.
Conclusion
There are great opportunities on the horizon for precision treatment of DS. Careful consideration will need to be given to genetic analysis and characterization of functional consequences of variants to allow for the development of improved animal models that represent these variants and appropriate selection of study participants. Development of reliable and valid outcome measures beyond seizures will improve our understanding of treatment responses across the lifespan. As these precision therapies are studied in clinical trials, there will be a need to determine who are the best candidates for each treatment approach as well as the risks and benefits of multiple treatments in a single patient. Finally, we cannot forget that there is a need for treatment options in DS patients with gain-of-function variants.
References
Wu, Y.W., et al., Incidence of Dravet Syndrome in a US Population. Pediatrics, 2015. 136(5): p. e1310-5.
Depienne, C., et al., Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J Med Genet, 2009. 46(3): p. 183-91.
Zuberi, S.M., et al., Genotype-phenotype associations in SCN1A-related epilepsies. Neurology, 2011. 76(7): p. 594-600.
Genton, P., R. Velizarova, and C. Dravet, Dravet syndrome: the long-term outcome. Epilepsia, 2011. 52 Suppl 2: p. 44-9.
Wolff, M., C. Casse-Perrot, and C. Dravet, Severe myoclonic epilepsy of infants (Dravet syndrome): natural history and neuropsychological findings. Epilepsia, 2006. 47 Suppl 2: p. 45-8.
Wirrell, E.C., et al., Optimizing the Diagnosis and Management of Dravet Syndrome: Recommendations From a North American Consensus Panel. Pediatr Neurol, 2017. 68: p. 18–34 e3.
Akiyama, M., et al., A long-term follow-up study of Dravet syndrome up to adulthood. Epilepsia, 2010. 51(6): p. 1043-52.
Takayama, R., et al., Long-term course of Dravet syndrome: a study from an epilepsy center in Japan. Epilepsia, 2014. 55(4): p. 528-38.
Wirrell, E.C. and R. Nabbout, Recent Advances in the Drug Treatment of Dravet Syndrome. CNS Drugs, 2019. 33(9): p. 867-881.
Cross, J.H., et al., Dravet syndrome: Treatment options and management of prolonged seizures. Epilepsia, 2019. 60 Suppl 3: p. S39-S48.
de Lange, I.M., et al., Outcomes and comorbidities of SCN1A-related seizure disorders. Epilepsy Behav, 2019. 90: p. 252-259.
Licheni, S.H., et al., Sleep problems in Dravet syndrome: a modifiable comorbidity. Dev Med Child Neurol, 2018. 60(2): p. 192-198.
Lagae, L., et al., Quality of life and comorbidities associated with Dravet syndrome severity: a multinational cohort survey. Dev Med Child Neurol, 2018. 60(1): p. 63-72.
Villas, N., M.A. Meskis, and S. Goodliffe, Dravet syndrome: Characteristics, comorbidities, and caregiver concerns. Epilepsy Behav, 2017. 74: p. 81-86.
Olivieri, G., et al., Cognitive-behavioral profiles in teenagers with Dravet syndrome. Brain Dev, 2016. 38(6): p. 554-62.
Connolly, M.B., Dravet Syndrome: Diagnosis and Long-Term Course. Can J Neurol Sci, 2016. 43 Suppl 3: p. S3-8.
Knupp, K.G., et al., Parental Perception of Comorbidities in Children With Dravet Syndrome. Pediatr Neurol, 2017. 76: p. 60-65.
Eschbach, K., et al., Growth and endocrine function in children with Dravet syndrome. Seizure, 2017. 52: p. 117-122.
Berg, A.T., et al., Nonseizure consequences of Dravet syndrome, KCNQ2-DEE, KCNB1-DEE, Lennox-Gastaut syndrome, ESES: A functional framework. Epilepsy Behav, 2020. 111: p. 107287.
