
Abstract
Parkinson disease (PD), as a slowly progressive neurodegenerative disorder, undergoes six neuropathological stages. The earliest clinical manifestation presents in the middle stage of the disorder pathologically, when 50% or more of the dopaminergic neurons have degenerated in the substantia nigra. This discrepancy between the early stage clinically and that pathologically has, in part, spurred the debate as to when it is best to initiate symptomatic therapy. The most well-studied monotherapeutic agents for PD in its early course include levodopa (the cornerstone of PD therapy), dopamine agonists, and monoamine oxidase inhibitors (MAOIs). With several options for initiating pharmacologic therapy, along with the heterogenous presentation of the disorder, an individualized approach is warranted. Careful deliberation must be done to optimize risk reduction while providing effective symptom control, taking the chronological age, comorbidities, social and financial disposition, work status, and both immediate- and long-term goals into consideration. Generally, treatment can be delayed in patients with mild symptoms and minimal functional impairment at any age. If treatment must be initiated, dopamine agonists and monoamine oxidase type B inhibitors can be used, especially in younger patients with milder disease. However, for older patients, those with moderate to severe PD symptoms, regardless of age, or for patients with greater comorbidities, levodopa generally remains the better choice. Eventually, regardless of initial therapy, studies have shown that most will eventually require levodopa therapy when symptoms become more disabling.
Similar content being viewed by others


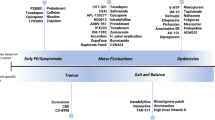
Parkinson disease (PD), as a slowly progressive neurodegenerative disorder [1,2,3], has been traditionally subdivided into “pre-symptomatic” and “symptomatic” phases. Non-motor manifestations often precede the more evident motor symptoms that are used to this day to diagnose probable PD [4, 5]. The pathological process develops “silently” and often requires years until motor symptoms finally emerge. PD undergoes six neuropathological stages, according to Braak staging, initially involving the deposition of intraneural inclusion bodies in the anterior olfactory nucleus and dorsal motor nucleus of the vagal nerve (stage 1), before advancing to denudation of melanoneurons in the substantia nigra (stage 3) [6, 7]. It is in this middle stage where patients start to have noticeable motor symptoms [6]. Therefore, more often than not, even the most proactive patients seeking consultation at the earliest clinical manifestation are, in fact, pathologically in the intermediate stages of their disorder [6, 8, 9], with 50% or more of the dopaminergic neurons degenerated in the substantia nigra [10, 11]. This discrepancy between what is “early” clinically and that pathologically, along with the variety of therapeutic options, has spurred the debate as to when exactly it is best to intervene and initiate the Parkinson patient with symptomatic therapy.
Early PD has been proposed to be further subdivided into preclinical, prodromal, and clinical stages [4]. The clinical stage comprises the presence of full parkinsonism spectrum of motor symptoms [4, 10, 12,13,14]. However, investigations of early PD symptomatology, with Hoehn and Yahr stage of 1 to 2, likewise showed non-motor symptoms as integral part of the phenomenology, particularly depression, apathy, and executive dysfunction [10, 12, 15, 16]. For the purpose of this review, since therapeutic strategies have yet to be defined in preclinical and prodromal stages, “early PD” in this chapter is operationally defined as having motor symptoms without the presence of motor fluctuations and features of advanced PD stages [16].
Dubbed as a pandemic in the recent years due to the exponential increase of the age-standardized prevalence rate, the quest for highly effective PD therapies continues at a relentless pace [17]. Hence, appropriate monotherapeutic agents for PD in its early course, such as levodopa, dopamine agonists, monoamine oxidase inhibitors (MAOIs), have been well studied. Other early symptomatic therapies include anticholinergic agents that have been used to control tremors, and amantadine to additionally prevent or suppress dyskinesias. Factors, including patient’s chronological age, life expectancy, comorbidities, degree of disability, finances, and possibly, ethnicity, are important in determining the best option for the patient’s initial therapy as well as long-term therapeutic strategy [18, 19]. Along with strategies to provide symptomatic relief in these patients, other relevant aims for early PD may include slowing down clinical progression, controlling non-motor symptoms, maintaining function in daily living activities, preventing motor complications, and minimizing side effects [20]. Thus, adherence to pharmacotherapy plays a key role in achieving adequate control of symptoms [21].
