
Abstract
Rapid eye movement (REM) sleep is implicated learning and memory (L/M) functions and hippocampal long-term potentiation (LTP). Here, we demonstrate that REM sleep deprivation (REMSD)-induced impairment of contextual fear memory in mouse is linked to a reduction in hexosamine biosynthetic pathway (HBP)/O-GlcNAc flux in mouse brain. In mice exposed to REMSD, O-GlcNAcylation, and O-GlcNAc transferase (OGT) were downregulated while O-GlcNAcase was upregulated compared to control mouse brain. Foot shock fear conditioning (FC) induced activation of protein kinase A (PKA) and cAMP response element binding protein (CREB), which were significantly inhibited in brains of the REMSD group. Intriguingly, REMSD-induced defects in L/M functions and FC-induced PKA/CREB activation were restored upon increasing O-GlcNAc cycling with glucosamine (GlcN) or Thiamet G. Furthermore, Thiamet G restored the REMSD-induced decrease in dendritic spine density. Suppression of O-GlcNAcylation by the glutamine fructose-6-phosphate amidotransferase (GFAT) inhibitor, 6‐diazo‐5‐oxo‐l‐norleucine (DON), or OGT inhibitor, OSMI-1, impaired memory function, and inhibited FC-induced PKA/CREB activation. DON additionally reduced the amplitude of baseline field excitatory postsynaptic potential (fEPSP) and magnitude of long-term potentiation (LTP) in normal mouse hippocampal slices. To our knowledge, this is the first study to provide comprehensive evidence of dynamic O-GlcNAcylation changes during the L/M process in mice and defects in this pathway in the brain of REM sleep-deprived mice. Our collective results highlight HBP/O-GlcNAc cycling as a novel molecular link between sleep and cognitive function.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sleep is an essential physiological process in all living organisms characterized by two main stages, specifically, nonrapid eye movement (non-REM) followed by a significantly shorter period of rapid eye movement (REM) [1]. Sleep deprivation, in particular, REM sleep deprivation (REMSD), impairs learning and memory (L/M) processes by affecting hippocampal synaptic plasticity, which is critical for learning acquisition and memory consolidation and dysregulation of the cAMP‐response element binding protein (CREB) pathway [2,3,4]. Furthermore, REMSD is reported to decrease long‐term potentiation (LTP) [5] while increasing long-term depression (LTD) of neuron activity in CA1 and dentate gyrus (DG) regions of the hippocampus [6]. Aging is associated with diverse sleep-associated pathology in both human and animal models. A few reports have shown that younger subjects are potentially more sensitive to the effects of sleep loss in humans [7], while the neurological damage induced by SD is more prevalent in the elderly. In healthy aged individuals, sleep-dependent memory consolidation and certain procedural tasks, such as motor sequence learning, are impaired [8]. Aged animals are also at greater risk for impaired glucose tolerance and diabetes [9, 10], which might lead susceptible to further stress resulting from SD. Although the destructive effect of SD on cognitive function is generally accepted [11,12,13], the effect of REMSD on memory functions remains a subject of controversy, and the effect of REMSD on memory functions remains a subject of controversy. For example, a number of reports suggest that loss of REM sleep in humans with brainstem lesions has no obvious impact on cognitive function [12, 14, 15]. REM sleep suppression has even been associated with improvement in memory [16,17,18,19]. Several studies have reported that extended wakefulness results in increased BDNF in the hippocampus, supporting a beneficial effect of SD on cognition [20, 21]. Therefore, the potential regulatory effects of REM sleep on L/M processes and underlying mechanisms thus remain to be clarified.
The mammalian brain depends on glucose as the main source of energy. Abnormalities in glucose metabolism have been shown to induce brain dysfunction or impair normal cognitive function. Accumulating evidence suggests that performance during memory tasks leads to a substantial decrease in glucose concentrations in extracellular fluid in rat [22] and glucose administration can enhance memory performance in healthy individuals [23]. A direct association between alterations in glucose metabolism and neurodegeneration has also been described whereby dysregulation of glucose metabolism is linked to Alzheimer’s disease (AD) progression and cognitive impairment [24, 25]. Although there are indications that hypoglycemic condition during sleep is associated with impairment of cognitive performance [26], limited studies to date have examined whether the sleep-associated memory function is linked to alterations in brain glucose metabolism. Furthermore, the issue of whether REMSD-induced impairment of memory consolidation can be reversed through regulating glucose metabolism in the brain is yet to be addressed.
Overall, ~3–5% intracellular glucose is converted into UDP-N-acetylglucosamine (UDP-GlcNAc), a substrate of O-linked-GlcNAc modification (O-GlcNAcylation) reactions [27]. The O-GlcNAcylation process participates in normal functioning of neurons, and its dysregulation contributes to the pathogenesis of various neurological disorders [28,29,30]. This process is regulated by two enzymes: O-GlcNAc transferase (OGT) that transfers GlcNAc residues to the hydroxyl group of serine or threonine of protein substrates and O-GlcNAcase (OGA) that catalyzes removal of GlcNAc [31]. OGT and OGA are particularly abundant in neurons enriched in presynaptic terminals and multiple O-GlcNAc-modified proteins in pre- and post-synapses have been identified [32, 33]. Impaired brain O-GlcNAcylation is one of the characteristic features of AD and inhibition of OGA to increase brain O-GlcNAcylation can alleviate AD pathology, protect against neurodegeneration and improve behavior [34,35,36,37]. However, other findings on O-GlcNAc function in the brain have been inconsistent, with reports that elevation of O-GlcNAcylation contributes to L/M impairment [38,39,40]. Therefore, establishing the functional role of O-GlcNAc cycling in brain remains an urgent challenge in the field.
In the current study, we have demonstrated dynamic regulation of O-GlcNAc cycling in association with OGT and OGA expression changes in mouse brain during L/M and compared findings in normal and REM sleep-deprived mice. Our data further validated the protective effect of elevating O-GlcNAcylation with glucosamine (GlcN) or Thiamet G on L/M dysfunction in REM sleep-deprived mice. A key finding was that cognitive impairment induced by REMSD is significantly associated with O-GlcNAc dysregulation, supporting the proposed theory that disruption of HBP signaling within the brain contributes to the progression of neuronal dysfunction.
Materials and Methods
Animals
Male C57BL/6 N mice 7 weeks old and weighing about 25 g were used for all experiments (SAMTACO, Osan, Korea). Mice were individually housed in ventilated polystyrene cages (15 cm × 30 cm) under a 12-h light–dark cycle at 24 ± 2 ℃. All animal experiments were conducted in accordance with policies and guidelines of Inha University and approved by Institutional Animal Care and Use Committee of Inha University in Incheon, Korea (INHA 190920–665).
Drug Treatment
PBS (control), 200 mg/kg GlcN [41], or 20 mg/kg Thiamet-G [42] was intraperitoneally administered to mice daily at 14:00. DMSO (0.1% as the control), 600 µg/kg DON [43], or 100 µg/kg OSMI-1 [44] was injected intraperitoneally 2 h before the passive avoidance test.
REM Sleep Deprivation Protocol
Naïve mice were subjected to the modified multiple platform method described previously [45]. Mice were individually housed in cages filled with water up to a depth of 1 cm containing 12 small cylindrical platforms (3.5 cm in diameter, 3 cm in height). Animals had access to food pellets and water ad libitum and were placed on platforms. Upon progression of REM sleep, they were awakened by falling in the water due to muscle atonia.