Nabbout, R., et al., Perception of impact of Dravet syndrome on children and caregivers in multiple countries: looking beyond seizures. Dev Med Child Neurol, 2019. 61(10): p. 1229-1236.
Cooper, M.S., et al., Mortality in Dravet syndrome. Epilepsy Res, 2016. 128: p. 43-47.
Shmuely, S., et al., Mortality in Dravet syndrome: A review. Epilepsy Behav, 2016. 64(Pt A): p. 69-74.
Jansson, J.S., T. Hallböök, and C. Reilly, Intellectual functioning and behavior in Dravet syndrome: A systematic review. Epilepsy Behav, 2020. 108: p. 107079.
Brown, A., et al., Cognitive, behavioral, and social functioning in children and adults with Dravet syndrome. Epilepsy Behav, 2020. 112: p. 107319.
Verheyen, K., et al., Independent walking and cognitive development in preschool children with Dravet syndrome. Dev Med Child Neurol, 2020.
Ceulemans, B., Overall management of patients with Dravet syndrome. Dev Med Child Neurol, 2011. 53 Suppl 2: p. 19-23.
Ishii, A., et al., Clinical implications of SCN1A missense and truncation variants in a large Japanese cohort with Dravet syndrome. Epilepsia, 2017. 58(2): p. 282-290.
Riva, D., et al., Progressive neurocognitive decline in two children with Dravet syndrome, de novo SCN1A truncations and different epileptic phenotypes. Am J Med Genet A, 2009. 149a(10): p. 2339–45.
Rilstone, J.J., et al., Dravet syndrome: seizure control and gait in adults with different SCN1A mutations. Epilepsia, 2012. 53(8): p. 1421-8.
Rodda, J.M., et al., Progressive gait deterioration in adolescents with Dravet syndrome. Arch Neurol, 2012. 69(7): p. 873-8.
Black, L. and D. Gaebler-Spira, Crouch Gait in Dravet Syndrome. Pediatr Neurol Briefs, 2016. 30(11): p. 42.
Claes, L., et al., De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet, 2001. 68(6): p. 1327-32.
Scheffer, I.E. and R. Nabbout, SCN1A-related phenotypes: Epilepsy and beyond. Epilepsia, 2019. 60 Suppl 3: p. S17-S24.
Catterall, W.A., Dravet Syndrome: A Sodium Channel Interneuronopathy. Curr Opin Physiol, 2018. 2: p. 42-50.
Berecki, G., et al., SCN1A gain of function in early infantile encephalopathy. Ann Neurol, 2019. 85(4): p. 514-525.
Loscher, W., et al., Drug Resistance in Epilepsy: Clinical Impact, Potential Mechanisms, and New Innovative Treatment Options. Pharmacol Rev, 2020. 72(3): p. 606-638.
Han, Z., et al., Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci Transl Med, 2020. 12(558).
Lim, K.H., et al., Antisense oligonucleotide modulation of non-productive alternative splicing upregulates gene expression. Nat Commun, 2020. 11(1): p. 3501.
Scharner, J. and I. Aznarez, Clinical Applications of Single-Stranded Oligonucleotides: Current Landscape of Approved and In-Development Therapeutics. Mol Ther, 2021. 29(2): p. 540-554.
Carvill, G.L., et al., Aberrant Inclusion of a Poison Exon Causes Dravet Syndrome and Related SCN1A-Associated Genetic Epilepsies. Am J Hum Genet, 2018. 103(6): p. 1022-1029.
Mistry, A.M., et al., Strain- and age-dependent hippocampal neuron sodium currents correlate with epilepsy severity in Dravet syndrome mice. Neurobiol Dis, 2014. 65: p. 1-11.
Liau, G., et al., TANGO OLIGONUCLEOTIDES FOR THE TREATMENT OF DRAVET SYNDROME: SAFETY, BIODISTRIBUTION, AND PHARMACOLOGY IN THE NON-HUMAN PRIMATE. AES 2019 Annual Meeting Abstract Database. AESnet.org., 2019. Abstr. 2.195.