Rationale for Early Levodopa Use
Dopamine replacement with levodopa has been, and continues to be, the mainstay of pharmacologic treatment for PD, at any stage of the illness from early to advanced, with clear and consistent evidence of improving disability and reducing mortality [22]. Levodopa has greatly impacted the treatment of PD, as survival of patients has increased by more than 12 years, in a 15-year follow-up, in the post-levodopa era, compared with the 9-year survival in the pre-levodopa era [23]. Although the advent of levodopa did not normalize the mortality rate as matched with non-PD patients, the relative increase in survival was attributed to the significant clinical improvement in the first 4 to 6 years following the initiation of levodopa. Continued levodopa benefit seen thereafter was associated with ongoing maintenance of activity of daily living. Thus, levodopa treatment provides substantial risk reduction for factors that may contribute to increased rate of mortality [24, 25]. As seen in an open-label trial comparing levodopa, dopamine agonists, and monoamine oxidase (MAO) type B inhibitors, newly diagnosed PD patients initiated with levodopa showed better 39-item Parkinson’s Disease Questionnaire (PDQ-39) mobility outcome scores and performed better in both general and disease-specific qualities of life measures [26]. As a prodrug of central dopamine, levodopa acts to ameliorate the motor symptoms of the disease as neurodegeneration progresses (i.e., progressive loss of dopaminergic neurons) [2, 3, 27, 28]. Levodopa was consistently shown to be superior to placebo and, more importantly, to most, if not all, other available PD medications, improving the motor Unified Parkinson’s Disease Rating Scale (UPDRS) score by a mean of 8.8 points (where the minimal clinically important difference in UPDRS is ≥ 3.25 or an improvement of 30% from baseline) (Table 1) [29,30,31,32,33,34,35]. The total UPDRS score improved by approximately 2 to 12 units, with the best result often seen when a dose of 600 mg/day or more is taken [34,35,37]. However, in Western Pacific studies, PD patients exhibit symptomatic benefits with a lower dose of levodopa, at 375 to 413 mg/day, compared with European and North American counterparts [19]. In summary, the early use of levodopa has been commonly advocated, based on its rapid and meaningful symptomatic benefit, along with a long-term mortality reduction. Levodopa may have the greatest impact on mortality when utilized during the early phase of the disease when most motor features are levodopa-responsive [23].
However, levodopa’s superiority in alleviating PD symptoms has been dampened in some patients with a higher propensity to developing medication-induced complications. Head-to-head comparisons between levodopa and dopamine agonists over a 2- to 5-year period uniformly showed a higher frequency, and at a sooner timepoint, of dopaminergic complications, particularly dyskinesia, among early PD patients randomized to levodopa [32]. Nonetheless, while the time-to-onset of dyskinesia was shorter with levodopa therapy [23, 29, 31, 38], this difference becomes non-significant at 3 years of follow-up, although the severity of dyskinesias remains lower in patients taking dopamine agonists, regardless of levodopa augmentation [29, 31]. On the other hand, the time-to-onset and severity of motor fluctuations (such as wearing off) did not differ between levodopa and dopamine agonists [23, 29].
For a period of time, the concept of early dyskinesia occurrence, often termed as levodopa-induced dyskinesia (LID), became a marker of accelerated disease progression and possible levodopa toxicity, questioning the practice of early levodopa initiation. However, increasing evidence has shown that additional variables are required for dyskinesias to develop, and not solely levodopa. The disease itself, along with duration and severity, plays a role. As an example, levodopa does not usually induce dyskinesia in “post-synaptic parkinsonian disorders” such as progressive supranuclear palsy or in regularly treated DRD. Levodopa’s short half-life, rather than its chemical composition, plays an even bigger role. Levodopa continuously administered via intestinal infusion reduces preexisting dyskinesia, even at higher doses, likely due to more favorable pharmacodynamic effects from more tonic stimulation of dopamine receptors. Thus, it is more appropriate to consider dyskinesia an artifact of the method of administration of levodopa rather than an intrinsic molecular effect of levodopa itself. A more accurate designation, therefore, for this treatment complication is levodopa-related dyskinesias, rather than LID [39].
To determine the effect of levodopa on the rate of PD progression, the Earlier versus Later Levodopa Therapy in Parkinson Disease (ELLDOPA) Study enrolled newly diagnosed PD patients into low and high doses of levodopa. A strong dose-dependent response benefit was established, where patients who were receiving the highest dose of levodopa had the best improvement in UPDRS motor scores. Moreover, during the washout period, while the subjects in levodopa groups expectedly worsened, the degree of deterioration was not as much as that noted in the placebo group. Therefore, no clinical evidence of accelerated worsening of PD in patients taking levodopa early in the disease was found in this study [28].
Cilia and colleagues [3] also investigated whether the occurrence of motor complications can primarily be associated to the duration of levodopa therapy versus disease progression per se. To answer this question, a large cohort of Ghanaian drug-naïve PD patients were matched with Italian drug-naïve PD patients and were followed until the development of motor fluctuations and dyskinesias. The reason for this cohort comparison was because Ghanaian PD patients generally received levodopa at a later period from the time of their PD diagnosis, compared to their Italian counterparts, thus allowing comparisons of “levodopa exposure versus disease duration” in the onset of motor fluctuations and dyskinesia. Interestingly, the study found that the severity and disease duration at the onset of motor fluctuations and dyskinesias were comparable between the 2 cohorts, regardless of timing of levodopa therapy. The main difference was that the earlier-treated Italians had a longer motor complication–free treatment period compared to the later-treated Ghanaians. Therefore, the delay in levodopa treatment among Ghanaian PD patients did not delay the onset of their motor fluctuations but only shortened their complication-free treatment period. In addition to disease duration, similar to previously reported risk factors, the younger age of onset, higher levodopa daily dose, and greater severity of symptoms were found to be great contributors to motor fluctuations. In conclusion, the study highlighted that the onset of motor complications and dyskinesias are likely to be influenced more by disease duration rather than by the timing of levodopa initiation [3].