Passive Avoidance Test
The passive avoidance test was employed to determine the ability of mice to learn and remember stimuli, such as foot shock, using a shuttle box avoidance system as described previously with some modifications [46]. The shuttle box (Jeungdo Bio&Plant Co, Ltd., Seoul, Korea) consisted of illuminated (bright) and dark compartments with a grid floor attached to an electric shock source. For the exploration, mice were placed in the bright compartment and the door between the two compartments kept open for 10 min. For assessment of learning, as soon as the animal had stepped into the dark compartment, the door between two compartments was closed and a single electric foot shock (0.7 mA, 1 s) delivered. The mouse was left in the dark compartment for an additional 1 min to allow the animal to associate the chamber with fear (foot shock) and subsequently returned to the home cage. One hour after the first foot shock, the mouse was placed again in the bright chamber and time taken to enter the dark compartment (step-through latency) recorded. The retention time to step-through for each mouse was recorded, and when mice had stepped into the dark compartment, a second electric shock was delivered. After a further hour, mice were again placed in the illuminated compartment to measure the step-through latency maximum of 300 s. Shorter latency periods indicate poor memory retention compared to longer latency periods.
MWM Test
To test spatial memory, mice were subjected to the Morris water maze test consisting of a learning session (for 5 days) and a memory retention test that took place 24 h after the final learning session [47]. GlcN (200 mg/kg i.p.) was injected daily from days 2 to 5 into REM sleep-deprived mice at AM 10:00. The water maze apparatus incorporated a circular tank made from stainless steel (90 cm in diameter and 45 cm in height) and filled to a depth of 35 cm with clear water (22–25 ℃) or opaque water produced by the addition of nontoxic white paint. Along the edge of the water tanks, four points were marked and the pool divided into four equal quadrants. For the pre-training water maze test, visible platform training was performed. A cylindrical white-colored platform (6 cm in diameter) was placed at the mid-point of the four quadrants 0.5 cm above the water surface. Animals were randomly released from four different quadrants, respectively, and allowed to swim to find the platform over a 2-min period. For hidden platform training on days 1 to 5 (P1–P5), the platform location was switched to a random position and submerged 1 cm below the water surface. Each mouse was started from different directions in a random manner. Training trials were conducted each day with a 30-min interval between trials and the latency to reach each platform recorded. The day after the final day (day 6) of the hidden platform test, a probe trial was conducted by removing the platform and allowing mice to explore the pool for 60 s. Mice were released from the opposite quadrant in which the platform was previously located. Swimming time spent in the four quadrants was recorded. The time (as a percentage) spent in the target quadrant was measured to evaluate memory retention.
Immunofluorescence Staining
For paraffin sectioning, mouse brains were fixed in 4% formaldehyde for 7 days, embedded in paraffin, and sectioned using a microtome. To obtain frozen sections, mouse brains were fixed with 4% PFA. We generated three serial coronal sections of mouse brain at −1.06, −1.76, and −2.06 mm relative to bregma with a microtome [48] or a cryostat (CM1800; Leica, Wetzlar, Germany). After mounting the matched section for each condition in the same slide, IF staining was performed using an identical setting. For immunofluorescence staining, sections were rinsed in 0.1% PBS-T (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, and 0.1% Tween 20; pH 7.6) and blocked in 10% normal goat serum (Jackson ImmunoResearch, West Grove, PA, USA) in 0.1% PBS-T. Subsequently, sections were incubated in solution containing antibodies for O-GlcNAc CTD110.6 (sc-59623), OGT (sc-32921;), C/EBP-β (sc-150), C/EBP-δ (sc-151), CREB (sc-377154), phospho-CREB (sc-81486) (Santa Cruz, Biotechnology, Santa Cruz, CA) and OGA (14,711–1-AP, proteintech, Chicago, IL), phospho-ERK (#9101S), ERK (#4695S) and PKA c α (#4782) (Cell Signaling Technology, Danvers, MA) in 1% PBS-T for 4 ℃ overnight. After washing, slides were incubated with secondary antibodies including goat anti-mouse or anti-rabbit IgG conjugated to Alexa 488 (Invitrogen, CA, USA). Nuclei were counterstained with DAPI (D9542) (Sigma-Aldrich, St. Louis, MO, USA). Slides were examined under a confocal LSM 510 META microscope (Carl Zeiss, Jena, Germany) and analyzed using ZEN Lite Edition software. The tissue edges were excluded from analysis and positive cells in confocal images (40 ×) counted and quantified using the ImageJ program.
OGA Activity Assay
Mouse brain tissues (hippocampus, cortex) were lysed in lysis buffer A (150 mM NaCl, 50 mM Tris-pH 8.0, 0.5% NP-40) containing protease and phosphatase inhibitors. Samples (100 μg) were mixed with OGA activity buffer (50 mM cacodylate, pH 6.4, 50 mM D-GalNAc, 0.3% BSA, 1 mM FD-GlcNAc) and incubated at 37 ℃ for 3 h. The reaction was stopped by addition 0.5 M Na2CO3. Absorbance was measured at an excitation wavelength of 485 nm and emission wavelength of 535 nm.
Golgi-Cox Staining
Golgi-Cox staining was performed as described previously with some modifications [49]. Isolated mouse brains were incubated in Golgi-Cox staining solution [5% mercuric chloride 50 ml + 5% potassium dichromate 50 ml + 5% potassium chromate 40 ml + 100 ml distilled water (D.W)] for 8 days at room temperature in the dark. After washing with D.W., samples were transferred to tissue protectant buffer and maintained at 4 ℃ in the dark for 8 days. Coronal sections of brain were sliced into 100 μm sections using a vibratome and applied to the slides. Samples were dehydrated using increasing concentrations of ethanol, followed by treatment with 5% sodium thiosulfate. Slides were examined under an Olympus IX 83 light microscope (Olympus, Tokyo, Japan) with a × 60 objective oil immersion lens and analyzed using ImageJ (NIH) software. Dendrites and neurons were randomly selected for analysis of dendrite length and spine density measured with the aid of Fiji ImageJ software as described previously [50].
Brain Slice Preparation and Electrophysiological Recordings
Male C57BL/6 N mice (3–5 weeks old) were briefly anesthetized by using isoflurane (100%). Isolated brains were stored in cold choline dissection buffer (containing 110 mM choline chloride, 11.6 mM Na-ascorbate, 3.1 mM pyruvate, 25 mM NaHCO3, 1.25 mM NaH2PO4, 2.5 mM KCl, 7 mM MgCl2, 0.5 mM CaCl2, and 25 mM glucose, bubbled with 95% O2 and 5% CO2). Coronal slices were prepared using a vibratome (VT 1000 s, Leica) and recovered in a chamber with artificial cerebrospinal fluid (aCSF; containing 1 mM NaH2PO4, 26.2 mM NaHCO3, 118 mM NaCl, 2.5 mM KCl, 11 mM glucose, 2 mM CaCl2, and 1 mM MgCl2) bubbled with 95% O2 and 5% CO2 in a 35 °C water bath for 45 min, then stored at room temperature before recordings. During recording, brain slices were transferred to the recording chamber perfused with aCSF containing 5 mM GlcN, 100 μM DON, or together and the temperature was maintained at 30–32 °C. Field excitatory postsynaptic potentials (fEPSPs) were obtained from Schaffer collateral pathway (CA3-to-CA1 synapses) using DAM80 and WinLTP 2.10 (University of Bristol), filtered at 3 kHz, and sampled at 20 kHz. Recording pipettes (resistance of 1–3 MΩ) were filled with aCSF. Electrical stimulations were provided by placing two bipolar electric stimulators (FHC) on the stratum radiatum and alternatively stimulating for 20 s. Long-term potentiation (LTP) was induced with four times theta burst stimulation (4 × TBS) comprising one stimulus train of 100 Hz (5 pulses) 20 times of 5-Hz intervals with an inter-train interval of 10 s. Low-frequency stimulation (LFS, 1 Hz; 600 pulses) was given to induce long-term depression (LTD).