Laux, L., et al., SAFETY AND PHARMACOKINETICS OF ANTISENSE OLIGONUCLEOTIDE STK-001 IN CHILDREN AND ADOLESCENTS WITH DRAVET SYNDROME: SINGLE ASCENDING DOSE DESIGN FOR THE OPEN-LABEL PHASE 1/2A MONARCH STUDY. AES 2020 Annual Meeting Abstract Database. AESnet.org., 2020. Abstr. 344.
Marini, C., et al., The genetics of Dravet syndrome. Epilepsia, 2011. 52 Suppl 2: p. 24-9.
Sadleir, L.G., et al., Not all SCN1A epileptic encephalopathies are Dravet syndrome: Early profound Thr226Met phenotype. Neurology, 2017. 89(10): p. 1035-1042.
Meisler, M.H., SCN8A encephalopathy: Mechanisms and models. Epilepsia, 2019. 60 Suppl 3: p. S86-S91.
Martin, M.S., et al., The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Hum Mol Genet, 2007. 16(23): p. 2892-9.
Lenk, G.M., et al., Scn8a Antisense Oligonucleotide Is Protective in Mouse Models of SCN8A Encephalopathy and Dravet Syndrome. Ann Neurol, 2020. 87(3): p. 339-346.
Wu, Z., H. Yang, and P. Colosi, Effect of genome size on AAV vector packaging. Mol Ther, 2010. 18(1): p. 80-6.
Ricobaraza, A., et al., High-Capacity Adenoviral Vectors: Expanding the Scope of Gene Therapy. Int J Mol Sci, 2020. 21(10).
Yu, F.H., et al., Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci, 2006. 9(9): p. 1142-9.
Liu, Y., et al., Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism. Ann Neurol, 2013. 74(1): p. 128-39.
AN, Y., et al., A GABA-Selective AAV Vector-Based Approach to Up-Regulate Endogenous Scn1a Expression Reverses Key Phenotypes in a Mouse Model of Dravet Syndrome. Molecular Therapy, 2019. 27(4S1): p. Abstract 915.
Belle, A., et al., ETX101, A GABAERGIC INTERNEURON SELECTIVE AAV-MEDIATED GENE THERAPY FOR THE TREATMENT OF SCN1A+ DRAVET SYNDROME: BIODISTRIBUTION AND SAFETY IN NON-HUMAN PRIMATES. AES 2020 Annual Meeting Abstract Database. AESnet.org., 2020. Abst. 391.
O'Malley, H.A. and L.L. Isom, Sodium Channel beta Subunits: Emerging Targets in Channelopathies. Annu Rev Physiol, 2015. 77: p. 481-504.
Bouza, A.A., et al., Sodium channel beta1 subunits participate in regulated intramembrane proteolysis-excitation coupling. JCI Insight, 2021 Feb 8;6(3):e141776. https://doi.org/10.1172/jci.insight.141776.
Niibori, Y., et al., Sexually Divergent Mortality and Partial Phenotypic Rescue After Gene Therapy in a Mouse Model of Dravet Syndrome. Hum Gene Ther, 2020. 31(5-6): p. 339-351.
Doudna, J.A. and E. Charpentier, Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science, 2014. 346(6213): p. 1258096.
Jinek, M., et al., A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science, 2012. 337(6096): p. 816-21.
Gasiunas, G., et al., Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A, 2012. 109(39): p. E2579-86.
Yamagata, T., et al., CRISPR/dCas9-based Scn1a gene activation in inhibitory neurons ameliorates epileptic and behavioral phenotypes of Dravet syndrome model mice. Neurobiol Dis, 2020. 141: p. 104954.
Colasante, G., et al., dCas9-Based Scn1a Gene Activation Restores Inhibitory Interneuron Excitability and Attenuates Seizures in Dravet Syndrome Mice. Mol Ther, 2020. 28(1): p. 235-253.