A retrospective analysis of all levodopa-treated patients utilized the “levodopa-sparing” approach with patients diagnosed at a young age to investigate the impact of early versus late levodopa administration [40]. Consistent with the other studies, a strong impact of disease duration on the onset of motor complications was found. In contrast to previous results, however, delaying levodopa seemed to increase the motor complication–free interval from PD onset, as exhibited in patients with the longest interval from PD onset to levodopa initiation. Nonetheless, the significant hazard ratio for early levodopa administration still gives a firm conclusion that the influence of disease duration is stronger than that of treatment duration [22]. Moreover, early PD patients who were left untreated showed deterioration on self-reported health status in less than a year, which improved with the initiation of levodopa. Therefore, although there is a lack of randomized evidence, a “watch and wait” strategy for early PD treatment may seem plausible, especially in milder disease, during the first year from symptom onset and diagnosis, but further treatment delay increases the risk of motor deterioration [41, 42].
While dyskinesias can be bothersome in some patients, most of them are not as noticeable to the patient, compared to the social impact to their caregiver. In the long-term Sydney study, almost all patients who were enrolled experienced dyskinesia by year 15, regardless of the initiated medication. However, the majority (54%) did not consider the dyskinesias to be bothersome enough to require treatment, and those that did, generally responded to pharmacological adjustments. Indeed, compared to dyskinesias, although prevalent, the more disabling long-term sequelae of PD include the emergence of cognitive deficits, gait instability, dysphagia, and autonomic disturbances, which do not respond to levodopa. These are considered the major causes of subsequent morbidity and mortality [23].
On the contrary, single-photon emission computed tomography (SPECT) studies utilizing β-CIT, suggesting presynaptic dopaminergic integrity, did not reflect the greater improvement in motor scores of patients randomized to early levodopa therapy. If anything, the rate of deterioration of uptake of β-CIT in the striatum was worse among patients initiated on levodopa earlier, which was in contradiction to the bigger improvement noted in their motor performance. These results, along with the earlier development of motor complications, have led to the suspicion of levodopa-induced neurotoxicity. However, despite contradicting imaging results, the consistent motor benefit derived from early levodopa use has been viewed as the strongest counter argument to neurotoxicity. It has been hypothesized that the greater decrease of striatal uptake in PD patients on levodopa was likely a pharmacologic effect on dopamine transporter that may have interfered and/or reduced the binding of the β-CIT ligand [28].
Finally, recently published data on levodopa use, whether initiated early or late, has only reinforced previous findings of significant motor benefit, especially in the earlier phase of levodopa therapy, whereas any difference in motor response among early versus delayed levodopa–treated patient disappeared in the long term [43]. On balance, cumulative evidence from previous and recent studies on the timing of levodopa initiation supports neither levodopa-induced neurotoxicity nor neuroprotection but consistently emphasizes the significant symptomatic benefit it provides.
Unfortunately, even with the weight of evidence that the levodopa does not cause neurotoxicity, earlier dissemination of such concept gave rise to levodopa phobia, causing delays in initiating levodopa. This pharmacophobia specific to levodopa have led some PD patients to delay or even avoid this treatment option [44]. Case reports have shown patients developing severe akinesia due to unwillingness of the patients or their clinicians to prescribe levodopa, in part because of widespread marketing of “levodopa-sparing strategies” [45]. These behaviors have contributed to poor levodopa compliance, further increasing reports of levodopa ineffectiveness and causing more deleterious effects such as immobility, contractures, and the rise of impulsive–compulsive disorders (in patients placed on high doses of dopamine agonists instead), among others. Clinically identifying these patients can be helpful in optimizing and adjusting PD treatment [44].
Why Levodopa-Sparing Strategies Persist
Evidence for Using Dopamine Agonist
As mentioned above, the caveat of long-term levodopa therapy consists of development of motor fluctuations, dyskinesias, and “wearing off” phenomenon. Although, as mentioned earlier, these complications are likely caused by several factors including drug pharmacokinetics, disease duration, severity, and duration of levodopa therapy, these are seen more often among levodopa responders [30, 46]. This has led to investigations for levodopa-sparing strategies, primarily utilizing dopamine agonists for early monotherapy [29, 30, 35]. Pramipexole, rotigotine, and ropinirole are non-ergot dopamine agonists which were studied to be effective treatments for early PD [30, 47, 48]. Other dopamine agonists, such as bromocriptine, pergolide, and cabergoline, were previously utilized as treatment but were discontinued, or largely underutilized, due to their serious adverse effects [23, 29, 49].