Western Blotting
For Western blotting, whole hippocampus was homogenated in RIPA buffer (150 mM NaCl, 1% Na-deoxycholate, 1% NP-40, 1 mM EDTA, 10 mM Tris, pH 7.4) with protease and phosphatase inhibitors, and 20 μM streptozotocin and centrifugated for 20 min at 13,200 rpm. Twenty micrograms of protein samples were separated by SDS-PAGE, transferred to Amersham Bioscieces™ Protan™ nitrocellulose membrane (GE Healthcare Life Science, Germany) and incubated in 5% skim milk or 5% BSA blocking buffer for 1 h at RT. Primary antibodies against anti-O-GlcNAc CTD 110.6 (sc-59623), OGT (sc-32921) (Santa Cruz Biotechnology, Santa Cruz, CA), OGA (14,711–1-AP), and anti-β-actin (66,009–1-Ig) (Proteintech, Chicago, IL) were probed overnight at 4 ℃, washed with TBS-T (50 mM, Tris, 150 mM NaCl, and 0.05% Tween 20; pH 7.6), incubated with horseradish peroxidase-conjugated secondary antibodies and detected by the Enhanced Chemiluminenscence (ECL) detection system (Promega Corp.). Densitometric quantification of protein bands were detected using ImageJ (NIH).
Hematoxylin and Eosin Staining
For hematoxylin and eosin staining, brains were fixed in 4% formaldehyde (Samchun, Korea) overnight at 4 °C, and wash with flow tap water for 1 h. Tissues were paraffinized and sectioned in 5 μm mounted onto coated slides (Matsunami Glass Ind., Ltd., Osaka, Japan). For staining, tissues were deparaffinized with 100% xylene (Duksan, Ansan, Korea) 5 min, and hydrated with gradient EtOH (Duksan, Ansan, Korea) order of 100%, 95%, 90%, and 70% each 1 min. Wash with distilled water for 1 min and fix with 4% formaldehyde for 5 min. After wash two times with PBS, stain the tissue with Harris hematoxylin solution (American MasterTech Scientific, CA, USA) for 2 min. Wash with flow tap water and counterstain in eosin solution (Sigma, MO, USA) for 30 s. The tissues were dehydrated through 95% and 100% alcohol and cleared by xylene for 5 min. After mounted with a mounting medium (Sigma), the slides were dry overnight at RT. Tissues were examined under a LS83 microscope (Olympus, Tokyo, Japan) and analyzed using cellSens Dimension and ImageJ software. For the analysis images, we analyzed 3 different fields in each tissue by counting the marker positive cell using ImageJ program.
Statistical Analysis
Data are expressed as means ± SEM. Statistical data were analyzed and graphs generated with Microsoft Excel. Comparisons were performed using one-way ANOVA and unpaired Student’s t test. Differences were considered significant at p < 0.05.
Results
REM Sleep Deprivation Impairs L/M Function and Decreases O-GlcNAc Flux in Mouse Brain
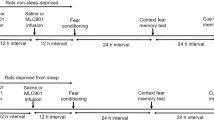
Using the modified multiple platform method, we induced REM sleep deprivation (REMSD) in mice for 24, 48, or 72 h and measured effects on foot shock fear conditioning (FC) and contextual fear memory (Fig. 1A). The step-through latency to enter the dark from the bright compartment was shorter in the 24-h REM sleep-deprived group than the control group and latency time was shorter with increase in REMSD time to 48 and 72 h (Fig. 1B). Next, we examined O-GlcNAc flux in the dentate gyrus (DG), CA1, and cortex regions of mouse brain (Fig. 1C, schematic diagram) between control and 72-h REMSD mouse groups by immunofluorescence (IF) staining. O-GlcNAcylation in DG, CA1, and cortex regions was significantly decreased in brain of the REMSD group relative to the control group (Fig. 1C, Suppl. Fig. 1A). In addition, the OGT level was decreased whereas the OGA level was increased in DG, CA1, and cortex regions of REM-deprived compared to control mice. Furthermore, OGA enzymatic activity was increased significantly in hippocampus region and less significantly in cortex region of REMSD mice (Fig. 1D). We reproduced the decrease in O-GlcNAcylation induced by REMSD via Western blot using a hippocampal brain preparation but OGT and OGA levels were not significantly changed by REMSD on Western blot (Suppl. Fig. 1B). Therefore, IF may be more effective than Western blot to determine regional changes of O-GlcNAc cycling. In order to examine the potential effects of dietary factors on REMSD-induced O-GlcNAc decrease, we compared the food consumption patterns of control and REM sleep-deprived mice. Consistent with previous findings [51,52,53], sleep deprivation was associated with increased food consumption (Suppl. Fig. 2A). Furthermore, the O-GlcNAc level was not significantly altered following 24 h starvation (Suppl. Fig. 2B), indicating little or no dietary effect on changes in O-GlcNAc.

REM sleep deprivation (REMSD)-induced changes in learning/memory (L/M) function and O-GlcNAc, OGT and OGA levels in mouse brain. A Schematic experimental design of REMSD and the passive avoidance test. Using the modified multiple platform method, REMSD was induced in mice for 24, 48, or 72 h. Learning and contextual fear memory (T1, T2, and T3) was measured via electric shock (fear conditioning) with 1-h intervals between tests. B Graph depicting retention times after first (T1), second (T2), and third (T3) electric stimulation in control, 24 (1 day), 48 (2 day), and 72 h (3 day) REM sleep-deprived (SD) mice (n = 8/group). C Representative confocal microscopy images (× 40) of O-GlcNAc (green), OGT (green), OGA (green), and merged immunofluorescence staining of dentate gyrus (DG), CA1, and cortex (Fig. 1C illustrations) of mouse brain at 72 h REMSD. Nuclei were counterstained with DAPI (blue). Enlarged images are presented in the white boxes. Scale bar = 50 μm. Graphs represent densitometric quantification of O-GlcNAc, OGT, and OGA (n = 4 ~ 12/group). D OGA enzymatic activity from total lysates of hippocampus and cortex. Absorbance values are presented as fold change relative to the control group (n = 3/group). All values are expressed as mean ± SEM. Student’s t test was used for statistical analysis; *p < 0.05, **p < 0.01 vs control group; N.S., not significant
Foot Shock FC Upregulates PKA/CREB, ERK, C/EBP β and δ Signaling and Increases O-GlcNAcylation in Brain of Control but not REM Sleep-Deprived Mice
To determine REMSD-induced changes in the signaling pathways of cognitive function, we examined the levels of the catalytic subunit of protein kinase A-alpha (PKAcα), phosphorylated CREB (p-CREB) and phosphorylated ERK (p-ERK) in brains of control and REM sleep-deprived mice after foot shock FC. FC triggered upregulation of both overall and nuclear levels of PKAcα and expression of p-CREB and p-ERK in brain samples of control mice, which was less significant for the REMSD group (Fig. 2A). While the ERK level was not altered by REMSD, the level of CREB protein was significantly decreased in the DG of REM sleep-deprived mouse brain compared to control, which was restored by GlcN (Suppl. Fig. 3). These results are consistent with the previous finding that sleep deprivation induces a decrease in hippocampal CREB mRNA [54]. Since CCAAT enhancer binding proteins (C/EBPs), C/EBP β, and C/EBP δ, co-localize with p-CREB to regulate long-term memory consolidation [55], we examined C/EBP β and C/EBP δ levels in both control and REM sleep-deprived mouse groups. Immunofluorescent staining data showed that FC induced strong upregulation of C/EBP β and δ, which was not apparent in the brain of REM sleep-deprive mice (Fig. 2B). Next, we examined HBP/O-GlcNAc flux after FC in brain samples of both normal and REM sleep-deprived groups. Interestingly, levels of O-GlcNAc, GFAT and OGT in DG, CA1 and cortex were significantly increased by FC in the control but not the REMSD group (Fig. 2C). OGA levels in brain were not significantly altered by FC in both control and REM-deprived mouse groups (data not shown).

