
Abstract
Many epigenetic regulators are involved in pain-associated spinal plasticity. Coactivator-associated arginine methyltransferase 1 (CARM1), an epigenetic regulator of histone arginine methylation, is a highly interesting target in neuroplasticity. However, its potential contribution to spinal plasticity–associated neuropathic pain development remains poorly explored. Here, we report that nerve injury decreased the expression of spinal CARM1 and induced allodynia. Moreover, decreasing spinal CARM1 expression by Fbxo3-mediated CARM1 ubiquitination promoted H3R17me2 decrement at the K+ channel promoter, thereby causing K+ channel epigenetic silencing and the development of neuropathic pain. Remarkably, in naïve rats, decreasing spinal CARM1 using CARM1 siRNA or a CARM1 inhibitor resulted in similar epigenetic signaling and allodynia. Furthermore, intrathecal administration of BC-1215 (a novel Fbxo3 inhibitor) prevented CARM1 ubiquitination to block K+ channel gene silencing and ameliorate allodynia after nerve injury. Collectively, the results reveal that this newly identified spinal Fbxo3-CARM1-K+ channel gene functional axis promotes neuropathic pain. These findings provide essential insights that will aid in the development of more efficient and specific therapies against neuropathic pain.
Similar content being viewed by others


Introduction
Evidence is starting to emerge in support of the involvement of histone epigenetic mechanisms at multiple loci in the dorsal horn and the relevance of these mechanisms in chronic pain processing [1]. For example, our previous studies revealed histone acetylation impacts the expression of nociceptive genes in the dorsal horn to modify chronic pain states [2, 3]. Histone methylation, another important process of histone modification, is also recognized as a key regulator of long-lasting changes in gene expression in the dorsal horn [4]. However, little is known about whether the histone methylation mechanism in the dorsal horn functions in the transcriptional regulation of nociceptive genes in chronic pain.
Histone arginine methylation, which is catalyzed by protein arginine methyltransferases (PRMTs), has emerged as an important histone methylation modification involved in gene regulation [5]. Coactivator-associated arginine methyltransferase 1 (CARM1), a member of the PRMT family, has been shown to mainly catalyze the asymmetric dimethylation of Arg17 and Arg26 in histone H3 (H3R17me2 and H3R26me2, respectively) as well as an additional methylation site, Arg42 (H3R42), for epigenetic transcriptional activation [6,7,8,9]. CARM1-mediated methylation inhibits neuronal differentiation [10]; conversely, inhibition of CARM1-mediated methylation promotes processes of learning/memory consolidation, including synaptogenesis [11], neuronal differentiation [10], and synaptic plasticity [12]. Emerging studies have suggested that the epigenetic modification of genes underlying pain-related spinal plasticity resembles that of learning/memory formation in brain regions [4, 13]. Although CARM1 in the dorsal root ganglion (DRG) is reported to be associated with the development of neuropathic pain [14], little is known about how inhibiting CARM1-mediated epigenetic transcriptional activation contributes to the spinal plasticity underlying neuropathic allodynia progression.
Accumulating evidence suggests that chronic pain generation results from specific dysfunction of the inhibitory system in the spinal cord [15]. Since voltage-gated potassium (Kv) activity generally inhibits sensory neuron excitability [16], reductions in Kv activity seem to be a hallmark of hyperexcitability associated with neuropathic pain [17,18,19]. Nociceptive plasticity and central sensitization associated with chronic pain conditions have been observed after inhibition of the Kv4.2-mediated current to increase neuronal excitability in dorsal horn neurons [20]. Decreased expression of Kv4.2 channels has been observed in preclinical models of neuropathic pain in the DRG neurons of animals subjected to sciatic nerve axotomy [21] or chronic constriction injury [22]. Kv1.4 channel expression is also similarly reduced in the ipsilateral DRG following spinal nerve ligation [23]. Other pain models, including pancreatitis, inflammatory bowel disease, and temporomandibular joint pain, have also shown decreases in Kv1.4 expression [24,25,26]. The mRNA levels of Kv1.4 and Kv4.2 were found to be significantly reduced in the DRG to increase afferent input to the spinal dorsal horn in diabetic neuropathy [27]. Moreover, downregulation of many genes encoding K+ channels in the DRG, including Kcna4 (Kv1.4 gene) and Kcnd2 (Kv4.2 gene), is crucially involved in chronic pain symptoms [28]. Interestingly, histone methylation is associated with the epigenetic silencing of K+ channel genes in neuropathic pain after nerve injury [28]. Considering that attenuation of the histone epigenetic coactivator CARM1 can decrease gene transcription, these data suggest that inhibition of CARM1 contributes to spinal plasticity underlying neuropathic pain through a decrease in histone methylation-associated transcription of K+ channels. In addition, a study has shown that the stability of the arginine methyltransferase CARM1 is regulated by the F-box family of proteins [29], an Skp1-CUL1-F box superfamily of E3. The F-box protein has been reported to be associated with synaptic plasticity in the CA1 region of the hippocampus [30]. Remarkably, our laboratory has linked the ubiquitination of Fbxo3 (a member of the F-box family) in dorsal horn neurons to the pathology of neuropathic pain [31,32,33]. Furthermore, ubiquitination-associated epigenetic regulation in the dorsal horn is crucial for plasticity-mediated neuropathic pain [34]. Taken together, these findings strongly suggest that spinal Fbxo3-mediated CARM1 ubiquitination inhibits CARM1-dependent histone methylation of the K+ channel gene to participate in neuropathic pain development.
Materials and Methods
Animal Model of Neuropathic Pain
Adult male and female Sprague-Dawley rats weighing 200–250 g were used for this study. All animals were housed under approved conditions with a 12-h/12-h light/dark cycle and with food and water available ad libitum. Spinal nerve ligation (SNL) in rats was used as a neuropathic pain model [31, 32, 35]. Briefly, rats were anesthetized with isoflurane (induction, 5%; maintenance, 2% in air). After an incision was made, the left L5 and L6 spinal nerves were carefully isolated from the surrounding tissue and then tightly ligated with 6-0 silk sutures. In the sham operation group, the surgical procedures were identical to the nerve-ligated animals, except the silk sutures were left unligated. The surgical preparation and experimental protocols were approved by the Institutional Review Board of Taipei Medical University, Taipei, Taiwan.
Behavioral Studies
To quantify mechanical allodynia, rats were placed in individual plastic boxes on a mesh floor and allowed to acclimate for 1 h. A series of calibrated von Frey filaments (Stoelting, Wood Dale, IL) were then applied to the plantar surface of the hind paws of animals to measure the paw withdrawal threshold (up-down method) according to a modification of a previously described method [36]. The motor function of animals was assessed in the rota-rod apparatus (Panlab Harvard Apparatus, Barcelona, Spain). For acclimatization, the animals were subjected to three training trials at 3–4-h intervals on 2 separate days. During the training sessions, the rod was set to accelerate from 4 to 30 rpm over a 180-s period. During test sessions, the performance times of rats were recorded up to a cut-off time of 180 s. Three measurements were obtained at intervals of 5 min and were averaged for each test.
Western Blot Analysis
Rats were deeply anesthetized, and dorsal horn (L4–5) samples were rapidly removed. The samples were homogenized in lysis buffer containing a mixture of 25 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, and protease inhibitor mixture (Roche). Samples were then put on ice for 1 h with shaking. Lysates were centrifuged at 14,000 rpm for 20 min at 4 °C. The supernatant was carefully collected, and the protein concentration was measured using a BCA protein assay reagent kit (Pierce, Rockford, IL, USA). Equal amount of protein was loaded and separated on SDS-PAGE (Bio-Rad) and transferred to PVDF membranes (Millipore). The membranes were blocked with 5% nonfat milk or BSA in TBS with 0.1% Tween-20 for 1 h, and incubated with primary antibodies at 4 °C for 1 h, following by peroxidase-conjugated secondary antibodies for 1 h at room temperature. Signals were visualized using enhanced chemiluminescence detection kit (ECL Plus, Millipore), and then capture by subjected to densitometric analysis using Science Lab 2003 software (Fuji, Tokyo, Japan). The primary antibodies were as follows: anti-CARM1 (rabbit, 1:1000, Millipore, Billerica, Massachusetts), anti-PRMT6 (rabbit, 1:1000, Cell Signaling, Beverly, MA, USA), anti-ub (rabbit, 1:1000, Millipore, Billerica, Massachusetts), anti-SKP2 (rabbit, 1:1000, Cell Signaling, Beverly, MA, USA), anti-Fbxo3 (rabbit, 1:1000, Abcam, Cambridge, USA), anti-Kv1.4 (rabbit, 1:1000, Abcam, Cambridge, USA), anti-Kv4.2 (rabbit, 1:1000, Abcam, Cambridge, USA, anti-H3R17me2 (rabbit, 1:1000, Abcam, Cambridge, USA), anti-H3 (rabbit, 1:1000, Abcam, Cambridge, USA) and anti-GAPDH (mouse, 1:2000, Santa Cruz Biotechnology, Santa Cruz, CA, USA). The secondary antibodies were as follows: goat anti-rabbit IgG (1:8000, Jackson ImmunoResearch, West Grove, PA, USA) and goat anti-mouse IgG (1:8000, Jackson ImmunoResearch) antibodies.
Co-precipitation Studies
Briefly, extractions of dorsal horn samples were incubated with a rabbit polyclonal antibody against CARM1 (rabbit, 1:1000, Genetex, Irvine, CA, USA) overnight at 4 °C. At 1:1 slurry protein agarose suspension (Millipore) was added to the protein immunocomplex, and the mixture was incubated at 4 °C for 2–3 h. Agarose beads were washed once with 1% (v/v) Triton X-100 in the immunoprecipitation buffer (50 mM Tris-HCl, pH 7.4, 5 mM EDTA, 0.02% (w/v) sodium azide), twice with 1% (v/v) Triton X-100 in immunoprecipitation buffer plus 300 mM NaCl, and three times with only immunoprecipitation buffer. The bound proteins were eluted in SDS-PAGE sample buffer at 95 °C. The proteins were then separated on SDS-PAGE, electrophoretically transferred to polyvinylidene difluoride membranes and detected using or mouse anti-ubiquitin antibody (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA).