Pramipexole has been proven to be an effective, safe, and well-tolerated monotherapeutic regimen, from 1.5 mg/day to an optimal dose of 4.5 mg/day in patients with early PD, improving the UPDRS motor scores of approximately 20%. When compared to placebo, even the lowest dose (1.5 mg/day) was significantly more effective [47]. Immediate-release (IR) pramipexole is prescribed in three times daily dosing, while extended-release (ER) pramipexole can be given once daily in a 1:1 conversion with IR pramipexole. Both formulae had similar sustained efficacy on early PD and were superior over placebo, as indicated by motor UPDRS, Clinical Global Impression of Improvement (CGI-I), Patient Global Impression of Improvement (PGI-I), and patient-reported adverse events. Switching from IR pramipexole to ER pramipexole has yielded no significant difference in motor UPDRS scores and adverse events as well [50,51,52].
Ropinirole has also been tested in early PD, with a generally well-tolerated therapeutic dose range of 8 to 24 mg per day [53]. In early PD, ropinirole has been proven to increase UPDRS motor scores, particularly resting tremors, when compared to placebo. In fact, its efficacy as monotherapy for resting tremors did not differ significantly with that of levodopa [53, 54]. IR ropinirole is given three times daily. Due to the hypothesis that a pulsatile stimulation of dopamine receptors may cause motor fluctuation, a prolonged-release (PR) ropinirole was developed, in 1:1 equivalent IR ropinirole dosing [55, 56]. The PGI-I was higher in patients given the PR ropinirole compared to IR ropinirole. Furthermore, those who were given the twice daily dosing of PR ropinirole had better PGI-I than those on single daily dose, reporting lesser “off” time and adverse events, later contributing to better compliance. Objectively, motor UPDRS scores were not significantly different between PR ropinirole dosing regimens [56]. Likewise, PR ropinirole was non-inferior to IR ropinirole in terms of motor improvement in early PD [57].
Another dopamine agonist, rotigotine, a transdermal patch with an optimal dose of 2 to 16 mg/24 h, was shown to be tolerated up to 6 years [58]. A study on early PD patients comparing rotigotine to placebo demonstrated 9.0 to 13.5 mg as the minimum effective dose range, with a dose–response improvement in UPDRS motor scores [59].
Piribedil, a D2 and D3 agonist, had been used for years as symptomatic treatment for PD. Studies were largely devoted to demonstrating its effect as a combination treatment with levodopa. As monotherapy, 150 to 300 mg/day decreased the UPDRS score with an effect size comparable to other dopamine agonists and superior over placebo. It was shown to be well tolerated in PD patients below 77 years of age [60]. As an early adjunct therapy to levodopa, 150 mg/day was generally tolerated and provided significant improvement in the UPDRS scores compared to levodopa with placebo adjunct [61].
On balance, however, compared to all dopamine agonists, levodopa was still found to be more efficacious in improving PD symptoms [35]. In PD patients who were diagnosed less than 7 years, levodopa had higher improvement in the total UPDRS scores compared to pramipexole (mean difference of 5.0, 95% confidence interval, p = < 0.001), largely contributed by motor and activities of daily living improvements. Even with levodopa supplementation in patients taking pramipexole, their UPDRS scores remained inferior to those taking levodopa alone [32, 35]. However, while the quality of life scores initially improved in both groups, they declined over time, with no significant difference between the groups at any given time [35]. Like pramipexole, patients on ropinirole had a lower improvement in motor scores compared to levodopa. However, while the frequency of responders among patients with a lower Hoehn and Yahr stage were comparable between ropinirole and levodopa, the responder frequency was higher with levodopa treatment among patients with a higher disease severity [62]. Pergolide, another dopamine agonist, has been reported to provide equivalent motor improvement to levodopa in the first year of treatment. However, the superiority of levodopa treatment over pergolide was demonstrated with longer treatment duration [29].
Ninety percent of patients who were taking pramipexole and 70% of patients on rotigotine patch necessitated levodopa therapy after 6 years. A similar trend was noted with bromocriptine, where patients required additional levodopa doses over time [23, 32, 58]. Early initiation of therapeutic dose of piribedil was also known to delay levodopa introduction and instituting levodopa rescue doses [60].
As described above, while the efficacy of levodopa to treat PD motor symptoms is consistently superior compared to dopamine agonists, its risk of developing dyskinesias has been just as consistent in randomized controlled as well as naturalistic studies [23, 29, 32, 63]. This finding was postulated to be an effect of the longer half-life of pramipexole, reducing pulsatile striatal dopamine stimulation.
On balance, adverse events were noted to be lower in levodopa as opposed to dopamine agonists, often limiting the ability to achieve their probable most effective dose. These agents cause higher incidence of somnolence, dizziness, nausea, postural hypotension, and peripheral edema. Sudden “sleep attacks” were not uncommonly reported as the most serious adverse events in patients on pramipexole, rotigotine, and ropinirole. Neuropsychiatric investigations in patients on these agents have also shown increased incidence of hallucinations compared to levodopa [23, 29, 32, 35, 58]. Piribedil caused gastrointestinal upset, including nausea, typically comparable to safety profile of dopamine agonists [60]. Moreover, pergolide has been shown to cause valvular heart disease [29], whereas skin reactions to rotigotine patch have been reported [58].