Changes in p-CREB, PKAcα, p-ERK C/EBP β and δ, GFPT, OGT, and O-GlcNAc in brain of control or REM sleep-deprived mice in response to fear conditioning (FC). A Representative confocal images (× 40) of p-CREB (green), PKAcα (green), p-ERK (green), and merged immunofluorescence staining of DG, CA1, and cortex of control (CON) and 72-h REM sleep-deprived (SD) mice at 6 h of third fear stimulation. Nuclei were counterstained with DAPI (blue). Enlarged images are presented in the white boxes. Scale bar = 50 μm. Graphs showing quantification of immunofluorescence of p-CREB, PKAcα, and p-ERK in DG, CA1, and cortex (n = 4/group). B Representative confocal images (× 40) of C/EBP β (green), C/EBP δ (green), and merged immunofluorescence staining of DG, CA1, and cortex in control (CON) and 72-h REM sleep-deprived (SD) mice at 6 h of third fear stimulation. Nuclei were counterstained with DAPI (blue). Scale bar = 50 μm. Enlarged images are presented in the white boxes. Graphs represent quantification of C/EBP β and C/EBP δ immunofluorescence in DG, CA1, and cortex (n = 4/group). C Representative confocal images (× 40) of GFPT (green), OGT (green), O-GlcNAc (green), and merged immunofluorescence staining of DG, CA1, and cortex in control (CON) or 72-h REM sleep-deprived (SD) mice at 6 h of third fear stimulation. Nuclei were counterstained with DAPI (blue). Enlarged images are presented in the white boxes. Scale bar = 50 μm. Graphs represent quantification of GFPT, OGT and O-GlcNAc immunofluorescence in DG, CA1, and cortex (n = 4/group). All values were expressed as the mean ± SEM. Statistical analysis was conducted with one-way ANOVA and Student’s t test; *p < 0.05, **p < 0.01 vs control group; #p < 0.05, ##p < 0.01 vs. FC CON group
GlcN or Thiamet-G Restores L/M Impairment Induced by REMSD
Since GlcN directly enters hexosamine biosynthetic pathway (HBP) and enhances UDP-GlcNAc, we investigated its effect on L/M impairment induced by REMSD. To this end, mice were subjected to a daily intraperitoneal injection (i.p.) of GlcN (200 mg/kg) during the REMSD period (Fig. 3A). Memory impairment in the REM sleep-deprived group was evident based on the shorter entry latency for the dark compartment compared to the control group. However, the GlcN-treated REMSD group spent a longer time in the bright compartment and increased step-through latency (Fig. 3B). The effect of GlcN on spatial L/M function was further examined using the Morris-water maze test (MWM). GlcN (200 mg/kg i.p.) was injected daily from days 2 to 5 into REM sleep-deprived mice at AM 10:00 and the MWM test performed (shown in the schematic experimental design, Fig. 3C). During the learning sessions, latency in reaching the escape platform was measured in seconds. In the first session, control and REM sleep-deprived mice reached the escape platform at equivalent times. In sessions 2, 3, 4, and 5, although both groups learnt to reach the escape platform more rapidly and their escape latency decreased progressively with number of sessions, the REMSD group took a longer time overall than the control group to reach the escape platform. The GlcN-treated REMSD group of mice displayed decreased latency to a similar degree as control mice (Fig. 3D). Spatial memory impairment in the REMSD group was also indicated by the decreased time spent in the target quadrant compared to the control group (Fig. 3E). Again, GlcN restored REMSD-dependent spatial memory impairment, as evident from the increase in time spent in the target quadrant compared to the REMSD group of mice (Fig. 3E). Next, we examined whether GlcN restores O-GlcNAc flux and L/M-associated molecular changes in brain of REM sleep-deprived mouse. Following three consecutive days of intraperitoneal GlcN (200 mg/kg) injection during the REMSD period, hippocampal O-GlcNAcylation was remarkably restored to control levels (Fig. 3F). Converse to O-GlcNAc changes, the REMSD-induced increase in OGA returned to control levels following GlcN treatment in DG and CA1 of mouse brain (Fig. 3F). GlcN-treated mice in the REMSD group displayed increased p-CREB and p-ERK to almost control levels in response to FC (Fig. 3F). To further explore the physiological function of OGA enhanced by REMSD, we examined the effect of the OGA inhibitor, Thiamet G, on REMSD-induced L/M impairment. REM sleep-deprived mice displayed decreased latency to enter the dark compartment compared to the control group. Notably, Thiamet G-treated REM sleep-deprived mice demonstrated increased step-through latency, clearly indicating a protective effect against REMSD-induced memory impairment (Fig. 3G). Moreover, Thiamet G upregulated O-GlcNAc in brain samples of both control and REMSD groups of mice (Fig. 3H). Loss of dendritic spines is a common alteration triggered by REMSD [56]. Our Golgi-Cox staining results demonstrated that REMSD induces a significant decrease in dendrite length and number of dendritic branches as well as spine density in the CA1 region of mouse brain (Fig. 3I, J, K). Administration of Thiamet G to the REMSD group promoted dendrite length, number of dendritic branches and dendritic spine density compared to mice subjected to REMSD only.

Effects of GlcN or Thiamet-G on REMSD-induced L/M dysfunction. A Schematic experimental design. Using the modified multiple platform method, REMSD was induced for 72 h with or without three daily GlcN (200 mg/kg i.p.) injections. Learning and contextual fear memory (T1, T2, and T3) were measured via application of electric shock (fear conditioning) after 72 h of REMSD (SD) with 1-h intervals between the tests. B Graph depicting retention time after first (T1), second (T2), and third (T3) electric stimulation in control, REM sleep-deprived (SD), and GlcN-treated SD mice (SD + GlcN) (n = 8/group). C Schematic experimental design of Morris water maze (MWM) test in control or REM sleep-deprived mice with or without three consecutive daily GlcN (200 mg/kg i.p.) injections. The training trial was conducted three times a day for five consecutive days (P1–P5). D Graph representing latency to reach the visible platform at days 0 (P1), 1 (P2), 2 (P3), 3 (P4), and 4 (P5) in the MWM tests of control, REM sleep-deprived (SD) and GlcN-treated SD mice (SD + GlcN) (n = 8/ group). E Representative swimming tracks during the probe test in the MWM. The graph represents the percentage of time spent in the target quadrant during the probe test (n = 8/group). F Representative confocal images (× 40) of O-GlcNAc (green), OGA (green), p-CREB (green), p-ERK, and merged immunofluorescence staining of DG, CA1, and cortex in control (CON), REM sleep-deprived (SD), and GlcN-treated SD mice (SD + GlcN) (n = 8 ~ 12/group). Nuclei were counterstained with DAPI (blue). Enlarged images are presented in the white boxes. Scale bar = 50 μm. Graphs represent quantification of immunofluorescence of O-GlcNAc, OGA, p-CREB, and p-ERK in DG, CA1, and cortex of control (C), SD (S), and SD + GlcN (SG) groups of mice (n = 8 ~ 12/group). G Graph depicting retention time after first (T1), second (T2), and third (T3) electric stimulation in control, Thiamet G (20 mg/kg)-treated, and REM sleep-deprived (SD) mice with or without Thiamet G (n = 8 ~ 12/group). The first retention time was measured at 72 h REMSD. H Representative confocal images (× 40) of O-GlcNAc (green), p-CREB, p-ERK, and merged immunofluorescence staining of DG, CA1, and cortex in control (CON), Thiamet G-treated (Thi-G), REM sleep-deprived (SD), and Thiamet G-treated SD mice (SD + Thi-G) (n = 8/group). Nuclei were counterstained with DAPI (blue). Enlarged images are presented in the white boxes. Scale bar = 50 μm. Graph showing quantification of immunofluorescence of O-GlcNAc in DG, CA1 and cortex (n = 4/group). H Representative images of dendrites with Golgi staining in hippocampal CA1 of control (CON) or REMSD (SD) group with or without daily Thiamet G (20 mg/kg) treatment. J, K Graphs showing quantitative measurement of total spine length and numbers of dendrites branches (J) and spine density (K) (n = 8/ group). All values were expressed as the mean ± SEM. Statistical analysis was conducted with one-way ANOVA and Student’s t test; *p < 0.05, **p < 0.01 vs control group; #p < 0.05, ##p < 0.01 vs. SD group
Reduced HBP/O-GlcNAc Flux Impairs L/M in Mouse
Next, we ascertained whether decreased HBP flux/O-GlcNAc cycling is associated with predisposition to mouse REMSD-induced L/M dysfunction. One approach to decrease O-GlcNAc levels is treatment with the GFAT inhibitor 6‐diazo‐5‐oxo‐l ‐norleucine (DON), which significantly blocks biosynthesis of UDP‐GlcNAc. O-GlcNAcylation can also be directly regulated by modulating OGT activity using the small molecule inhibitor OSMI-1. Interestingly, both DON and OSMI-1 induced impairment of L/M function in mice (Fig. 4A, B). As expected, DON and OSMI-1 inhibited basal and FC-induced O-GlcNAcylation in DG, CA1 and cortex regions of mouse brain (Fig. 4C and Suppl. Fig. 4). Histological examination of hematoxylin and eosin (H&E)-stained sections revealed no significant changes in cell density by OSMI-1 in the hippocampal area (Suppl. Fig. 5). The L/M-associated signaling molecules p-CREB and p-ERK were strongly activated by FC in CA1 and cortex region of control mice. Both DON and OSMI-1 suppressed FC-induced stimulation of p-CREB and p-ERK (Fig. 4D).