Immunofluorescence Analysis
Rats were deeply anesthetized and then transcardially perfused with PBS followed by 4% paraformaldehyde in PBS (pH 7.4). The L4–5 segments of the spinal cord were removed and postfixed for 4 h at 4 °C and then dehydrated in gradient sucrose at 4 °C. Transverse spinal cord sections (30 μm) were cut on a cryostat and mounted on glass slides. The sections were blocked with PBS containing 5% BSA and 0.1% Triton X-100 for 1 h at room temperature. Subsequently, the sections were incubated with rabbit anti-CARM1 (1:200, Genetex, Irvine, CA, USA), together with mouse monoclonal anti-neuronal nuclear antigen (NeuN, a neuronal marker, 1:500, Millipore, Billerica, Massachusetts), mouse anti-glial fibrillary acidic protein (GFAP, an astrocyte marker; 1:1000, Millipore, Billerica, Massachusetts), or mouse anti-integrin αM (OX-42, a microglial marker; 1:500, Santa Cruz Biotechnology, Santa Cruz, CA, USA), overnight at 4 °C. The sections were then incubated with mixture of Alexa Fluor 488- or/594-conjugated secondary antibodies (1:1500, Invitrogen, USA) for 1 h at room temperature, or DAPI (1:1500, Invitrogen, USA) for 2 min at room temperature. When examining the interaction between CARM1 (1:400, Genetex, Irvine, CA, USA), H3R17me2 (rabbit, 1:1000, Cell Signaling, Beverly, MA, USA) and Kv1.4 or Kv4.2 (rabbit, 1:400, Abcam, Cambridge, USA), the specific antibodies were mixed with 10X reaction buffer (Mix-n-Stain, Biotium, Hayward, CA, USA) at a ratio of 1:10. Then, the solution was transferred to a vial containing dye (CF, Biotium) and incubated in the dark (30 min, room temperature). Then, the sample sections were sequentially incubated (overnight, 4 °C) with diluted solutions, and washed 5 times between each incubation. The sections were subsequently rinsed in PBS, and coverslips were applied. After excitation, the fluorescent markers were easily detected using a camera-coupled device (X-plorer; Diagnostic Instruments, Inc., USA) using fluorescence microscopy (LEICA DM2500, Germany).
Chromatin Immunoprecipitation-qPCR
Chromatin immunoprecipitation (ChIP) was performed using a ChIP Kit (Millipore, Billerica, Massachusetts) according to a modified protocol from the manufacturer. Dissected dorsal horn (L4–5) samples were cut into small pieces (1–2 mm3) using razor blades, treated with fresh 1% paraformaldehyde in PBS buffer, and gently agitated for 10 min at room temperature to crosslink proteins to DNA. Subsequently, the tissues were washed and re-suspended in lysis buffer, and the lysates were sheared by sonication to generate chromatin fragments with an average length of 200–1000 bp. One percent of the sonicated chromatin was saved as an input control for qPCR. The chromatin was then immunoprecipitated for 2 h at room temperature with anti-CARM1 (rabbit, 1:1000, Millipore, Billerica, Massachusetts), anti-H3R17me2 (rabbit, 1:1000, Genetex, Irvine, CA, USA) or an equivalent amount of control IgG. The protein-DNA immunocomplexes were precipitated overnight using protein G magnetic beads at 4 °C. After the beads were washed, they were re-suspended in ChIP elution buffer, incubated with proteinase K at 62 °C for 2 h, and then incubated at 95 °C for 10 min to reverse the protein-DNA crosslinks. ChIP signals were quantified via a SYBR Green method quantitative PCR analysis on a QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific) or a 7500 Real-Time PCR System (Applied Biosystems, Carlsbad, CA). The specific primer pairs for the Kcna4 and Kcnd2 promoter region are described below.
-
Kcna4: 5′-GAAGCAGCACCAAGCTATCC-3′ and 5′- CCCACGACTGCTTAGCTTCT-3′.
-
Kcnd2: 5′- CTGGCTTTTGGGAAGGTGAC-3′ and 5′-CATACCTGGCAAAGTACGCG-3′.
Quantitative PCR
Animals were deeply anesthetized with isoflurane and the dorsal horn (L4–5) tissues were rapidly removed and completely submerged in a sufficient volume of RNAlater solution (Ambion, Thermo Fisher Scientific, Waltham, USA) overnight at 4 °C to allow thorough penetration of the tissue and then transferred to 80 °C. Total RNA was isolated under RNase-free conditions. Total RNA was extracted from the dorsal horn using using RNA isolation kits (74,106; Qiagen, Valencia, CA, USA) the TRIzol-chloroform total RNA extraction system and treated with DNase I (Invitrogen, Carlsbad, CA). cDNA was prepared by using the Superscript III first-strand synthesis kit and treated by RNase H (Invitrogen, Carlsbad, CA). Reverse transcription was performed using complementary DNA reverse transcription kits (205,311; Qiagen, Valenica, CA). Real-time PCR was performed on a QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific) or 7500 Real-Time PCR system (Applied Biosystems, Carlsbad, CA). TaqMan Universal PCR Master Mix (2X) and TaqMan gene expression assay probes for target genes were GAPDH (Rn99999916_s1, Applied Biosystems, Carlsbad, CA), Kv1.4 (Rn02532059_s1, IDT, Coralville, IA, USA), and Kv4.2 (Rn00581941_m1, IDT, Coralville, IA, USA). Reactions (total volume, 20 ul) were performed by incubating at 95 °C for 20 s, followed by 40 cycles of 1 s at 95 °C and 20 s at 60 °C. Relative mRNA levels were calculated according to the 2 −△△CT method [37]. All CT values were normalized to GAPDH.
Intrathecal Catheter Implantation
The protocol for implantation of intrathecal cannulae was as described in our previous study [32]. To surgically implant an intrathecal catheter, we anesthetized rats with isoflurane (induction, 5%; maintenance, 2% in air) and implanted PE-10 catheter in the dorsal aspect of lumbar enlargement of the spinal cord of rats. Once the catheter was in place, the outer part of the catheter was plugged and immobilized onto the skin on closure of the wound. After 3 days of recovery, animal with any sign of neurological deficits were discarded and excluded from further experiments.
Small Interfering RNA
The siRNA targeting CARM1 (5′-AAUAUGUGGAAUACGGGAA-3′) and the universal negative control siRNA (missense siRNA, 5′-UGAUAUUACCCUGAAUAUG-3′) were purchased from Dharmacon (Lafayette, CO, USA). On the day of injection, siRNA was mixed with the transfection reagent iFect (Neuromics, Edina, MN) to a final concentration (100 ng, 10 μL) according to the protocol for intrathecal injection, were administered intrathecally for once daily for 4 consecutive days (daily from days 0 to 3 of naïve rat). Twenty-four hours after the last injection (day 4 after injection), the dorsal horn (L4–5) samples were harvested and used for qPCR, ChIP-qPCR, and Western blot analyses. Behavioral studies were tested every day until sample harvested (form day 0 to day 4).
Spinal Slice Preparations and Electrophysiological Recordings
Under anesthesia with isoflurane (5% for induction, 2% for maintenance in oxygen), the rat lumbar spinal cord was quickly removed and placed in cutting buffer bubbled with 95% O2/5% CO2. The cutting buffer consisted of the following (in mm): 234 sucrose, 3.6 KCl, 1.2 MgCl2, 2.5 CaCl2, 1.2 NaH2PO4, 12 glucose, and 25 NaHCO3. Transverse spinal cord slices (400 μm) were cut at the L5–L6 level by vibratome (DTK1000, Dosaka). After dissection, slices were equilibrated in artificial CSF (aCSF) at room temperature for ≥ 1 h before recording. The aCSF consisted of the following (in mm): 117 NaCl, 4.5 KCl, 2.5 CaCl2, 1.2 MgCl2, 1.2 NaH2PO4, 25 NaHCO3, and 11.4 dextrose bubbled with 95% O2/5% CO2, pH 7.4. The Kv currents were recorded in the spinal dorsal horn neurons. The lamina II area of the dorsal horn was identified by a translucent band in the superficial dorsal horn on an upright fixed-stage IR-DIC microscope (BX51WI, Olympus, Tokyo, Japan). Spinal lamina I and outer lamina II were selected for recordings, as previously described [38]. Glass pipettes (resistance, 6–8 MΩ) were pulled and filled with an internal solution containing the following (in mm): 125 K+ gluconate, 5 KCl, 5 BAPTA, 0.5 CaCl2, 10 HEPES, 5 MgATP, and 0.33 GTP-Tris, pH 7.3, 280 mOsm/L. All electrophysiological signals were acquired using an Axon setup (Molecular Devices). The whole-cell Kv current was recorded by using a series of depolarizing voltages from − 70 to 60 mV (400-ms pulse duration) in 10-mV increments at 2-s intervals. Signals were sampled by pCLAMP 9.2 via an amplifier (Axopatch 200B) and an analog-to-digital converter (Digidata 1322A), filtered at 2–5 kHz, digitized at 10 kHz, and stored for off-line analysis.
Drugs and Drug Administration
The TP 064 (CARM1 inhibitor; 10, 30, 300 nM, 10 μL; Tocris Bioscience, Bristol, UK) and the vehicle solution were intrathecal administered by single bolus injection in naïve rats. For the time course experiment, we measured the paw withdrawal threshold at every 1 h interval until 6 h after intrathecal injection. Four hours after the injection, the dorsal horn (L4–5) samples were harvested and used for Western blot analyses, ChIP-qPCR, and electrophysiological recordings. The BC-1215 (a novel inhibitor of Fbxo3 activity; 100 nM, 10 μL, Merck) and the vehicle solution were intrathecal administered by single bolus injection in SNL rats. Two or 4 h after the injection the dorsal horn (L4–5) samples were harvested and used for qPCR. Four hours after the injection the dorsal horn (L4–5) samples were harvested and used for Western blot analyses, IP, ChIP-qPCR, electrophysiological recordings and behavioral tests.