Evidence for Using MAOIs
Dopamine is predominantly centrally metabolized by the MAO type B inhibitor. Consequently, the initial appeal for the use of MAOIs in early PD was driven by the hypothesis that the hydrogen peroxide metabolite produced in dopamine metabolism was responsible for the formation of hydroxyl radical, resulting in selective and progressive loss of nigral dopaminergic neurons [64]. Based on this concept, selegiline (deprenyl), a monoamine oxidase type B inhibitor, had been thought to have antioxidative properties [65, 66]. Selegiline has been shown to improve motor symptoms in early PD patients during the first year of treatment, often requiring additional levodopa for long-term use [67]. The disease-modifying investigation on selegiline (and α-tocopherol—the DATATOP trial carried out by the Parkinson Study Group) showed that selegiline significantly delayed the onset of initiation of l-dopa. However, the pattern of motor improvement while on selegiline and motor decline on withdrawal of the drug, suggesting that the delay of initiation of l-dopa is likely due to its mild symptomatic effects, rather than from any neuroprotective benefit [68,69,70]. The use of selegiline with levodopa became even more controversial when the results of a trial raised the possibility of increased mortality when receiving both medications. Nonetheless, most other trials have not supported this finding [71, 72].
Rasagiline, a potent irreversible MAO type B inhibitor, has also been proven to be effective as monotherapy in early PD at well-tolerated doses of 1 mg/day [73,74,75,76]. Moreover, results of rasagiline monotherapy studies (TEMPO and LARGO trials), followed by its long-term extension, raised the possibility of a disease-modifying effect, leading to the execution of a large, multinational trial, termed the ADAGIO study [74, 75, 77]. In this study, the immediate versus delayed initiation of 1 mg and 2 mg of rasagiline was compared to determine disease-modifying benefits of the drug. The hypothesis was that if, indeed, rasagiline produced disease-modifying effects, the motor performance of patients given rasagiline earlier would remain superior to those who received it later by the end of 3 years, whereas if rasagiline only provided symptomatic benefit, patients who received the drug later would have caught up in their motor performance to those who received it earlier. Interestingly, the study’s parameters for “disease modification” were met by patients who received 1 mg of rasagiline earlier compared to those treated later, whereas the parameters for symptomatic benefit alone were met by patients who were randomized to 2 mg of rasagiline [75]. Rasagiline posed generally mild and infrequent adverse events [74]. The major adverse events usually seen in patients on dopamine agonists, such as somnolence, peripheral edema, and hallucinations, were seldom seen in the patients on rasagiline [75, 76]. On balance, the conflicting results between patients randomized to 1 mg of rasagiline versus those randomized to 2 mg failed to answer whether the drug has disease-modifying properties.
Safinamide is the latest approved MAOI that has both dopaminergic and non-dopaminergic actions. It was initially tested for the treatment of PD patients experiencing motor fluctuations, either as an add-on to levodopa or as a combination with dopamine agonist. In early PD, safinamide 1.0 mg/kg given solely or in combination significantly improved motor symptoms [78]. Further, adding 100 mg of safinamide to a stable dose of single dopamine agonist improved motor symptoms and quality of life (measured by PDQ-39) significantly compared to placebo [78, 79]. The same dose of safinamide if given on a longer term (over 18 months) lowered the rate of intervention (increased dose and/or addition of other PD drug classes) in early PD patients [80].
Evidence for Other Classes of Drugs
Other classes of drugs tested for early PD include non-dopaminergic agents. One of these is amantadine, a NMDA antagonist, which showed a satisfactory relief from symptoms in early PD [81]. In de novo patients, it also reduced the risk of development of dyskinesia. Its antagonistic activity on NMDA receptor avoids a direct effect on postsynaptic dopaminergic receptor, reducing the pulsatile stimulation as seen in patients on levodopa [82].
For tremor-predominant, younger PD patients, anticholinergics, such as benztropine, and trihexyphenidyl and tricyclic antidepressants have been shown to be effective, primarily in reducing tremors. The utility of these drugs, however, have been limited by their lack of meaningful benefit on rigidity and bradykinesia and, more importantly, the high prevalence of anticholinergic side effects including dry mouth, dry eyes, urinary hesitancy, psychosis, and cognitive impairment, especially in the elderly [81].
Early PD management has recently evolved due to the introduction of new agents as options for initiating pharmacologic therapy, facilitating individualized treatment practice. As alternatives to the cornerstone of PD treatment, levodopa, careful selection of patients appropriate for these drugs must be done to ensure risk reduction while providing effective symptom control, particularly in treatment-naive patients. Treatment-naïve patients, who were never exposed to any pharmacologic treatment, can be classified as mild, moderate, and severe. In addition, factors, such as chronological age, life expectancy, comorbidities, quality of life, current social and physical capabilities, and profession, should be taken into consideration in selecting the appropriate regimen. Likewise, individualized treatment of early PD patients must include plans to promote and monitor adherence. Although levodopa poses excellent symptomatic relief, the risk of early drug-related adverse effects can be concerning for younger, actively performing patients in the community. These patients have longer years of life expectancy, hence requiring good symptom relief and necessitating longer complication-free years, without compromising compliance. Generally, treatment can be delayed in patients with mild symptoms with minimal functional impairment at any age. This is due to the lack of evidence of disease progression modification by current medications and a way to prevent early occurrence of medication-induced complications. Dopamine agonists and MAO type B inhibitors, which have roles in early PD as either monotherapy or adjunct therapy, can be initiated in these patients, arbitrarily, younger than 70 years of age. For patients with moderate to severe PD symptoms requiring more potent symptom control and/or with comorbidities limiting the use of dopamine agonists and MAO type B inhibitors or for older patients regardless of their disease severity, levodopa is the best choice. Levodopa can be slowly titrated up to achieve maximal symptomatic effect. Regardless of initial therapy, eventually, most if not all patients will need levodopa therapy later in the course of their disease, when symptoms become more disabling [18, 21, 26, 83].