Effects of DON and OSMI-1 on L/M function and O-GlcNAc, p-CREB and p-ERK levels in mouse brain. A, B Graphs showing retention times after first (T1), second (T2), and third electric stimulation (T3) in control, DON (600 μg/kg i.p. injection)-treated (A), and OSMI-1 (100 μg/kg i.p. injection)-treated (B) mice. The first retention time was measured at 2 h of DON or OSMI-1 injection (n = 6/group). C, D Representative confocal images (× 40) of O-GlcNAc (green, C), p-CREB, and p-ERK (green, D) immunofluorescence staining of DG, CA1 and cortex regions in control (CON), DON-treated (DON), or OSMI-1-treated (OSMI) mice at 6 h of FC. Nuclei were counterstained with DAPI (blue). Enlarged images are presented in the white boxes. Scale bar = 50 μm. Graphs showing quantification of immunofluorescence of O-GlcNAc (C), p-CREB, and p-ERK (D) in DG, CA1, and cortex of mouse brain (n = 4 ~ 12/group). All values were expressed as the mean ± SEM. Statistical analysis was conducted with one-way ANOVA and Student’s t test; *p < 0.05, **p < 0.01 vs control group; #p < 0.05, ##p < 0.01 vs. FC group
Hippocampal Synaptic Plasticity Changes Induced by DON With or Without GlcN
To investigate whether changes in O-GlcNAc cycling directly alter synaptic transmission of the hippocampus, we recorded fEPSPs from the Schaffer collateral pathway during bath application of 100 μM DON or with 5 mM GlcN. DON triggered an acute decrease in the slope of fEPSPs (Fig. 5A) whereas application of GlcN had minimal effect (Fig. 5B). Next, we examined whether synaptic plasticity, a measure of learning at the cellular level, was altered by DON in the presence and absence of GlcN. Upon induction of LTP via introduction of 4 × TBS, bath application of DON failed to block or facilitate the induction of LTP per se (Fig. 5C). Instead, the levels of LTP tended to decrease upon DON treatment although the impact of DON on magnitude of LTP was marginally significant. Following co-application of DON and GlcN, LTP was successfully induced and the average hippocampal LTP level was comparable to that of control (Fig. 5D). Induction of LTD has been established as critical for learning [57]. LTD triggered via low-frequency stimulation (LFS) remained intact following DON treatment as well as co-treatment with DON + GlcN (Fig. 5E). Both DON and DON + GlcN only exerted a minimal impact on the magnitude of LTD, if any (Fig. 5F), suggesting that HBP/O-GlcNAc flux selectively regulates the induction of LTP but not LTD in the hippocampus.

Regulation of hippocampal synaptic plasticity by DON with or without GlcN. A Effect of DON on field excitatory postsynaptic potential (fEPSP) in hippocampal slices (n = 7, 78.3 ± 6.5%, p < 0.05). B Effect of GlcN on levels of synaptic activity measured as slope of fEPSPs in hippocampal slices (n = 11, 94 ± 5.0%, p > 0.2). C Effect of DON with or without GlcN on hippocampal LTP induction by 4xTBS (Ctrl., 150.1 ± 10.9%, p < 0.01; 100 μM DON, 121.6 ± 7.2%, p < 0.05; 100 μM DON + 5 mM GlcN, 148.6 ± 9.5%, p < 0.005). D Graph showing the magnitude of LTP by DON with or without GlcN (one-way ANOVA, p = 0.09, post hoc LSD). E Effect of DON with or without GlcN on hippocampal LTD induction by LFS (Ctrl., 71.1 ± 6.2%, p < 0.05; DON, 58.1 ± 10.8%, p < 0.05; DON + GlcN, 70.4 ± 8.4%, p < 0.05). F Graph presenting comparison of LTD magnitude (one-way ANOVA, p > 0.5). Scale bar, 20 ms, 0.2 mV
Discussion
Elucidation of the molecular aspects of impaired cognitive function induced by sleep deprivation (SD) is important in view of the increasing work burden and sleep disorder issues in modern society. A recent study by our group using a zebrafish model demonstrated that SD induces cognitive dysfunction potentially through dysregulation of neuronal HBP/O-GlcNAc flux [58]. As an extension of previous research, we explored the functional importance of dynamic brain O-GlcNAc cycling for cognitive function and compared normal and REM sleep-deprived mouse groups in the current study. While accumulating studies have shown the involvement of dysregulated O-GlcNAcylation in inducing neurodegeneration [29, 30, 37, 59], the issue of whether hypo-O-GlcNAcylation is causal in brain dysfunction remains uncertain. Here, we have provided the first direct evidence of elevated O-GlcNAc cycling during the learning and memory (L/M) process and shown that decreased flux into this pathway (through reciprocal regulation of OGT and OGA levels) is linked to cognitive dysfunction using a mouse REM sleep deprivation (REMSD) model. Furthermore, our results support parallel regulation of brain O-GlcNAcylation with molecular alterations in the L/M-associated pathways, such as PKA/CREB, ERK, and C/EBP-β /C/EBP-δ signaling.