Statistical Analysis
All the data in this study were analyzed using Sigma Plot 10.0 (Systat Software) or Prism 6.0 (GraphPad) and are expressed as the mean ± S.E.M. Statistical comparison was performed by one-way ANOVA (using Bonferroni correction for post hoc analysis), two-way ANOVA (using Bonferroni correction for post hoc analysis), or unpaired Student’s t test where appropriate. Significance was set at p < 0.05.
Results
SNL Decreases Spinal CARM1 Expression
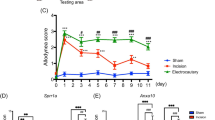
To examine the role of spinal CARM1 in the development of neuropathic pain, we used SD rats with spinal nerve ligation (SNL), an animal model mimicking neuropathic injury [35, 36]. Our SNL procedure successfully induced tactile allodynia, as evidenced by decrements in the withdrawal threshold of the ipsilateral hind paw of rats on days 3, 7, 14, and 21 after operation (Fig. 1b). SNL decreased the expression of CARM1 in the ipsilateral dorsal horn on days 3, 7, 14, and 21 after operation with a time course that matched the SNL-induced allodynia (Fig. 1c), indicating that spinal CARM1 protein expression was inversely correlated with SNL-induced allodynia. Further analysis revealed that SNL decreased the expression of CARM1 in the ipsilateral dorsal horn at day 7 in both gender, and there was no significant difference in CARM1 protein expression in either gender, suggesting that there was no gender difference in this region after SNL (Fig. 1d). To determine the cellular distribution of CARM1 in the dorsal horn, we next labeled spinal cord slices dissected at day 7 after operation (time-point SNL rats displayed maximal behavioral allodynia and lowest spinal CARM1 protein expression) using specific antibodies. SNL decreased CARM1 immunofluorescence in the ipsilateral dorsal horn on day 7 after operation (Fig. 1e), and double-labeling revealed that CARM1 immunofluorescence colocalized with neuronal, but not microglial or astrocyte, markers (Fig. 1e). Collectively, these results indicated that SNL provokes behavioral allodynia, accompanied by decreased CARM1 expression selectively in ipsilateral dorsal horn neurons.

SNL downregulates CARM1 expression in the dorsal horn accompanied by behavioral allodynia. (a) Diagram of the timeline of this experiment. (b) The time course of SNL-decreased the ipsilateral paw withdrawal threshold after operation. Sham, sham operation group. SNL, spinal nerve ligation. IPSI, ipsilateral. CONTRA, contralateral. **p < 0.01 versus Sham IPSI. #p < 0.05, ##p < 0.01 versus SNL IPSI day − 1. Each group has 6 rats. Two-way ANOVA, group, F(3, 20) = 63.74, p < 0.0001; time, F(4, 80) = 2.986, p = 0.0237; interaction, F(12, 80) = 2.860, p = 0.0025. (c) Representative Western blot and statistical analyses (normalized to GAPDH) demonstrating the time course of SNL-decreased CARM1 expression in the ipsilateral dorsal horn after operation. IB, Immunoblotting. I, ipsilateral. C, contralateral. **p < 0.01 versus Sham IPSI. ##p < 0.01 versus SNL IPSI day − 1. Each group has 5 rats. Two-way ANOVA, group, F(3, 16) = 14.69, p < 0.0001; time, F(4, 64) = 2.581, p = 0.0455; interaction, F(12, 64) = 6.141, p < 0.0001. (d) Representative Western blot and statistical analyses (normalized to GAPDH) demonstrating SNL-decreased CARM1 expression in the ipsilateral dorsal horn at day 7 after operation in male and female rats. **p < 0.01 versus male IPSI − 1 or female IPSI − 1. Each group has 4 rats. Unpaired t tests. (e) In the ipsilateral dorsal horn, SNL (SNL 7D) decreased CARM1 immunofluorescence (red, 2, 5), which colocalized with NeuN (green, 7, a neuronal marker) not OX-42 (green, 8, a microglial marker) or GFAP (green, 9, an astrocytic marker) at day 7 after operation. Scale bar = 50 μm. Thickness = 30 μm. **p < 0.01 versus Sham IPSI. ##p < 0.01 versus SNL IPSI day − 1. Each group has 6 rats. Unpaired t tests
Knockdown of Spinal CARM1 Expression Provokes Tactile Allodynia
Based on the above observations, we next used siRNA targeting the CARM1 gene to validate the role of spinal CARM1 in neuropathic pain. CARM1 siRNA (100 ng; 10 μL; once daily for 4 days) reduced CARM1 expression but not PRMT6 expression in the dorsal horn of naïve rats at day 4 after injection (Fig. 2b, c). Considering that PRMT6 has similar sequences of CARM1 in the PRMT type I family, we confirmed the efficacy and specificity of our knockdown protocols. The naïve and CARM1 siRNA-treated groups exhibited no significant differences in motor performance, as measured by the rota-rod test (Fig. 2d), indicating that neither our procedures nor CARM1 siRNA itself led to motor deficits in rats. Intriguingly, CARM1 siRNA provoked allodynia in naïve animals (Fig. 2e). These data provide a genetic basis to support the notion that downregulated CARM1 expression is an essential factor for the spinal machinery underlying the development of tactile allodynia.

Focal knockdown of spinal CARM1 expression elicits no motor deficits but provokes behavioral allodynia. (a) Diagram of the timeline of this experiment. (b) Representative Western blot and statistical analysis (normalized to GAPDH) demonstrating that intrathecal administration of CARM1 siRNA (CARM1 siRNA; 100 ng; 10 μL; once daily for 4 days) but not missense siRNA (MS siRNA, 100 ng, 10 μL) or polyethylenimine (PEI, a transfection reagent, 10 μL) led to decrease in CARM1 levels in the dorsal horn of naïve rats at day 4 after injection. IB, Immunoblotting. **p < 0.01 versus Naïve. ##p < 0.01 versus MS siRNA. Each group has 5 rats. One-way ANOVA, F(3, 16) = 15.35, p < 0.0001. (c) Representative Western blot and statistical analysis (normalized to GAPDH) demonstrating that intrathecal administration of CARM1 siRNA (100 ng; 10 μL; once daily for 4 days) did not decrease in PRMT6 levels in the dorsal horn of naïve rats at day 4 after injection. Each group has 5 rats. One-way ANOVA, F(3, 16) = 0.2426, p = 0.8654. (d) Rota-rod test demonstrating no statistical difference in the motor performance among Naïve, PEI, MS siRNA, and CAM1 siRNA groups (100 ng; 10 μL; once daily for 4 days). The gray bar at the bottom indicates the duration of the reagents’ intrathecal administration. Each group has 6 rats. Two-way ANOVA, group, F(3, 20) = 1.842, p = 0.1720; time, F(4, 80) = 1.222, p = 0.3082; interaction, F(12, 80) = 0.9595, p = 0.4939. (e) Results of the von Frey test demonstrating that administering naïve rats with CARM1 siRNA (100 ng; 10 μL; once daily for 4 days) decreased the withdrawal threshold of the hind paw at days 1, 2, 3, and 4 after the injection. The gray bar at the bottom indicates the duration of the reagents’ intrathecal administration. *p < 0.05, **p < 0.01 versus Naïve. ##p < 0.01 versus MS siRNA. Each group has 6 rats. Two-way ANOVA, group, F(3, 20) = 32.47, p < 0.0001; time, F(4, 80) = 5.394, p = 0.0007; interaction, F(12, 80) = 4.405, p < 0.0001
SNL Facilitates Spinal Fbxo3-Dependent CARM1 Ubiquitination
CARM1 downregulation by ubiquitination-dependent CARM1 degradation is considered a critical factor in the development of diabetic nephropathy [39]. A study has shown that the stability of CARM1 is regulated by the F-box superfamily of E3 proteins, including S-phase kinase-associated protein 2 (SKP2; also known as Fbxl1) [40]. Considering that our laboratory has linked spinal Fbxo3 (a member of the F-box family)-dependent ubiquitination to neuropathic pain pathology [31, 32], we further speculated that spinal CARM1 downregulation contributes to neuropathic allodynia by F-box family-dependent CARM1 ubiquitination. SNL predictably increased CARM1 ubiquitination in the ipsilateral, not contralateral, dorsal horn on day 7 after operation (Fig. 3b). Considering that the observed CARM1 ubiquitination changes reflect only the ipsilateral side, subsequent examinations were conducted in the ipsilateral side. SNL increased the abundance of Fbxo3 in the ipsilateral dorsal horn on day 7 after operation (Fig. 3c). Unpredictably, SKP2 expression in the ipsilateral dorsal horn was not affected on day 7 after SNL (Fig. 3c). The results of the von Frey test demonstrated that administering BC-1215 (a novel activity inhibitor of Fbxo3; 100 nM, 10 μL) to SNL rats at 4 h after injection reversed SNL-induced behavioral allodynia on day 7 after operation (Fig. 3d). Interestingly, administering BC-1215 to SNL rats at 4 h after injection reversed not only SNL-enhanced CARM1 ubiquitination but also SNL-decreased CARM1 expression in the ipsilateral dorsal horn on day 7 after operation (Fig. 3e, f). In addition, we observed that administering CARM1 siRNA (100 ng; 10 μL; once daily for 4 days) to naïve rats did not affect the abundance of Fbxo3 in the dorsal horn on day 4 after injection (Fig. 3g), indicating that Fbxo3 is upstream of CARM1. Collectively, these results suggest that SNL-decreased CARM1 expression participates in the development of neuropathic pain by Fbxo3-dependent CARM1 ubiquitination in the dorsal horn.