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
References
Przuntek H, Müller T, Riederer P. Diagnostic staging of Parkinson’s disease: Conceptual aspects. J Neural Transm. 2004;111(2):201–16.
Jankovic J. Parkinson’s disease: Clinical features and diagnosis. J Neurol Neurosurg Psychiatry. 2008;79(4):368–76.
Cilia R, Akpalu A, Sarfo FS, Cham M, Amboni M, Cereda E, et al. The modern pre-levodopa era of Parkinson’s disease: Insights into motor complications from sub-Saharan Africa. Brain. 2014;137(10):2731–42.
Postuma RB, Berg D, Stern M, Poewe W, Marek K, Litvan I. CME MDS Clinical Diagnostic Criteria for Parkinson’s Disease Centrality of Motor Syndrome—Parkinsonism and PD Criteria Benchmark—The Expert Examination. 2015;30(12).
Goldman JG, Postuma R. Premotor and nonmotor features of Parkinson’s disease. Curr Opin Neurol. 2014;27(4):434–41.
Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004;318(1):121–34.
Przedborski S. Inflammation and Parkinson’s disease pathogenesis. Mov Disord. 2010;25(Suppl.1):78–82.
Braak H, Del K, Rüb U, Vos RAI De, Jansen ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. 2003;24:197–211.
Braak H, Sastre M, Bohl JRE, Vos RAI De, Del K. Parkinson’s disease: lesions in dorsal horn layer I, involvement of parasympathetic and sympathetic pre- and postganglionic neurons. 2007;421–9.
Rodriguez-Oroz MC, Jahanshahi M, Krack P, Litvan I, Macias R, Bezard E, et al. Initial clinical manifestations of Parkinson’s disease: features and pathophysiological mechanisms. Lancet Neurol. 2009;8(12):1128–39.
Noyce AJ, Lees AJ, Schrag AE. The prediagnostic phase of Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2016;87(8):871–8.
Getz SJ, Levin B. Cognitive and Neuropsychiatric Features of Early Parkinson’s Disease. Arch Clin Neuropsychol. 2017;32(7):769–85.
Berg D, Lang AE, Postuma RB, Maetzler W, Deuschl G, Gasser T, et al. Changing the research criteria for the diagnosis of Parkinson’s disease: Obstacles and opportunities. Lancet Neurol [Internet]. 2013;12(5):514–24. Available from: https://doi.org/10.1016/S1474-4422(13)70047-4
Berg D, Postuma RB, Bloem B, Chan P, Dubois B, Gasser T, et al. Time to redefine PD? Introductory statement of the MDS Task Force on the definition of Parkinson’s disease. Mov Disord. 2014;29(4):454–62.
Löhle M, Ramberg CJ, Reichmann H, Schapira AHV. Early versus delayed initiation of pharmacotherapy in Parkinson’s disease. Drugs. 2014;74(6):645–57.
Hristova AH, Koller WC. Early Parkinson’s disease: What is the best approach to treatment. Drugs and Aging. 2000;17(3):165–81.
Dorsey ER, Sherer T, Okun MS, Bloem BR. The Emerging Evidence of the Parkinson Pandemic. J Parkinsons Dis [Internet]. 2018;8:3–8. Available from: https://content.iospress.com/download/journal-of-parkinsons-disease/jpd181474?id=journal-of-parkinsons-disease%2Fjpd181474
Schapira AHV. Treatment options in the modern management of Parkinson disease. Arch Neurol. 2007;64(8):1083–8.
Lim SY, Tan AH, Ahmad-Annuar A, Klein C, Tan LCS, Rosales RL, et al. Parkinson’s disease in the Western Pacific Region. Lancet Neurol [Internet]. 2019;18(9):865–79. Available from: https://doi.org/10.1016/S1474-4422(19)30195-4
Stocchi F, Vacca L, Radicati FG. How to optimize the treatment of early stage Parkinson’s disease. 2015;1–7.
Straka I, Minar M, Valkovic P. Clinical aspects of adherence to pharmacotherapy in Parkinson disease: A PRISMA-compliant systematic review. 2018;(June).
Poewe W, Antonini A, Zijlmans JC, Burkhard PR, Vingerhoets F. Levodopa in the treatment of Parkinson’s disease: an old drug still going strong. Clin Interv Aging. 2010;5:229–38.