In our experiments, restoration of O-GlcNAc flux in REM sleep-deprived mice by GlcN, a direct HBP stimulator, or Thiamet G, an OGA inhibitor, promoted repair of REMSD-induced defects in L/M. Although IP-injected GlcN and Thiamet G systemically regulate O-GlcNAc cycling in organs other than the brain, including lung, liver, and skeletal muscle [41, 42], both our current and previous findings [58] strongly and consistently support an emerging link between increased brain OGA expression and SD-associated memory impairment. The detrimental role of OGA in normal brain function has been reported in earlier studies showing that hyper-O-GlcNAcylation induced by an OGA inhibitor in the 5xFAD amyloid-β mouse model decreases AD pathogenesis and improves cognition [34, 60, 61]. However, conflicting results demonstrating a beneficial effect of OGA on brain function have also been obtained. For instance, Thiamet G was shown to interfere with novel object recognition and placement in rats [62] and partial knockout of the OGA gene in Oga+/− mice displaying increased O-GlcNAcylation induced impairment of spatial L/M, along with defects in synaptic plasticity in mice [39]. One potential explanation for the differences in OGA function in Oga+/− mice from our current or previous reports is HAT-like activity or unidentified non-catalytic function of OGA [63]. In this case, chemical regulation and genetic modification of OGA may generate different physiological results. A plausible reason for the inconsistent effects of Thiamet G on cognitive function may be differences in activity between normal and REM sleep-deprived conditions. Given that O-GlcNAcylation could be reduced in our REMSD model via regulation of OGA, OGT and other potential genes, the effects of Thiamet G under hypo-O-GlcNAcylated REM sleep-deprived conditions could vary from those under normal conditions. Additionally, we cannot rule out the possibility of species-specific differences in effects of Thiamet G in mouse and rat. The positive influence of Thiamet G on the L/M process was further verified by an increase in dendritic spine density in REM sleep-deprived mice. The dendritic plastic structures of spines are important for L/M processes [64] and loss or altered morphology of dendritic spines in brain of sleep-deprived mouse has been reported [56, 65]. The Thiamet G-induced increase in total dendritic length and branched spines and spine numbers in hippocampus of REM sleep-deprived mice reflects potential involvement of O-GlcNAc cycling in synapse strength or plasticity. In fact, pharmacological modulation of O-GlcNAc by GlcN, PUGNAc, or Thiamet G specifically alters electrophysiological read outs of synaptic plasticity, while not affecting basal synaptic transmission [62]. Although it has been suggested that hippocampal synaptic activity and L/M be regulated by O-GlcNAcylation of synaptic proteins [32, 66], the issue of whether hypo-O-GlcNAcylation of synaptic proteins is responsible for exacerbating synaptic transmission or one of the consequences of metabolic dysregulation occurring in progression of cognitive dysfunction remains to be clarified. Our experiments additionally showed that DON reduced baseline fEPSP amplitude and decreased the magnitude of LTP in normal mouse hippocampal slices, revealing another mechanistic link between decreased O-GlcNAc flux and reduced synaptic transmission. These results are consistent with previous reports that reduction in O‐GlcNAc levels induced by DON or alloxan blocks L/M function and LTP, respectively [38, 67]. Intriguingly, in contrast to our finding that GlcN restores DON-induced decrease in LTP magnitude, an earlier study showed that GlcN triggers depression of baseline transmission and interferes with LTP [62]. In addition, Oga+/− mice with increased O-GlcNAcylation in brain display impairment of spatial learning, LTP, and LTD [68]. These conflicting results may be attributed to the potential importance of homeostatic O-GlcNAc cycling in synaptic function: although either hypo-O-GlcNAc induced by DON or hyper-O-GlcNAc flux induced by GlcN may interfere with LTP, combined treatment with DON and GlcN may lead to normal O-GlcNAc cycling and restoration of normal LTP. This finding highlights the importance of maintaining the O-GlcNAc level for L/M. Additionally, inhibition of O-GlcNAc using DON selectively suppressed the magnitude of LTP to control levels, thereby shifting bi-directional hippocampal plasticity towards less potentiation.
The importance of maintaining normal O-GlcNAc flux in the L/M process was further verified based on the deleterious effects of DON and OSMI, GFAT and OGT inhibitors, respectively, on L/M function in mice. Both DON and OSMI abrogated the increase in O-GlcNAc flux and activation of CREB and ERK in response to FC, suggesting that upon FC stimulation, increase in O-GlcNAc flux is necessary for CREB and ERK activation. Given that PKA/CREB signaling is regulated by both O-GlcNAcylation and phosphorylation of catalytic subunit of PKA (PKAcs) and CREB [67], the issue of whether FC and/or REMSD directly regulate O-GlcNAcylation of CREB or PKAcs to modulate phosphorylation or localization of these proteins remains to be clarified. Further mass spectrometry analyses of dynamic O-GlcNAcylation and phosphorylation changes of those proteins should aid in elucidation of the association between O-GlcNAc cycling and CREB/ERK signaling pathways.
Examination of memory functions in normal and REM sleep-deprived mice after genetic manipulation of GFAT, OGT or OGA may provide valuable evidence on the significance of HBP/O-GlcNAcylation in cognition. Previous research showed that O-GlcNAc protein modifications are associated in the regulation of axonal filopodia and branching. O-GlcNAc modification by PUGNAC, an alternative OGA inhibitor, was involved in the regulation of cAMP-induced axon branches [69]. Genetic modulation of OGT by brain-specific K/O is reported to induce widespread neuronal cell death, neurodegeneration, and neuroinflammation [70]. In addition, neuron-specific Ogt KO in mice resulted in severe defects and early postnatal death [71]. Oga knockout was shown to induce delayed brain differentiation and neurogenesis as well as abnormal proliferation, accompanied by developmental delay [72]. Furthermore, genetic manipulation often induces simultaneous compensatory changes to maintain O-GlcNAc homeostasis. For example, OGA protein levels are downregulated upon OGT knockdown or knockout [73, 74]. Conversely, upon OGA suppression, OGT protein levels are downregulated [75]. Consistently, an earlier study by our group demonstrated that stable knockdown of OGA via shRNA in macrophage cells led to concomitant downregulation of OGT [41]. Therefore, comprehensive assessment of the compensatory mechanisms controlling OGT and OGA expression or activity is necessary to determine the relevance of HBP/O-GlcNAc modulation through genetic alterations.
In conclusion, our results confirm that maintenance of normal O-GlcNAc cycling plays a central role in the L/M process and provide novel evidence that defects in O-GlcNAc homeostasis contribute to REMSD-induced L/M impairment. Indications of dynamic O-GlcNAc changes occurring within days of REMSD or even hours of FC support the underlying theory that protein O-GlcNAcylation is a dynamically active process that can directly control brain function. Further research on profiling and comparing O-GlcNAcylated proteins in the brain under physiological or pathological conditions is warranted to establish the molecular targets of neurological disease.
Abbreviations
- SD:
-
Sleep deprivation
- O-GlcNAc:
-
O-linked β-N-acetylglucosamine
- OGT:
-
O-GlcNAc transferase
- OGA:
-
O-GlcNAcase
- GlcN:
-
Glucosamine
- DG:
-
Dendate gyrus
- GFAT:
-
Glutamine-fructose-6-phosphate transaminase
- C/EBP:
-
CCAAT-enhancer-binding protein
- CREB:
-
CAMP response element-binding protein
- ERK:
-
Extracellular signal regulated kinase
- PKA:
-
Protein kinase A
- MWM:
-
Morris water maze
- FC:
-
Fear conditioning
- TBS:
-
Theta burst stimulation
- LFS:
-
Low-frequency stimulation
References
Stickgold R. Sleep: off-line memory reprocessing. Trends in cognitive sciences, 2(12), 484-492 (1998).
Havekes R, Meerlo P, Abel T. Animal studies on the role of sleep in memory: from behavioral performance to molecular mechanisms. Current topics in behavioral neurosciences, 25, 183-206 (2015).
Alhaider IA, Aleisa AM, Tran TT, Alkadhi KA. Sleep deprivation prevents stimulation-induced increases of levels of P-CREB and BDNF: protection by caffeine. Molecular and cellular neurosciences, 46(4), 742-751 (2011).
McDermott CM, LaHoste GJ, Chen C, Musto A, Bazan NG, Magee JC. Sleep deprivation causes behavioral, synaptic, and membrane excitability alterations in hippocampal neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience, 23(29), 9687-9695 (2003).
Ishikawa A, Kanayama Y, Matsumura H, Tsuchimochi H, Ishida Y, Nakamura S. Selective rapid eye movement sleep deprivation impairs the maintenance of long-term potentiation in the rat hippocampus. The European journal of neuroscience, 24(1), 243-248 (2006).