SNL induces spinal Fbxo3-dependent CARM1 ubiquitination to induce behavioral allodynia. (a) Diagram of the timeline of this experiment. (b) SNL increased CARM1 ubiquitination in the ipsilateral, not contralateral, dorsal horn at day 7 after the operation (SNL 7D). I, ipsilateral. C, contralateral. IB, immunoblotting. Sham 7D, sham operation at day 7. SNL 7D, SNL operation at day 7. **p < 0.01 versus Sham 7D IPSI. ##p < 0.01 versus SNL 7D CONTRA. Each group has 5 rats. Unpaired t tests. (c) Representative Western blot and statistical analyses (normalized to GAPDH) demonstrating SNL increased Fbxo3, not SKP2, expression in the ipsilateral dorsal horn on day 7 after operation. **p < 0.01 versus Sham 7D. Each group has 5 rats. Unpaired t tests. (d)–(f) Intrathecal BC-1215 (100 nM, 10 μL, at 4 h after injection) into SNL rats significantly increased the ipsilateral paw withdrawal threshold in accompanied with a decreased ubiquitination and increased expression of CARM1 protein in the ipsilateral dorsal horn. *p < 0.05, **p < 0.01, versus SNL 7D. ##p < 0.01 versus SNL 7D+Veh. Each group has 6 (d), 5 (e), or 4 (f) rats. (d) One-way ANOVA, F(2, 15) = 21.89, p < 0.0001. (e) One-way ANOVA, F(2, 12) = 24.68, p < 0.0001. (f) One-way ANOVA, F(2, 9) = 9.971, p = 0.0052. (g) Intrathecal injection of CARM1 siRNA (CARM1 siRNA; 100 ng; 10 μL; once daily for 4 days) into naïve rats did not affect the expression levels of Fbxo3 protein in the dorsal horn at day 4 after injection. Each group has 5 rats. One-way ANOVA, F(2, 12) = 1.601, p = 0.2419
SNL-Enhanced Spinal Fbxo3/CARM1 Signaling Modifies Epigenetic Silencing of K+ Channel Genes
CARM1 is a well-known histone epigenetic modification marker for transcriptional activation [6, 7]. Notably, histone epigenetic process-associated impairment of K+ channel genes (Kcna4 and Kcnd2) is observed in neuropathic pain [28]. Moreover, regulation of K+ channels is mediated by ubiquitination in the nervous system [41]. Considering that attenuation of the histone epigenetic coactivator CARM1 decreased gene transcription, we hypothesized that spinal Fbxo3/CARM1 signaling contributes to neuropathic pain hypersensitivity by decreasing the transcription of Kcna4 and Kcnd2. Supporting our hypothesis, RT-PCR revealed that SNL decreased the mRNA expression of Kcna4 and Kcnd2 in the ipsilateral dorsal horn, which was markedly inhibited by BC-1215 (100 nM, 10 μL) at 4 h after injection (Fig. 4b). Further analysis revealed that administering BC-1215 (100 nM, 10 μL) to SNL rats at 2 h after injection also reversed SNL decreased the mRNA expression of Kcna4 not Kcnd2 in the ipsilateral dorsal horn on day 7 after operation (Fig. 4c). Western blot analyses revealed that SNL also decreased the protein expression of Kv1.4 (encoded by Kcna4) and Kv4.2 (encoded by Kcnd2) in the ipsilateral dorsal horn, which were markedly inhibited by BC-1215 (100 nM, 10 μL) at 4 h after injection (Fig. 4d). Moreover, ChIP-qPCR assay revealed that SNL decreased the amounts of CARM1 antibody-recognized Kcna4 and Kcnd2 promoter fragments in the ipsilateral dorsal horn samples; these effects were reversed by BC-1215 (100 nM, 10 μL) at 4 h after injection (Fig. 4e). Finally, we performed electrophysiological experiments to determine whether Fbxo3 inhibition could restore the Kv currents reduced by SNL in dorsal horn neurons. We conducted whole-cell patch-clamp Kv current recordings in acutely dissociated ipsilateral dorsal horn neurons from Sham 7D rats, SNL 7D rats, and SNL 7D rats administered intrathecal BC-1215 (100 nM, 10 μL) at 4 h after injection. The peak amplitudes of potassium currents in the ipsilateral spinal dorsal horn neurons were significantly lower in SNL 7D rats than in Sham 7D rats (Fig. 4f). Intrathecal treatment with BC-1215 significantly restored the potassium currents of the ipsilateral spinal dorsal horn neurons in SNL rats (Fig. 4f). Collectively, these data indicate that Fbxo3/CARM1 ubiquitination signaling causes epigenetic silencing of the Kcna4 and Kcnd2 genes in the dorsal horn, leading to the development of neuropathic pain.

SNL induces Fbxo3-dependent CARM1 ubiquitination to decrease K+ channel expression in the dorsal horn. (a) Diagram of the timeline of this experiment. (b) Intrathecal BC-1215 (100 nM, 10 μL, at 4 h after injection) reversed SNL-decreased expression levels of Kcna4 and Kcnd2 mRNA in the ipsilateral dorsal horn. **p < 0.01 versus Sham 7D. ##p < 0.01 versus SNL 7D. ++p < 0.01 versus SNL 7D. Each group has 5 rats. Kcna4, one-way ANOVA, F(3, 16) = 20.95, p < 0.0001. Kcnd2, one-way ANOVA, F(3, 16) = 36.06, p < 0.0001. (c) Intrathecal BC-1215 (100 nM, 10 μL, at 2 h after injection) reversed SNL-decreased expression levels of Kcna4, not Kcnd2, mRNA in the ipsilateral dorsal horn. *p < 0.05, **p < 0.01 versus SNL 7D+Veh 4 h. Each group has 5 rats. Kcna4, one-way ANOVA, F(2, 12) = 8.808, p = 0.0044. Kcnd2, one-way ANOVA, F(2, 12) = 5.412, p = 0.0211. (d) Intrathecal BC-1215 (100 nM, 10 μL, at 4 h after injection) reversed SNL-decreased expression levels of Kv1.4 and Kv4.2 in the ipsilateral dorsal horn. **p < 0.01 versus Sham 7D. #p < 0.05, ##p < 0.01 versus SNL 7D. ++p < 0.01 versus SNL 7D+Veh. Each group has 5 rats. Kv1.4, one-way ANOVA, F(3, 16) = 16.73, p < 0.0001. Kv4.2, one-way ANOVA, F(3, 16) = 14.87, p < 0.0001. (e) Intrathecal BC-1215 (100 nM, 10 μL, at 4 h after injection) reversed SNL-decreased abundance of Kcna4 and Kcnd2 promoter fragments immunoprecipitated by CARM1-specific antibodies in the ipsilateral dorsal horn. **p < 0.01 versus Sham 7D. #p < 0.05, ##p < 0.01 versus SNL 7D. ++p < 0.01 versus SNL 7D+Veh. Each group has 5 rats. Kv1.4, one-way ANOVA, F(3, 16) = 26.59, p < 0.0001. Kv4.2, one-way ANOVA, F(3, 16) = 42.97, p < 0.0001. (f) Representative traces of K+ currents recorded from dorsal horn neurons dissected 7 days after Sham (Sham 7D), SNL operation (SNL 7D) and SNL operation with BC-1215 (SNL 7D + BC-1215, 100 nM, 10 μL, at 4 h after injection). **p < 0.01 versus Sham 7D. #p < 0.05, ##p < 0.01 versus SNL 7D. Each group has 7–8 rats. Two-way ANOVA, group, F(2, 19) = 3.919, p = 0.0376; time, F(15, 285) = 198.1, p < 0.0001; interaction, F(30, 285) = 5.086, p < 0.0001
Focal Knockdown of CARM1 Diminishes Spinal CARM1-Activated K+ Channel Genes to Induce Allodynia
To directly determine whether diminished CARM1-dependent activation of K+ channel genes in the dorsal horn induces tactile allodynia, we used the CARM1 siRNA approach to validate our findings. Intriguingly, administering CARM1 siRNA (100 ng; 10 μL; once daily for 4 days) to naïve rats decreased the mRNA and protein expression levels of Kcna4 (Kv1.4) and Kcnd2 (Kv4.2) in the dorsal horn on day 4 after injection (Fig. 5b, c). Furthermore, ChIP-qPCR demonstrated that administering CARM1 siRNA (100 ng; 10 μL; once daily for 4 days) to naïve rats significantly decreased CARM1 antibody-recognized Kcna4 and Kcnd2 promoter fragments in the dorsal horn samples on day 4 after injection (Fig. 5d). Together, these findings suggest that CARM1 has an important function in the transcription of K+ channel genes in the dorsal horn and that impeding spinal CARM1-dependent activation of Kcna4 and Kcnd2 transcription can affect neuropathic pain.