Hely MA, Morris JGL, Reid WGJ, Trafficante R. Sydney Multicenter Study of Parkinson’s Disease: Non-L-Dopa–Responsive Problems Dominate at 15 Years. 2005;20(2):190–9.
Morgan JC, Currie LJ, Harrison MB, Bennett JP, Trugman JM, Wooten GF. Mortality in levodopa-treated Parkinson’s disease. Parkinsons Dis. 2014;2014.
Uitti RJ, Ahlskog JE, Maraganore DM, Muenter MD, Atkinson EJ, Cha RH, et al. Levodopa therapy and survival in idiopathic parkinson’s disease: Olmsted county project. Neurology. 1993;43(10):1918–26.
Gray R, Ives N, Rick C, Patel S, Gray A, Jenkinson C, et al. Long-term effectiveness of dopamine agonists and monoamine oxidase B inhibitors compared with levodopa as initial treatment for Parkinson’s disease (PD MED): A large, open-label, pragmatic randomised trial. Lancet [Internet]. 2014;384(9949):1196–205. Available from: https://doi.org/10.1016/S0140-6736(14)60683-8
Sethi KD. The impact of levodopa on quality of life in patients with Parkinson disease. Neurologist. 2010;16(2):76–83.
Walton-Hadlock JL, Fahn S, Keiburtz K, Tanner CM. Levodopa and the progression of Parkinson’s disease [4] (multiple letters). N Engl J Med. 2005;352(13):1386.
Oertel WH, Wolters E, Sampaio C, Gimenez-roldan S, Bergamasco B, Dujardin M, et al. Pergolide Versus Levodopa Monotherapy in Early Parkinson’s Disease Patients: The PELMOPET Study. 2006;21(3):343–53.
Adler CH, Sethi KD, Hauser RA, Davis TL, Hammerstad JP, Bertoni J, et al. Ropinirole for the treatment of early Parkinson’s disease. Neurology. 1997;49(2):393–9.
Sethi KD, O’Brien CF, Hammerstad JP, Adler CH, Davis TL, Taylor RL, et al. Ropinirole for the treatment of early Parkinson disease: A 12-month experience. Arch Neurol. 1998;55(9):1211–6.
Holloway R, Marek K, Biglan K, Dick A, Fahn S, Julian-Baros E, et al. Long-term effect of initiating Pramipexole vs Levodopa in early Parkinson disease. Arch Neurol. 2009;66(5):563–70.
Horváth K, Aschermann Z, Ács P, Deli G, Janszky J, Komoly S, et al. Minimal clinically important difference on the Motor Examination part of MDS-UPDRS. Park Relat Disord. 2015;21(12):1421–6.
Fahn S. Does levodopa slow or hasten the rate of progression of Parkinson’s disease? J Neurol. 2005;252(SUPPL. 4):37–42.
Holloway RG. Pramipexole vs levodopa as initial treatment for Parkinson Disease: A 4-year randomized controlled trial. Arch Neurol. 2004;61(7):1044–53.
Growdon JH, Kieburtz K, McDermott MP, Panisset M, Friedman JH. Levodopa improves motor function without impairing cognition in mild nondemented Parkinson’s disease patients. Neurology. 1998;50(5):1327–31.
Morgan J, Sethi KD. Levodopa and the progression of Parkinson’s disease. Curr Neurol Neurosci Rep. 2005;5(4):261–2.
Blindeauer K. A Controlled Trial of Rotigotine Monotherapy in Early Parkinson’s Disease. Arch Neurol. 2003;60(12):1721–8.
Espay AJ, Lang AE. Common myths in the use of levodopa in Parkinson disease: When clinical trials misinform clinical practice. JAMA Neurol. 2017;74(6):633–4.
Yahalom G, Cohen OS, Warmann-Alaluf N, Shabat C, Strauss H, Elincx-Benizri S, et al. The impact of early versus late levodopa administration. J Neural Transm. 2017;124(4):471–6.
Grosset D, Taurah L, Burn DJ, MacMahon D, Forbes A, Turner K, et al. A multicentre longitudinal observational study of changes in self reported health status in people with Parkinson’s disease left untreated at diagnosis. J Neurol Neurosurg Psychiatry. 2007;78(5):465–9.
Asimakopoulos P, Caslake R, Harris CE, Gordon JC, Taylor KSM, Counsell C. Changes in quality of life in people with Parkinson’s disease left untreated at diagnosis. 2008;716–8.
Verschuur CVM, Suwijn SR, Boel JA, Post B, Bloem BR, Van Hilten JJ, et al. Randomized delayed-start trial of levodopa in Parkinson’s disease. N Engl J Med. 2019;380(4):315–24.
Titova N, Levin O, Katunina E, Chaudhuri KR. ‘Levodopa Phobia’: a review of a not uncommon and consequential phenomenon. npj Park Dis [Internet]. 2018;(February):1–5. Available from: https://doi.org/10.1038/s41531-018-0067-z
Mccarron MO, Choudhari KA. Clinical/Scientific Notes Aneurysmal subarachnoid leak with normal CT and CSF spectrophotometry. 2005;1–7.