Tadavarty R, Rajput PS, Wong JM, Kumar U, Sastry BR. Sleep-deprivation induces changes in GABA(B) and mGlu receptor expression and has consequences for synaptic long-term depression. PloS one, 6(9), e24933 (2011).
Philip P, Taillard J, Sagaspe P et al. Age, performance and sleep deprivation. Journal of sleep research, 13(2), 105-110 (2004).
Pace-Schott EF, Germain A, Milad MR. Effects of sleep on memory for conditioned fear and fear extinction. Psychological bulletin, 141(4), 835 (2015).
Gunasekaran U, Gannon M. Type 2 diabetes and the aging pancreatic beta cell. Aging (Albany NY), 3(6), 565 (2011).
Gong Z, Muzumdar RH. Pancreatic function, type 2 diabetes, and metabolism in aging. International journal of endocrinology, 2012 (2012).
Siegel JM. The REM sleep-memory consolidation hypothesis. Science (New York, N.Y.), 294(5544), 1058–1063 (2001).
Rasch B, Born J. About sleep's role in memory. Physiological reviews, 93(2), 681-766 (2013).
Datta S, Mavanji V, Ulloor J, Patterson EH. Activation of phasic pontine-wave generator prevents rapid eye movement sleep deprivation-induced learning impairment in the rat: a mechanism for sleep-dependent plasticity. The Journal of neuroscience : the official journal of the Society for Neuroscience, 24(6), 1416-1427 (2004).
Lavie P, Pratt H, Scharf B, Peled R, Brown J. Localized pontine lesion: nearly total absence of REM sleep. Neurology, 34(1), 118-120 (1984).
Vertes RP, Siegel JM. Time for the sleep community to take a critical look at the purported role of sleep in memory processing. Sleep, 28(10), 1228–1229; discussion 1230–1223 (2005).
Rasch B, Pommer J, Diekelmann S, Born J. Pharmacological REM sleep suppression paradoxically improves rather than impairs skill memory. Nature neuroscience, 12(4), 396-397 (2009).
Gillin JC, Buchsbaum M, Wu J, Clark C, Bunney Jr W. Sleep deprivation as a model experimental antidepressant treatment: findings from functional brain imaging. Depression and anxiety, 14(1), 37-49 (2001).
Jiang Y, Zhu J. Effects of sleep deprivation on behaviors and abnormal hippocampal BDNF/miR-10B expression in rats with chronic stress depression. International journal of clinical and experimental pathology, 8(1), 586 (2015).
Hines D, Schmitt L, Hines R, Moss S, Haydon P. Antidepressant effects of sleep deprivation require astrocyte-dependent adenosine mediated signaling. Translational psychiatry, 3(1), e212-e212 (2013).
Fujihara H, Sei H, Morita Y, Ueta Y, Morita K. Short-term sleep disturbance enhances brain-derived neurotrophic factor gene expression in rat hippocampus by acting as internal stressor. Journal of Molecular Neuroscience, 21(3), 223-231 (2003).
Mahboubi S, Nasehi M, Imani A et al. Benefit effect of REM-sleep deprivation on memory impairment induced by intensive exercise in male wistar rats: with respect to hippocampal BDNF and TrkB. Nat Sci Sleep, 11, 179-188 (2019).
McNay EC, Gold PE. Age-related differences in hippocampal extracellular fluid glucose concentration during behavioral testing and following systemic glucose administration. The journals of gerontology. Series A, Biological sciences and medical sciences, 56(2), B66–71 (2001).
Sünram-Lea SI, Foster JK, Durlach P, Perez C. The effect of retrograde and anterograde glucose administration on memory performance in healthy young adults. Behavioural brain research, 134(1-2), 505-516 (2002).
Mergenthaler P, Lindauer U, Dienel GA, Meisel A. Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends in neurosciences, 36(10), 587-597 (2013).
Launer LJ, Miller ME, Williamson JD et al. Effects of intensive glucose lowering on brain structure and function in people with type 2 diabetes (ACCORD MIND): a randomised open-label substudy. The Lancet. Neurology, 10(11), 969-977 (2011).
Jauch-Chara K, Hallschmid M, Gais S et al. Hypoglycemia during sleep impairs consolidation of declarative memory in type 1 diabetic and healthy humans. Diabetes care, 30(8), 2040-2045 (2007).
Milewski S. Glucosamine-6-phosphate synthase--the multi-facets enzyme. Biochimica et biophysica acta, 1597(2), 173-192 (2002).
Morris M, Knudsen GM, Maeda S et al. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nature neuroscience, 18(8), 1183-1189 (2015).
Jacobsen KT, Iverfeldt K. O-GlcNAcylation increases non-amyloidogenic processing of the amyloid-beta precursor protein (APP). Biochemical and biophysical research communications, 404(3), 882-886 (2011).
Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America, 101(29), 10804-10809 (2004).
Vocadlo DJ. O-GlcNAc processing enzymes: catalytic mechanisms, substrate specificity, and enzyme regulation. Current opinion in chemical biology, 16(5-6), 488-497 (2012).
Cole RN, Hart GW. Cytosolic O-glycosylation is abundant in nerve terminals. Journal of neurochemistry, 79(5), 1080-1089 (2001).
Khidekel N, Ficarro SB, Peters EC, Hsieh-Wilson LC. Exploring the O-GlcNAc proteome: direct identification of O-GlcNAc-modified proteins from the brain. Proceedings of the National Academy of Sciences of the United States of America, 101(36), 13132-13137 (2004).
Yuzwa SA, Shan X, Jones BA et al. Pharmacological inhibition of O-GlcNAcase (OGA) prevents cognitive decline and amyloid plaque formation in bigenic tau/APP mutant mice. Molecular neurodegeneration, 9, 42 (2014).
Kim C, Nam DW, Park SY et al. O-linked beta-N-acetylglucosaminidase inhibitor attenuates beta-amyloid plaque and rescues memory impairment. Neurobiology of aging, 34(1), 275-285 (2013).
Yuzwa SA, Vocadlo DJ. O-GlcNAc modification and the tauopathies: insights from chemical biology. Current Alzheimer research, 6(5), 451-454 (2009).
Hastings NB, Wang X, Song L et al. Inhibition of O-GlcNAcase leads to elevation of O-GlcNAc tau and reduction of tauopathy and cerebrospinal fluid tau in rTg4510 mice. 12(1), 39 (2017).
Tallent MK, Varghis N, Skorobogatko Y et al. In vivo modulation of O-GlcNAc levels regulates hippocampal synaptic plasticity through interplay with phosphorylation. The Journal of biological chemistry, 284(1), 174-181 (2009).
Yang YR, Song S, Hwang H et al. Memory and synaptic plasticity are impaired by dysregulated hippocampal O-GlcNAcylation. Scientific reports, 7, 44921 (2017).
Yang YR, Song M, Lee H et al. O-GlcNAcase is essential for embryonic development and maintenance of genomic stability. Aging cell, 11(3), 439-448 (2012).
Hwang JS, Kim KH, Park J et al. Glucosamine improves survival in a mouse model of sepsis and attenuates sepsis-induced lung injury and inflammation. The Journal of biological chemistry, 294(2), 608-622 (2019).
Andrés-Bergós J, Tardio L, Larranaga-Vera A, Gómez R, Herrero-Beaumont G, Largo R. The increase in O-linked N-acetylglucosamine protein modification stimulates chondrogenic differentiation both in vitro and in vivo. Journal of Biological Chemistry, 287(40), 33615-33628 (2012).
Silva-Aguiar RP, Bezerra NC, Lucena MC et al. O-GlcNAcylation reduces proximal tubule protein reabsorption and promotes proteinuria in spontaneously hypertensive rats. Journal of Biological Chemistry, 293(33), 12749-12758 (2018).