CARM1 knockdown impeded CARM1 coupling at K+ gene to decrease dorsal horn K+ channel expression. (a) Diagram of the timeline of this experiment. (b) Intrathecal CARM1 siRNA (CARM1 siRNA; 100 ng; 10 μL; once daily for 4 days) into naïve rats decreased the expression levels of Kcna4 and Kcnd2 mRNA in the dorsal horn at day 4 after injection. IB, Immunoblotting. **p < 0.01 versus Naïve. ##p < 0.01 versus MS siRNA. Each group has 5 rats. Kcna4, one-way ANOVA, F(2, 12) = 21.3, p = 0.0001. Kcnd2, one-way ANOVA, F(2, 12) = 17.04, p = 0.0003. (c) Intrathecal CARM1 siRNA (CARM1 siRNA; 100 ng; 10 μL; once daily for 4 days) into naïve rats decreased the expression levels of Kv1.4 and Kv4.2 proteins in the dorsal horn at day 4 after injection. IB, Immunoblotting. **p < 0.01 versus Sham 7D. #p < 0.05, ##p < 0.01 versus MS siRNA. Each group has 5 rats. Kv1.4, one-way ANOVA, F(2, 12) = 8.698, p = 0.0046. Kv4.2, one-way ANOVA, F(2, 12) = 10.28, p = 0.0025. (d) Intrathecal CARM1 siRNA (CARM1 siRNA; 100 ng; 10 μL; once daily for 4 days) into naïve rats decreased abundances of Kcna4 and Kcnd2 promoter fragments immunoprecipitated by CARM1-specific antibodies in the dorsal horn at day 4 after injection. **p < 0.01 versus Naïve. #p < 0.05, ##p < 0.01 versus MS siRNA. Each group has 5 rats. Kcna4, one-way ANOVA, F(2, 12) = 23.13, p < 0.0001. Kcnd2, one-way ANOVA, F(2, 12) = 7.398, p = 0.0081
SNL-Enhanced Fbxo3/CARM1 Signaling Modifies the Epigenetic Silencing of Spinal K+ Channel Genes by Decreasing H3R17me2 at the Promoter
CARM1, which belongs to the protein arginine methyltransferase family, has been demonstrated to mainly catalyze H3R17me2 for epigenetic transcriptional activation [6, 7]. Therefore, we next determined whether SNL-enhanced Fbxo3/CARM1 signaling affects H3R17me2 at the Kcna4 and Kcnd2 promoters to mediate neuropathic allodynia development. Consistent with behavioral allodynia, decreased CARM1 expression, increased Fbxo3 expression and enhanced CARM1 ubiquitination in the ipsilateral dorsal horn were observed on day 7 after operation; SNL indeed decreased the levels of H3R17me2 in the ipsilateral dorsal horn on day 7 after operation (Fig. 6b), and this effect was blunted by administering BC-1215 (100 nM, 10 μL, at 4 h) to SNL rats. Intrathecal administration of CARM1 siRNA (100 ng; 10 μL; once daily for 4 days) to naïve rats also decreased the expression levels of H3R17me2 in the dorsal horn on day 4 after injection (Fig. 6c). Moreover, ChIP-qPCR demonstrated that SNL decreased the amounts of H3R17me2 antibody-recognized Kcna4 and Kcnd2 promoter fragments in the ipsilateral dorsal horn samples; these effects were reversed by treating the SNL rats with BC-1215 (100 nM, 10 μL, at 4 h; Fig. 6d). In addition, intrathecal administration of CARM1 siRNA (100 ng; 10 μL; once daily for 4 days) to naïve rats markedly decreased the amounts of H3R17me2 antibody-recognized Kcna4 and Kcnd2 promoter fragments in the dorsal horn samples on day 4 after injection (Fig. 6e). Furthermore, the images of spinal slices dissected on postoperative day 7 revealed SNL-decreased CARM1-positive, H3R17me2-positive, Kv1.4-positive, Kv4.2-positive, CARM1/H3R17me2/Kv1.4 triple-labeled puncta and CARM1/H3R17me2/Kv4.2 triple-labeled puncta in the ipsilateral dorsal horn, which were markedly reversed by BC-1215 administration (100 nM, 10 μL, at 4 h; Fig. 7b–d). Thus, these data indicate that the SNL-induced downregulation of the transcription of Kcna4 and Kcnd2 in the dorsal horn is associated with a decrease in the enrichment of CARM1-dependent H3R17me2 by Fbxo3/CARM1 signaling.

SNL induces Fbxo3/CARM1 signaling-mediated epigenetic silencing H3R17me2 at K+ channel genes promoter in dorsal horn. (a) Diagram of the timeline of this experiment. (b) BC-1215 (100 nM, 10 μL, at 4 h after injection) reversed SNL-decreased expression levels of H3R17me2 in the ipsilateral dorsal horn. IB, Immunoblotting. **p < 0.01 versus Sham 7D. #p < 0.05 versus SNL 7D. +p < 0.05 versus SNL 7D+Veh. Each group has 5 rats. One-way ANOVA, F(3, 16) = 11.26, p = 0.0003. (c) Intrathecal CARM1 siRNA (CARM1 siRNA; 100 ng; 10 μL; once daily for 4 days) decreased the level of H3R17me2 in the ipsilateral dorsal horn at day 4 after injection. *p < 0.05 versus Naïve. ##p < 0.01 versus MS siRNA. Each group has 5 rats. One-way ANOVA, F(2, 12) = 8.306, p = 0.0054. (d) Intrathecal BC-1215 (100 nM, 10 μL, at 4 h after injection) reversed SNL-decreased the Kcna4 and Kcnd2 promoter fragments immunoprecipitated by H3R17me2-specific antibodies in the ipsilateral dorsal horn of SNL rats. **p < 0.01 versus Sham 7D. #p < 0.05 versus SNL 7D. +p < 0.05 versus SNL 7D+Veh. Each group has 5 rats. Kcna4, one-way ANOVA, F(3, 16) = 19.34, p < 0.0001. Kcnd2, one-way ANOVA, F(3, 16) = 25.5, p < 0.0001. (e) Intrathecal CARM1 siRNA (CARM1 siRNA; 100 ng; 10 μL; once daily for 4 days) decreased the Kcna4 and Kcnd2 promoter fragments immunoprecipitated by H3R17me2-specific antibodies in the dorsal horn of naïve rats at day 4 after injection. *p < 0.01 versus Naïve. #p < 0.05, ##p < 0.01 versus MS siRNA. Each group has 5 rats. Kcna4, one-way ANOVA, F(2, 12) = 8.836, p = 0.0044. Kcnd2, one-way ANOVA, F(2, 12) = 7.645, p = 0.0072

BC-1215 reverses SNL-decreased CARM1, H3R17me2, Kv1.4, Kv4.2, CARM1/H3R17me2/Kv1.4 colocalization and CARM1/H3R17me2/Kv4.2 colocalization in dorsal horn. (a) Diagram of the timeline of this experiment. (b) Immunofluorescence images analysis demonstrating intrathecal BC-1215 (100 nM, 10 μL, at 4 h after injection) reversed SNL (SNL 7D) decreased CARM1-positive (red), H3R17me2-positive (green), Kv1.4-positive (blue) and CARM1/H3R17me2/Kv1.4 triple-labeled (white) immunofluorescence ipsilateral dorsal horn at day 7 after operation. Scale bar = 50 μm. Thickness = 30 μm. (c) Immunofluorescence images analysis demonstrating intrathecal BC-1215 (100 nM, 10 μL, at 4 h after injection) reversed SNL (SNL 7D) decreased CARM1-positive (red), H3R17me2-positive (green), Kv4.2-positive (blue) and CARM1/H3R17me2/Kv4.2 triple-labeled (white) immunofluorescence ipsilateral dorsal horn at day 7 after operation. Scale bar = 50 μm. Thickness = 30 μm. (d) Representative (b) and (c) statistical analyses **p < 0.01 versus Sham 7D. #p < 0.05 ##p < 0.01 versus SNL 7D. Each group has 6 rats. (b) CARM1, one-way ANOVA, F(2, 15) = 54.24, p < 0.0001. (b) H3R17me3, one-way ANOVA, F(2, 15) = 30.4, p < 0.0001. (b) Kv1.4, one-way ANOVA, F(2, 15) = 28.9, p < 0.0001. (b) Merge, one-way ANOVA, F(2, 15) = 11.56, p = 0.0009. (c) CARM1, one-way ANOVA, F(2, 15) = 15.52, p = 0.0002. (c) H3R17me3, one-way ANOVA, F(2, 15) = 21.63, p < 0.0001. (c) Kv4.2, one-way ANOVA, F(2, 15) = 35.1, p < 0.0001. (c) Merge, one-way ANOVA, F(2, 15) = 8.134, p = 0.0041
Inhibition of CARM1-Mediated Histone Methylation at the K+ Channel Gene Promoter in the Dorsal Horn Induces Pain Hypersensitivity
To further provide pharmacological evidence supporting the induction of tactile allodynia by inhibition of spinal CARM1-mediated arginine methyltransferase activity at the K+ channel gene, we next examined the effects of intrathecal injection of TP 064, a selective CARM1 inhibitor, into naïve rats. Compared to the vehicle solution, intrathecal administration of TP 064 (10, 30, and 300 nM, 10 μL) to naïve rats dose-dependently induced behavioral allodynia in the hind paw at 2–6 h after injection (Fig. 8b). Western blot analyses further revealed that intrathecal administration of TP 064 (300 nM, 10 μL) to naïve rats markedly inhibited the expression levels of H3R17me2, Kv1.4 and Kv4.2, but not Fbxo3 or H3, in the dorsal horn at 4 h after injection (Fig. 8c). Notably, intrathecal administration of TP 064 (300 nM, 10 μL) to naïve rats significantly decreased CARM1 and H3R17me2 antibody-recognized Kcna4 and Kcnd2 promoter fragments in the dorsal horn samples at 4 h after injection (Fig. 8d, e), indicating that TP 064 treatment led to the loss of CARM1 and H3R17me2 binding at the Kcna4 and Kcnd2 promoters. In addition, we conducted whole-cell patch-clamp Kv current recordings with dorsal horn neurons from naïve rats and naïve rats acutely treated with TP 064 (300 nM, 10 μL, at 4 h). The peak amplitudes of potassium currents in spinal dorsal horn neurons were significantly lower in the naïve rats treated with TP 064 at 4 h after injection (Fig. 8f). These observations indicate that TP 064 induced allodynia by disrupting CARM1-mediated H3R17me2 at the Kcna4 and Kcnd2 promoters in the dorsal horn.