Ogiwara, I., et al., Na(v)1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci, 2007. 27(22): p. 5903–14.
Wilson, J.M., Lessons learned from the gene therapy trial for ornithine transcarbamylase deficiency. Mol Genet Metab, 2009. 96(4): p. 151-7.
Grimm, D., et al., Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature, 2006. 441(7092): p. 537-41.
Bevan, A.K., et al., Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol Ther, 2011. 19(11): p. 1971-80.
Chand, D., et al., Hepatotoxicity following administration of onasemnogene abeparvovec (AVXS-101) for the treatment of spinal muscular atrophy. J Hepatol, 2021. 74(3): p. 560-566.
Feldman, A.G., et al., Subacute Liver Failure Following Gene Replacement Therapy for Spinal Muscular Atrophy Type 1. J Pediatr, 2020. 225: p. 252–258 e1.
Hinderer, C., et al., Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum Gene Ther, 2018. 29(3): p. 285-298.
Stevens, D., et al., Onasemnogene Abeparvovec-xioi: Gene Therapy for Spinal Muscular Atrophy. Ann Pharmacother, 2020. 54(10): p. 1001-1009.
Waldrop, M.A., et al., Gene Therapy for Spinal Muscular Atrophy: Safety and Early Outcomes. Pediatrics, 2020. 146(3).
Darras, B.T., et al., An Integrated Safety Analysis of Infants and Children with Symptomatic Spinal Muscular Atrophy (SMA) Treated with Nusinersen in Seven Clinical Trials. CNS Drugs, 2019. 33(9): p. 919-932.
Jason, T.L., J. Koropatnick, and R.W. Berg, Toxicology of antisense therapeutics. Toxicol Appl Pharmacol, 2004. 201(1): p. 66-83.
Chan, J.H., S. Lim, and W.S. Wong, Antisense oligonucleotides: from design to therapeutic application. Clin Exp Pharmacol Physiol, 2006. 33(5-6): p. 533-40.
Connock, M., et al., Will the US$5 million onasemnogene abeparvosec treatment for spinal muscular atrophy represent 'value for money' for the NHS? A rapid inquiry into suggestions that it may be cost-effective. Expert Opin Biol Ther, 2020. 20(7): p. 823-827.
Dangouloff, T., et al., Systematic literature review of the economic burden of spinal muscular atrophy and economic evaluations of treatments. Orphanet J Rare Dis, 2021. 16(1): p. 47.
Campbell, J.D., et al., Assessing the impact of caring for a child with Dravet syndrome: Results of a caregiver survey. Epilepsy Behav, 2018. 80: p. 152-156.
Jensen, M.P., et al., The humanistic and economic burden of Dravet syndrome on caregivers and families: Implications for future research. Epilepsy Behav, 2017. 70(Pt A): p. 104-109.
Whittington, M.D., et al., The direct and indirect costs of Dravet Syndrome. Epilepsy Behav, 2018. 80: p. 109-113.
Acknowledgements
The authors acknowledge the expert graphic design contributions of Jennifer Fairman for Figs. 1 and 2.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Disclosure
A portion of Dr. Isom’s work was funded by a research grant to the University of Michigan from Stoke Therapeutics. Dr. Knupp has received research funding from Stoke Therapeutics; Encoded Inc.; Zogenix Inc.; Eisai; and West Pharmaceuticals, and consulting funding from Stoke Therapeutics; Encoded Inc.; Zogenix Inc.; Biocodex; GW Pharmaceuticals; and West Pharmaceuticals.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Isom, L.L., Knupp, K.G. Dravet Syndrome: Novel Approaches for the Most Common Genetic Epilepsy. Neurotherapeutics 18, 1524–1534 (2021). https://doi.org/10.1007/s13311-021-01095-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-021-01095-6