Zhang J, Yu P, Hua F et al. Sevoflurane postconditioning reduces myocardial ischemia reperfusion injury-induced necroptosis by up-regulation of OGT-mediated O-GlcNAcylated RIPK3. Aging (Albany NY), 12(24), 25452 (2020).
Suchecki D, Duarte Palma B, Tufik S. Sleep rebound in animals deprived of paradoxical sleep by the modified multiple platform method. Brain research, 875(1-2), 14-22 (2000).
Rao Barkur R, Bairy LK. Evaluation of passive avoidance learning and spatial memory in rats exposed to low levels of lead during specific periods of early brain development. International journal of occupational medicine and environmental health, 28(3), 533-544 (2015).
D'Hooge R, De Deyn PP. Applications of the Morris water maze in the study of learning and memory. Brain research. Brain research reviews, 36(1), 60–90 (2001).
Paxinos G, Franklin KB. Paxinos and Franklin's the mouse brain in stereotaxic coordinates (Academic press, 2019).
Zaqout S, Kaindl AM. Golgi-Cox Staining Step by Step. Frontiers in neuroanatomy, 10, 38 (2016).
Orlowski D, Bjarkam CR. A simple reproducible and time saving method of semi-automatic dendrite spine density estimation compared to manual spine counting. J Neurosci Methods, 208(2), 128-133 (2012).
Mavanji V, Teske JA, Billington CJ, Kotz CM. Partial sleep deprivation by environmental noise increases food intake and body weight in obesity-resistant rats. Obesity, 21(7), 1396-1405 (2013).
Greer SM, Goldstein AN, Walker MP. The impact of sleep deprivation on food desire in the human brain. Nature communications, 4(1), 1-7 (2013).
Crispim CA, Zalcman I, Dáttilo M et al. The influence of sleep and sleep loss upon food intake and metabolism. Nutrition Research Reviews, 20(2), 195-212 (2007).
Guzman-Marin R, McGinty D. Sleep deprivation suppresses adult neurogenesis: clues to the role of sleep in brain plasticity. Sleep and Biological Rhythms, 4(1), 27-34 (2006).
Taubenfeld SM, Wiig KA, Monti B, Dolan B, Pollonini G, Alberini CM. Fornix-dependent induction of hippocampal CCAAT enhancer-binding protein [beta] and [delta] Co-localizes with phosphorylated cAMP response element-binding protein and accompanies long-term memory consolidation. The Journal of neuroscience : the official journal of the Society for Neuroscience, 21(1), 84-91 (2001).
Raven F, Meerlo P, Van der Zee EA, Abel T, Havekes R. A brief period of sleep deprivation causes spine loss in the dentate gyrus of mice. Neurobiol Learn Mem, 160, 83-90 (2019).
Kemp A, Manahan-Vaughan D. Hippocampal long-term depression and long-term potentiation encode different aspects of novelty acquisition. Proceedings of the National Academy of Sciences of the United States of America, 101(21), 8192-8197 (2004).
Lee Y, Park J, Kim SM et al. Sleep deprivation impairs learning and memory by decreasing protein O-GlcNAcylation in the brain of adult zebrafish. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 34(1), 853-864 (2020).
Yuzwa SA, Vocadlo DJ. O-GlcNAc and neurodegeneration: biochemical mechanisms and potential roles in Alzheimer's disease and beyond. Chemical Society reviews, 43(19), 6839-6858 (2014).
Kim C, Nam DW, Park SY et al. O-linked β-N-acetylglucosaminidase inhibitor attenuates β-amyloid plaque and rescues memory impairment. Neurobiology of aging, 34(1), 275-285 (2013).
Zhu Y, Shan X, Safarpour F et al. Pharmacological Inhibition of O-GlcNAcase Enhances Autophagy in Brain through an mTOR-Independent Pathway. ACS Chem Neurosci, 9(6), 1366-1379 (2018).
Taylor EW, Wang K, Nelson AR et al. O-GlcNAcylation of AMPA receptor GluA2 is associated with a novel form of long-term depression at hippocampal synapses. The Journal of neuroscience : the official journal of the Society for Neuroscience, 34(1), 10-21 (2014).
Toleman C, Paterson AJ, Whisenhunt TR, Kudlow JE. Characterization of the histone acetyltransferase (HAT) domain of a bifunctional protein with activable O-GlcNAcase and HAT activities. The Journal of biological chemistry, 279(51), 53665-53673 (2004).
Sala C, Segal M. Dendritic spines: the locus of structural and functional plasticity. Physiological reviews, 94(1), 141-188 (2014).
Raven F, Van der Zee EA, Meerlo P, Havekes R. The role of sleep in regulating structural plasticity and synaptic strength: Implications for memory and cognitive function. Sleep Med Rev, 39, 3-11 (2018).
Kanno T, Yaguchi T, Nagata T, Mukasa T, Nishizaki T. Regulation of AMPA receptor trafficking by O-glycosylation. Neurochemical research, 35(5), 782-788 (2010).
Xie S, Jin N, Gu J et al. O-GlcNAcylation of protein kinase A catalytic subunits enhances its activity: a mechanism linked to learning and memory deficits in Alzheimer's disease. Aging cell, 15(3), 455-464 (2016).
Yang YR, Song S, Hwang H et al. Memory and synaptic plasticity are impaired by dysregulated hippocampal O-GlcNAcylation. Scientific reports, 7(1), 1-9 (2017).
Francisco H, Kollins K, Varghis N, Vocadlo D, Vosseller K, Gallo G. O‐GLcNAc post‐translational modifications regulate the entry of neurons into an axon branching program. Developmental neurobiology, 69(2‐3), 162-173 (2009).
Wang AC, Jensen EH, Rexach JE, Vinters HV, Hsieh-Wilson LC. Loss of O-GlcNAc glycosylation in forebrain excitatory neurons induces neurodegeneration. Proceedings of the National Academy of Sciences, 113(52), 15120-15125 (2016).
O'Donnell N, Zachara NE, Hart GW, Marth JD. Ogt-dependent X-chromosome-linked protein glycosylation is a requisite modification in somatic cell function and embryo viability. Molecular and cellular biology, 24(4), 1680-1690 (2004).
Olivier-Van Stichelen S, Wang P, Comly M, Love DC, Hanover JA. Nutrient-driven O-linked N-acetylglucosamine (O-GlcNAc) cycling impacts neurodevelopmental timing and metabolism. Journal of Biological Chemistry, 292(15), 6076-6085 (2017).
Kazemi Z, Chang H, Haserodt S, McKen C, Zachara NE. O-linked beta-N-acetylglucosamine (O-GlcNAc) regulates stress-induced heat shock protein expression in a GSK-3beta-dependent manner. The Journal of biological chemistry, 285(50), 39096-39107 (2010).
Burén S, Gomes AL, Teijeiro A et al. Regulation of OGT by URI in Response to Glucose Confers c-MYC-Dependent Survival Mechanisms. Cancer cell, 30(2), 290-307 (2016).
Zhang Z, Tan EP, VandenHull NJ, Peterson KR, Slawson C. O-GlcNAcase Expression is Sensitive to Changes in O-GlcNAc Homeostasis. Frontiers in endocrinology, 5, 206 (2014).
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Funding
This work was supported by National Research Foundation (NRF) of Korea Grants, NRF-2020R1A2C2013345 and research grant of Inha University.
Author information
Authors and Affiliations
Contributions
IO Han designed the study and supervised the project; SM Kim conducted experiments, wrote the manuscript, and analyzed data; JW Park, HJ Sung, and TD Thi Tran performed experiments and analyzed data; CH Chung and SJ Zhang analyzed electrophysiology recording.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kim, SM., Zhang, S., Park, J. et al. REM Sleep Deprivation Impairs Learning and Memory by Decreasing Brain O-GlcNAc Cycling in Mouse. Neurotherapeutics 18, 2504–2517 (2021). https://doi.org/10.1007/s13311-021-01094-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-021-01094-7