TP 064 induces allodynia by increasing spinal Fbxo3/CARM1/H3R17me2/ K+ channel signaling. (a) Diagram of the timeline of this experiment. (b) Intrathecal administration of TP 064 (10, 30, and 300 nM, 10 μL) decreased the paw withdrawal threshold of naïve rats. *p < 0.05, **p < 0.01 versus Naïve. #p < 0.05, ##p < 0.01 versus Naïve+Veh. Each group has 6 rats. Two-way ANOVA, group, F(4, 25) = 86.28, p < 0.0001; time, F(6, 150) = 20.8, p < 0.0001; interaction, F(24, 150) = 3.014, p < 0.0001. (c) Intrathecal TP 064 (300 nM, 10 μL, at 4 h after injection) inhibited the expression levels of H3R17me2, Kv1.4 and Kv4.2, but not Fbxo3 and H3 in the dorsal horn. **p < 0.01 versus naïve. #p < 0.05, ##p < 0.01 versus Naïve+Veh. Each group has 5 rats. Fbxo3, one-way ANOVA, F(2, 12) = 0.5578, p = 0.5866. H3R17me2, one-way ANOVA, F(2, 12) = 30.42, p < 0.0001. H3, one-way ANOVA, F(2, 12) = 0.04733, p = 0.9540. Kv1.4, one-way ANOVA, F(2, 12) = 9.952, p = 0.0028. Kv4.2, one-way ANOVA, F(2, 12) = 11.83, p = 0.0014. (d), (e) Intrathecal administration of TP 064 (300 nM, 10 μL, at 4 h after injection) significantly decreased the Kcna4 and Kcnd2 promoter fragments immunoprecipitated by CARM1 and H3R17me2-specific antibodies in the dorsal horn. **p < 0.01 versus naïve. Each group has 5 rats. (d) CARM1, one-way ANOVA, F(2, 12) = 22.47, p < 0.0001. (d) H3R17me2, one-way ANOVA, F(2, 12) = 14.52, p = 0.0006. (e) CARM1, one-way ANOVA, F(2, 12) = 9.195, p = 0.0038. (e) H3R17me2, one-way ANOVA, F(2, 12) = 15.78, p = 0.0006. (f) Representative traces of K+ currents recorded from dorsal horn neurons dissected from naïve (Naïve) and naïve with TP 064 (TP 064, 300 nM, 10 μL, at 4 h after injection) *p < 0.05, **p < 0.01 versus Naïve. Each group has 8 rats. Two-way ANOVA, group, F(1, 14) = 5.371, p = 0.0376; time, F(15, 210) = 64.76, p < 0.0001; interaction, F(15, 210) = 6.525, p < 0.0001
Discussion
Chronic pain is a public health problem that leads to patient debilitation and a decrease in quality of life [1]. Our results reveal that decreased CARM1-mediated H3R17me2 at the K+ channel promoter of dorsal horn neurons underlies the development of neuropathic allodynia. We further characterized that SNL-induced CARM1 downregulation by Fbxo3-related CARM1 ubiquitination, and this signaling modified the epigenetic silencing of K+ channel genes in the dorsal horn. Importantly, we demonstrated that interrupting Fbxo3-mediated CARM1 ubiquitination using BC-1215 is a target for relieving neuropathic allodynia. These results link histone arginine methylation to F-box family-dependent ubiquitination in the dorsal horn, which underlies neuropathic pain, and provide novel insight into the spinal mechanism mediating allodynia. Thus, these findings indicate a possible medical epigenetic and ubiquitination strategy for neuropathic relief by targeting the Fbxo3-CARM1-H3R17me2-K+ channel functional axis in the dorsal horn.
The PRMT-induced arginine methylation of synaptic proteins has attracted interest in recent years [42]. PRMTs catalyze the methylation of arginine residues in proteins, including those of types I, II, and III [43], which are present in neuronal cells [42, 44]. CARM1, also known as PRMT4, is expressed in neurons and functions as a posttranslational coactivator [12, 45]. Notably, transcription of CARM1 is downregulated following the induction of differentiation through neurotrophic signaling in neurons [46]. Pharmacological inhibition of CARM1 activity increased dendritic arborization and spine maturation in hippocampal neurons [11]. Genetic knockdown of CARM1 expression results in precocious dendritic maturation, with increased spine width and density at sites along dendrites and the induction of mushroom-type spines in hippocampal neurons [12]. These works link CARM1 to forms of activity-dependent plasticity in brain regions. Considering that the current study revealed that CARM1 participates in the spinal plasticity underlying the pathophysiology of behavioral allodynia, our findings are supported by works investigating the epigenetic modification of genes underlying pain-related spinal plasticity, which resemble those of learning/memory formation in brain regions [4, 13, 47]. In addition, a proteome-wide approach demonstrated that PRMT8 is involved in synaptic maturation and is potentially an epigenetic modulator of developmental neuroplasticity [48]. PRMT8 has been identified as an important molecule in regulating neuron function and cognition in the mammalian brain because PRMT8 conditional knockout mice exhibit impaired hippocampal-dependent fear learning [49]. Therefore, the possible involvement of other spinal PRMT candidates in neuropathic pain, including PRMT8, needs to be clarified further.
Dysregulation and aberrant expression of PRMTs are associated with various disease states [44]. Endometriosis is associated with infertility and debilitating chronic pain, and downregulation of CARM1 protein expression is detected in ectopic endometrium [50]. Similarly, CARM1 expression in podocytes is diminished in rats with streptozotocin-induced diabetic neuropathy [39]. Consistent with these studies, we observed that SNL-decreased spinal CARM1 expression mediates the maintenance of behavioral allodynia. Nevertheless, a study showed that peripheral nerve injury induced the upregulation of the mRNA and protein expression of CARM1; conversely, a CARM1 inhibitor or knockdown attenuated the induction and maintenance of neuropathic pain [14]. Although the detailed mechanism is unclear, several potential causes may underlie this discrepancy between their results and ours. First, in contrast to the current study, which investigated the histone arginine methylation pattern in L4–5 spinal dorsal horn, their study focused on L4 DRG. Because a study has shown that pain-associated protein expression varies in different levels of DRG after spared nerve injury-induced neuropathic pain model [51]; and compelling evidence studies has indicated the sensory impulses of the DRG, which project onto the spinal cord, are required for neuropathic pain [52]. These evidence indicated that protein expression varies in different levels of DRG could results distinct protein changes in the center nerve system; and therefore underlies the discrepancy in CARM1 expression. Moreover, studies have demonstrated inconsistent changes in protein expression in response to neuropathic insults between the DRG and spinal cord [28, 53]. Furthermore, their study used unilateral or bilateral L4 SNL whereas our study used unilateral L5–6 SNL, if nerve injury of multiple and unique segments accounts for the discrepancies between our study and the above mentioned study. Finally, it worth notice that the animals used in these study are different, i.e., the above mentioned study used C57BL/6 J mice, whereas our study used SD rats. It is well recognized pain-associated modification of molecules in the nervous system differs among animal strains and species [54], indicating strains/species difference might explain the discrepancies between these studies. Nevertheless, the specific mechanisms underlying these discrepancies require further investigation.
On the other hand, CARM1 is thought to be a coactivator of inflammatory mediators such as NF-κB [55] and has been implicated in the loss of spinal motor neurons in spinal muscular atrophy [45]. It is worth noting that inflammation confers dual effects on nociceptive processing [56], and inflammatory mediators have a role in pain and analgesia [57]. Moreover, some results have reported that CARM1 methyltransferase activity is dispensable for the expression of a subset of NF-κB target genes [55, 58], indicating that CARM1 has a methyltransferase-independent role in NF-κB regulation. Furthermore, CARM1 is best known as a transcriptional regulator through its methylation of histones and transcription factors/co-regulators but has also emerged as a factor that influences posttranscriptional processes, including alternative pre-mRNA splicing and mRNA stability. Sanchez et al. have reported that CARM1 is abnormally upregulated in spinal muscular atrophy, leading to promoting nonsense-mediated mRNA decay through a mechanism that is independent of its methyltransferase activity [45]. Considering our results and the above literature, we suggest that CARM1 negatively regulates synaptic gene expression by methyltransferase activity. Further investigations will be required to determine the underlying mechanism of CARM1 in neuropathic pain that is independent of its methyltransferase activity.
Notably, selective CARM1 activators are available for research use [59]. The current study revealed that decreases in spinal CARM1 provokes SNL-induced behavioral allodynia; conversely, these effects were induced by intrathecally administering CARM1 siRNA or TP 064 to naïve rats. Thus, we suggest that pharmaceutical enhancement of CARM1 activity would reverse mechanical allodynia after SNL. However, selective CARM1 activators have been identified in vitro [59]. Therefore, the ability of selective CARM1 activators to enhance the catalytic activity of CARM1 in SNL rats need to be clarified further.
Evidence is starting to emerge in support of the involvement of epigenetic mechanisms, such as DNA methylation, histone modification and noncoding RNA, at multiple loci relevant to pain processing [1]. Our previous studies have demonstrated that DNA demethylation, histone acetylation and histone monoubiquitination control pain-related gene expression in the dorsal horn under chronic pain conditions [2, 3, 34, 60, 61]. Histone modifications such as methylation, acetylation, phosphorylation, SUMOylation, and ubiquitylation have been uncovered thus far [62,63,64]. Notably, transcriptional regulation by histone methylation is more complex than that by histone acetylation. Histone methylation can be correlated with either gene activation or repression depending on histone residues (lysine or arginine), specific loci, or distinctive methylation patterns (e.g., asymmetric or symmetric) [65,66,67,68]. CARM1 mainly catalyzes H3R17me2, H3R26me2, and H3R42 for epigenetic transcriptional activation [6,7,8,9]. Notably, a recent study has shown through comparison of kcat/KM values that methylation of H3R17 is preferred over that of H3R26 [69]. Thus, we explored the CARM1-mediated H3K17me2 of the gene promoter and how it contributes to pain development in this study. Furthermore, CARM1 catalyzes mono- and asymmetrical dimethylation on R17 and R26 sites in histone H3 and non-histone proteins, including CBP/p300, SRC3, and RNA polymerase II (RNAPII) [70,71,72,73]. Our previous results have linked pain pathophysiology to activating spinal RNAPII phosphorylation [60, 74, 75], and the possibility that the CARM1-mediated H3R17me2 of the K+ channel promoter contributes to neuropathic allodynia development through RNAPII function in the dorsal horn warrants further study.
The dynamics of protein ubiquitination, which modifies protein expression and function [76], critically involve E3-ubiquitin ligase (E3) [77]. Emerging evidence suggests that E3-mediated ubiquitination is involved in various activity-dependent plasticity events, including dendritic spine maturation, long-term potentiation (LTP) in hippocampal neurons, and learning performance [78, 79]. Interestingly, studies have revealed that E3-mediated ubiquitination contributes to chronic pain by regulating the levels of synaptic proteins, thereby modifying synaptic efficacy and plasticity [80, 81]. Recent studies have identified SKP2, a member of the F-box family of E3 proteins, as associated with synaptic plasticity in the hippocampus CA1 region [30]. SKP2-dependent ubiquitination of target proteins has been demonstrated to regulate the neuronal plasticity of primary neurons [82]. Notably, a study has shown that the stability of the arginine methyltransferase CARM1 is regulated by SKP2 [40] and upregulates SKP2-dependent ubiquitination in spinal cord neurons following peripheral nerve injury [83]. Considering that our previous results have linked spinal F-box family-dependent ubiquitination to chronic pain pathology [31,32,33] and that spinal ubiquitination-associated epigenetics participates in neuropathic pain development [34], the above evidence motivated us to explore the effect of SKP2-related CARM1 ubiquitination on spinal plasticity, which participates in the development and maintenance of chronic pain. However, SPK2 expression in the ipsilateral dorsal horn was surprisingly unaffected on day 7 after SNL. Recent findings have demonstrated that SKP2-dependent CARM1 ubiquitination is a crucial nuclear event in autophagy induction after nutrient starvation [40]. Notably, one study suggested that autophagy dysfunction contributes to neuropathic pain development [84]. However, spinal autophagy is differently modulated in distinct animal models of neuropathic pain [85]. These reasons might underlie the discrepancy between our study and the abovementioned study.
Glia activation-associated spinal plasticity has recently been recognized as a crucial modulator of neuropathic pain [86]. Chronic constriction injury was found to induce microglial and astrocyte activation in the dorsal horn [86]. In addition, persistent production of glial factors has been reported to maintain the sensitization of dorsal horn neurons in spinal cord injury rats [87]. In the current study, Fbxo3/CARM1 signaling was found to play an important role in neuropathic pain, including CARM1-positive puncta, which were immunoreactive predominantly in ipsilateral dorsal horn neurons, not in microglia and astrocytes. Moreover, our previous studies have linked spinal Fbxo3-dependent ubiquitination to neuropathic pain pathology, which has been reported to be present primarily in dorsal horn neurons [31, 32]. Therefore, we presumably by different neuropathic pain model but possibly may diffident pathways in neuron, microglia, or astrocytes.
Conclusions
In this study, we demonstrate that nerve injury decreases CARM1 by Fbxo3-mediated CARM1 ubiquitination in the dorsal horn, subsequently decreasing H3R17me2-dependent K+ channel gene transcription/expression in the dorsal horn, which underlies tactile allodynia development. We demonstrate that attenuation of spinal CARM1-induced K+ channel gene transcription is required for the development of neuropathic allodynia. These results may provide a novel therapeutic approach for chronic neuropathic pain.
Abbreviations
- PRMTs:
-
Protein arginine methyltransferases
- CARM1:
-
Coactivator-associated arginine methyltransferase 1
- H3R17me2:
-
Asymmetric dimethylation of Arg17 in histone H3
- H3R26me2:
-
Asymmetric dimethylation of Arg17 and Arg26 in histone H3
- DRG:
-
Dorsal root ganglion
- SNL:
-
Spinal nerve ligation
- ChIP:
-
Chromatin immunoprecipitation
- CCI:
-
Chronic constriction injury
- RNAPII:
-
RNA polymerase II
- E3:
-
E3-ubiquitin ligase
- LTP:
-
Long-term potentiation
- SKP2:
-
S-phase kinase-associated protein 2
- SCF:
-
Skp1-CUL1-F box
References
Denk F, McMahon SB. Chronic pain: emerging evidence for the involvement of epigenetics. Neuron 2012;73435-444.
Lin TB, Hsieh MC, Lai CY, et al. Melatonin relieves neuropathic allodynia through spinal MT2-enhanced PP2Ac and downstream HDAC4 shuttling-dependent epigenetic modification of hmgb1 transcription. J Pineal Res 2016;60:263-276.
Lin TB, Hsieh MC, Lai CY, et al. Modulation of nerve Injury-induced HDAC4 cytoplasmic retention contributes to neuropathic pain in rats. Anesthesiology 2015;123:199-212.
Liang L, Lutz BM, Bekker A, Tao YX. Epigenetic regulation of chronic pain. Epigenomics 2015;7:235-245.
Kim DI, Park MJ, Lim SK, et al. High-glucose-induced CARM1 expression regulates apoptosis of human retinal pigment epithelial cells via histone 3 arginine 17 dimethylation: role in diabetic retinopathy. Arch Biochem Biophys 2014;560:36-43.
Ma H, Baumann CT, Li H, et al. Hormone-dependent, CARM1-directed, arginine-specific methylation of histone H3 on a steroid-regulated promoter. Curr Biol 2001;11:1981-1985.
Schurter BT, Koh SS, Chen D, et al. Methylation of histone H3 by coactivator-associated arginine methyltransferase 1. Biochemistry 2001;40:5747-5756.
Bauer UM, Daujat S, Nielsen SJ, Nightingale K, Kouzarides T. Methylation at arginine 17 of histone H3 is linked to gene activation. EMBO Rep 2002;3:39-44.
Casadio F, Lu X, Pollock SB, et al. H3R42me2a is a histone modification with positive transcriptional effects. Proc Natl Acad Sci U S A 2013;110:14894-14899.
Colombrita C, Silani V, Ratti A. ELAV proteins along evolution: back to the nucleus? Mol Cell Neurosci 2013;56:447-455.
Lim CS, Alkon DL. Protein kinase C stimulates HuD-mediated mRNA stability and protein expression of neurotrophic factors and enhances dendritic maturation of hippocampal neurons in culture. Hippocampus 2012;22:2303-2319.
Lim CS, Alkon DL. Inhibition of coactivator-associated arginine methyltransferase 1 modulates dendritic arborization and spine maturation of cultured hippocampal neurons. J Biol Chem 2017;292:6402-6413.
Rahn EJ, Guzman-Karlsson MC, David Sweatt J. Cellular, molecular, and epigenetic mechanisms in non-associative conditioning: implications for pain and memory. Neurobiol Learn Mem 2013;105:133-150.
Mo K, Xu H, Gong H, et al. Dorsal Root Ganglia Coactivator-associated Arginine Methyltransferase 1 Contributes to Peripheral Nerve Injury-induced Pain Hypersensitivities. Neuroscience 2018;394:232-242.
Woolf CJ, Mannion RJ. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet 1999;353:1959-1964.
Wulff H, Castle NA, Pardo LA. Voltage-gated potassium channels as therapeutic targets. Nat Rev Drug Discov 2009;8:982-1001.
Tsantoulas C, Zhu L, Yip P, Grist J, Michael GJ, McMahon SB. Kv2 dysfunction after peripheral axotomy enhances sensory neuron responsiveness to sustained input. Exp Neurol 2014;251:115-126.
Caylor J, Reddy R, Yin S, et al. Spinal cord stimulation in chronic pain: evidence and theory for mechanisms of action. Bioelectron Med 2019;5:12.
Vicario N, Turnaturi R, Spitale FM, et al. Intercellular communication and ion channels in neuropathic pain chronicization. Inflamm Res 2020;69:841-850.
Hu HJ, Alter BJ, Carrasquillo Y, Qiu CS, Gereau RWt. Metabotropic glutamate receptor 5 modulates nociceptive plasticity via extracellular signal-regulated kinase-Kv4.2 signaling in spinal cord dorsal horn neurons. J Neurosci 2007;27:13181-13191.
Ishikawa K, Tanaka M, Black JA, Waxman SG. Changes in expression of voltage-gated potassium channels in dorsal root ganglion neurons following axotomy. Muscle Nerve 1999;22:502-507.
Kim DS, Choi JO, Rim HD, Cho HJ. Downregulation of voltage-gated potassium channel alpha gene expression in dorsal root ganglia following chronic constriction injury of the rat sciatic nerve. Brain Res Mol Brain Res 2002;105:146-152.
Rasband MN, Park EW, Vanderah TW, Lai J, Porreca F, Trimmer JS. Distinct potassium channels on pain-sensing neurons. Proc Natl Acad Sci U S A 2001;98:13373-13378.
Takeda M, Tanimoto T, Nasu M, Matsumoto S. Temporomandibular joint inflammation decreases the voltage-gated K+ channel subtype 1.4-immunoreactivity of trigeminal ganglion neurons in rats. Eur J Pain 2008;12:189-195.
Zhu Y, Colak T, Shenoy M, et al. Transforming growth factor beta induces sensory neuronal hyperexcitability, and contributes to pancreatic pain and hyperalgesia in rats with chronic pancreatitis. Mol Pain 2012;8:65.
Chen J, Winston JH, Sarna SK. Neurological and cellular regulation of visceral hypersensitivity induced by chronic stress and colonic inflammation in rats. Neuroscience 2013;248:469-478.
Cao XH, Byun HS, Chen SR, Cai YQ, Pan HL. Reduction in voltage-gated K+ channel activity in primary sensory neurons in painful diabetic neuropathy: role of brain-derived neurotrophic factor. J Neurochem 2010;114:1460-1475.
Laumet G, Garriga J, Chen SR, et al. G9a is essential for epigenetic silencing of K(+) channel genes in acute-to-chronic pain transition. Nat Neurosci 2015;18:1746-1755.
Lough L, Sherman D, Ni E, Young LM, Hao B, Cardozo T. Chemical probes of Skp2-mediated p27 ubiquitylation and degradation. Medchemcomm 2018;9:1093-1104.
Zhang Y, Yao W, Qiu J, Qian W, Zhu C, Zhang C. The involvement of down-regulation of Cdh1-APC in hippocampal neuronal apoptosis after global cerebral ischemia in rat. Neurosci Lett 2011;505:71-75.
Lai CY, Ho YC, Hsieh MC, et al. Spinal Fbxo3-Dependent Fbxl2 Ubiquitination of Active Zone Protein RIM1alpha Mediates Neuropathic Allodynia through CaV2.2 Activation. J Neurosci 2016;36:9722-9738.
Lin TB, Hsieh MC, Lai CY, et al. Fbxo3-Dependent Fbxl2 Ubiquitination Mediates Neuropathic Allodynia through the TRAF2/TNIK/GluR1 Cascade. J Neurosci 2015;35:16545-16560.
Hsieh MC, Ho YC, Lai CY, et al. Spinal TNF-alpha impedes Fbxo45-dependent Munc13-1 ubiquitination to mediate neuropathic allodynia in rats. Cell Death Dis 2018;9:811.
Lai CY, Hsieh MC, Ho YC, et al. Spinal RNF20-Mediated Histone H2B Monoubiquitylation Regulates mGluR5 Transcription for Neuropathic Allodynia. J Neurosci 2018;38:9160-9174.
Chung JM, Kim HK, Chung K. Segmental spinal nerve ligation model of neuropathic pain. Methods Mol Med 2004;99:35-45.
Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 1994;53:55-63.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001;25:402-408.
Zhou HY, Chen SR, Chen H, Pan HL. Opioid-induced long-term potentiation in the spinal cord is a presynaptic event. J Neurosci 2010;30:4460-4466.
Kim D, Lim S, Park M, et al. Ubiquitination-dependent CARM1 degradation facilitates Notch1-mediated podocyte apoptosis in diabetic nephropathy. Cell Signal 2014;26:1774-1782.
Shin HJ, Kim H, Oh S, et al. AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature 2016;534:553-557.
Ekberg J, Schuetz F, Boase NA, et al. Regulation of the voltage-gated K(+) channels KCNQ2/3 and KCNQ3/5 by ubiquitination. Novel role for Nedd4-2. J Biol Chem 2007;282:12135-12142.
Guo A, Gu H, Zhou J, et al. Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol Cell Proteomics 2014;13:372-387.
Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer 2013;13:37-50.
Morales Y, Caceres T, May K, Hevel JM. Biochemistry and regulation of the protein arginine methyltransferases (PRMTs). Arch Biochem Biophys 2016;590:138-152.
Sanchez G, Bondy-Chorney E, Laframboise J, et al. A novel role for CARM1 in promoting nonsense-mediated mRNA decay: potential implications for spinal muscular atrophy. Nucleic Acids Res 2016;44:2661-2676.
Hubers L, Valderrama-Carvajal H, Laframboise J, Timbers J, Sanchez G, Cote J. HuD interacts with survival motor neuron protein and can rescue spinal muscular atrophy-like neuronal defects. Hum Mol Genet 2011;20:553-579.
Lin TB, Lai CY, Hsieh MC, et al. Inhibiting MLL1-WDR5 interaction ameliorates neuropathic allodynia by attenuating histone H3 lysine 4 trimethylation-dependent spinal mGluR5 transcription. Pain 2020;161:1995-2009.
Lee PK, Goh WW, Sng JC. Network-based characterization of the synaptic proteome reveals that removal of epigenetic regulator Prmt8 restricts proteins associated with synaptic maturation. J Neurochem 2017;140:613-628.
Penney J, Seo J, Kritskiy O, et al. Loss of protein arginine methyltransferase 8 alters synapse composition and function, resulting in behavioral defects, J Neurosci 2017;37:8655-8666.
Baumann C, Olson M, Wang K, Fazleabas A, De La Fuente R. Arginine methyltransferases mediate an epigenetic ovarian response to endometriosis. Reproduction 2015;150:297-310.
Wang F, Ma SB, Tian ZC, et al. Nociceptor-localized cGMP-dependent protein kinase I is a critical generator for central sensitization and neuropathic pain. Pain 2020 (in press).
Haroutounian S, Ford AL, Frey K, et al. How central is central poststroke pain? The role of afferent input in poststroke neuropathic pain: a prospective, open-label pilot study. Pain 2018;159:1317-1324.
Chen SR, Cai YQ, Pan HL. Plasticity and emerging role of BKCa channels in nociceptive control in neuropathic pain. J Neurochem 2009;110:352-362.
Hoffman AF, Macgill AM, Smith D, Oz M, Lupica CR. Species and strain differences in the expression of a novel glutamate-modulating cannabinoid receptor in the rodent hippocampus. Eur J Neurosci 2005;22:2387-2391.
Kim JH, Yoo BC, Yang WS, Kim E, Hong S, Cho JY. The role of protein arginine methyltransferases in inflammatory responses. Mediat Inflamm 2016;2016:4028353.
Liou JT, Liu FC, Mao CC, Lai YS, Day YJ. Inflammation confers dual effects on nociceptive processing in chronic neuropathic pain model. Anesthesiology 2011;114:660-672.
Zhang P, Yang M, Chen C, Liu L, Wei X, Zeng S. Toll-like receptor 4 (TLR4)/opioid receptor pathway crosstalk and impact on opioid analgesia, immune function, and gastrointestinal motility. Front Immunol 2020;11:1455.
Jayne S, Rothgiesser KM, Hottiger MO. CARM1 but not its enzymatic activity is required for transcriptional coactivation of NF-kappaB-dependent gene expression. J Mol Biol 2009;394:485-495.
Castellano S, Spannhoff A, Milite C, et al. Identification of small-molecule enhancers of arginine methylation catalyzed by coactivator-associated arginine methyltransferase 1. J Med Chem 2012;55:9875-9890.
Hsieh MC, Ho YC, Lai CY, et al. Bromodomain-containing protein 4 activates voltage-gated sodium channel 1.7 transcription in dorsal root ganglia neurons to mediate thermal hyperalgesia in rats. Anesthesiology 2017;127:862-877.
Hsieh MC, Lai CY, Ho YC, et al. Tet1-dependent epigenetic modification of BDNF expression in dorsal horn neurons mediates neuropathic pain in rats. Sci Rep 2016;6:37411.
Berger SL. The complex language of chromatin regulation during transcription. Nature 2007;447:407-412.
Kouzarides T. Chromatin modifications and their function. Cell 2007;128:693-705.
Patel DJ, Wang Z. Readout of epigenetic modifications. Annu Rev Biochem 2013;82:81-118.
Bernstein BE, Humphrey EL, Erlich RL, et al. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc Natl Acad Sci U S A 2002;99:8695-8700.
Santos-Rosa H, Schneider R, Bannister AJ, et al. Active genes are tri-methylated at K4 of histone H3. Nature 2002;419:407-411.
Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 2001;410:116-120.
Feng Y, Wang J, Asher S, et al. Histone H4 acetylation differentially modulates arginine methylation by an in Cis mechanism. J Biol Chem 2011;286:20323-20334.
Jacques SL, Aquino KP, Gureasko J, et al. CARM1 preferentially methylates H3R17 over H3R26 through a random kinetic mechanism. Biochemistry 2016;55:1635-1644.
Sims RJ, 3rd, Rojas LA, Beck DB, et al. The C-terminal domain of RNA polymerase II is modified by site-specific methylation. Science 2011;332:99-103.
Feng Q, Yi P, Wong J, O’Malley BW. Signaling within a coactivator complex: methylation of SRC-3/AIB1 is a molecular switch for complex disassembly. Mol Cell Biol 2006;26:7846-7857.
Xu W, Chen H, Du K, et al. A transcriptional switch mediated by cofactor methylation. Science 2001;294:2507-2511.
Yadav N, Lee J, Kim J, et al. Specific protein methylation defects and gene expression perturbations in coactivator-associated arginine methyltransferase 1-deficient mice. Proc Natl Acad Sci U S A 2003;100:6464-6468.
Lai CY, Hsieh MC, Ho YC, et al. Growth arrest and DNA-damage-inducible protein 45beta-mediated DNA demethylation of voltage-dependent t-type calcium channel 3.2 subunit enhances neuropathic allodynia after nerve injury in rats. Anesthesiology 2017;126:1077-1095.
Hsieh MC, Peng HY, Ho YC, et al. Transcription Repressor Hes1 contributes to neuropathic pain development by modifying CDK9/RNAPII-dependent spinal mGluR5 transcription. Int J Mol Sci 2019;20(17):4177.
Tanaka Y, Tanaka N, Saeki Y, et al. c-Cbl-dependent monoubiquitination and lysosomal degradation of gp130. Mol Cell Biol 2008;28:4805-4818.
Berndsen CE, Wolberger C. New insights into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol 2014;21:301-307.
Sun J, Liu Y, Jia Y, et al. UBE3A-mediated p18/LAMTOR1 ubiquitination and degradation regulate mTORC1 activity and synaptic plasticity. Elife 2018;7:e37993.
Gao J, Marosi M, Choi J, et al. The E3 ubiquitin ligase IDOL regulates synaptic ApoER2 levels and is important for plasticity and learning. Elife 2017;6:e29178.
Edwards IJ, Dallas ML, Poole SL, et al. The neurochemically diverse intermedius nucleus of the medulla as a source of excitatory and inhibitory synaptic input to the nucleus tractus solitarii. J Neurosci 2007;27:8324-8333.
Gautam V, Trinidad JC, Rimerman RA, Costa BM, Burlingame AL, Monaghan DT. Nedd4 is a specific E3 ubiquitin ligase for the NMDA receptor subunit GluN2D. Neuropharmacology 2013;74:96-107.
Boix-Perales H, Horan I, Wise H, et al. The E3 ubiquitin ligase skp2 regulates neural differentiation independent from the cell cycle. Neural Dev 2007;2:27.
Shi S, Cheng C, Zhao J, et al. Expression of p27kip1 and Skp2 in the adult spinal cord following sciatic nerve injury. J Mol Neurosci 2007;32:64-71.
Liu X, Zhu M, Ju Y, Li A, Sun X. Autophagy dysfunction in neuropathic pain. Neuropeptides 2019;75:41-48.
Berliocchi L, Maiaru M, Varano GP, et al. Spinal autophagy is differently modulated in distinct mouse models of neuropathic pain. Mol Pain 2015;11:3.
Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci 2007;10:1361-1368.
Hulsebosch CE. Gliopathy ensures persistent inflammation and chronic pain after spinal cord injury. Exp Neurol 2008;214:6-9.
Acknowledgments
This research was supported by the Ministry of Science and Technology, Taipei, Taiwan: MOST 108-2320-B-038-028 to Dr. TB Lin, MOST 108-2320-B-715-002-MY3 and 108-2314-B-715-004-MY3 to Dr. HY Peng, MOST 108-2320-B-715-004-MY3 to Dr. YC Ho, and MOST 109-2320-B-715-005 to Dr. MC Hsieh; by the Mackay Memorial Hospital: MMH-MM-10705, MMH-MM-10803, and MMH-MM-10902 to Dr. HY Peng and MMH-MM-108-87, MMH-MM-10814, and MMH-MM-10913 to Dr. PS Yang; and by the Department of Medicine, Mackay Medical College: 1071B16, 1081B03, and 1091B01 to Dr. HY Peng; 1071B17, 1081B28, and 1082B03 to Dr. YC Ho; and 1091A01 to Dr. MC Hsieh.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(PDF 2460 kb)
Rights and permissions
About this article

Cite this article
Hsieh, MC., Ho, YC., Lai, CY. et al. Blocking the Spinal Fbxo3/CARM1/K+ Channel Epigenetic Silencing Pathway as a Strategy for Neuropathic Pain Relief. Neurotherapeutics 18, 1295–1315 (2021). https://doi.org/10.1007/s13311-020-00977-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-020-00977-5