Manson A, Stirpe P, Schrag A. Levodopa-induced-dyskinesias clinical features, incidence, risk factors, management and impact on quality of life. J Parkinsons Dis. 2012;2(3):189–98.
Kieburtz K. Safety and efficacy of pramipexole in early Parkinson disease: A randomized dose-ranging study. J Am Med Assoc. 1997;278(2):125–30.
Rascol O, Zesiewicz T, Chaudhuri KR, Asgharnejad M, Surmann E, Dohin E, et al. A Randomized Controlled Exploratory Pilot Study to Evaluate the Effect of Rotigotine Transdermal Patch on Parkinson’s Disease–Associated Chronic Pain. 2016;56(7):852-861
Orlando N, Grimal R, Study C. Cabergoline in the treatment of early Parkinson’s disease: 1997;48;363-368
Jost WH, Eisenreich W, Sommer B, Hartter S. Pramipexole extended release: A novel treatment option in Parkinson’s disease. Parkinsons Dis. 2010;2010.
Hauser RA, Schapira AH V, Rascol O, Barone P, Mizuno Y, Salin L, et al. Randomized, Double-Blind, Multicenter Evaluation of Pramipexole Extended Release Once Daily in Early Parkinson’s Disease. 2010;25(15):2542–9.
Poewe W, Rascol O, Barone P, Hauser RA, Mizuno Y, Haaksma M, et al. Extended-release pramipexole in early Parkinson disease A 33-week randomized controlled trial. 2011.
Korczyn AD, Thalamas C, Adler CH. Dosing with ropinirole in a clinical setting. Acta Neurol Scand. 2002;106(4):200–4.
Schrag A, Keens J, Warner J. Ropinirole for the treatment of tremor in early Parkinson’s disease. Eur J Neurol. 2002;9(3):253–7.
Nashatizadeh M, Lyons KE, Pahwa R. A review of ropinirole prolonged release in Parkinson’s disease. Clin Interv Aging. 2009;4(1):179–86.
Yun JY, Kim H, Lee J, Kim YE, Kim JS, Kim J, et al. Comparison of once-daily versus twice-daily combination of Ropinirole prolonged release in Parkinson’s disease. 2013.
Stocchi F, Hersh BP, Scott BL, Nausieda PA, Stocchi F, Hersh BP, et al. Ropinirole 24-hour prolonged release and ropinirole immediate release in early Parkinson’s disease: a randomized, double-blind, non-inferiority crossover study. 2008;7995.
Giladi N, Boroojerdi B, Surmann E. The safety and tolerability of rotigotine transdermal system over a 6-year period in patients with early-stage Parkinson’s disease. 2013;1321–9.
Kamp C, Zhang L, Wooten F, Frei K. A Controlled Trial of Rotigotine Monotherapy in Early Parkinson’s Disease. 2003;(August 2017).
Rascol O, Dubois B, Caldas AC, Senn S, Signore S Del, Lees A, et al. Early Piribedil Monotherapy of Parkinson’s Disease: A Planned Seven-Month Report of the REGAIN Study. 2006;21(12):2110–5.
Glatz K, Schuhan C, Sommer C, Berger C, Schwaninger M, Hartmann M, et al. Severe Forward Flexion of the Trunk in Parkinson’s Disease: Focal Myopathy of the Paraspinal Muscles Mimicking Camptocormia. 2003;18(4):408–51.
Rascol O, Brooks J, Brunt ZR, Korczyn AD, Poewe WH. Ropinirole in the Treatment of Early Parkinson’s Disease: A 6-Month Interim Report of a 5-Year Levodopa-Controlled Study. 1998;13(1).
Rascol O, Brooks D, Korczyn A, et al. A Five-Year Study of the Incidence of Dyskinesia in Patients with Early Parkinson’s Disease who were Treated with Ropinirole or Levodopa. 2000;342(20);1485-1491
Dias V, Junn E, Mouradian MM. The Role of Oxidative Stress in Parkinson’s Disease. 2013;3:461–91.
Miyasaki JM, Martin W, Suchowersky O, Weiner WJ, Lang AE. Practice parameter: Initiation of treatment for Parkinson’s disease: An evidence-based review: Report of the quality standards subcommittee of the American Academy of Neurology. Neurology. 2002;58(1):11–7.
LeWitt PA, Taylor DC. Protection Against Parkinson’s Disease Progression: Clinical Experience. Neurotherapeutics. 2008;5(2):210–25.
Pang S, Bodian CA. Selegiline Use to Prevent Progression of Parkinson’s Disease. 1989;46;1275-1279
Gary PH. JBM. SCT et al. The New England Journal of Medicine Downloaded from nejm.org on April 1, 2015. For personal use only. No other uses without permission. Copyright © 1990 Massachusetts Medical Society. All rights reserved. New English J Med. 1990;323(16):1120–3.
Parkinson Study Group, Datatop: A multicenter controlled clinical trial in early parkinson’s disease: Parkinson study group. Arch Neurol. 1989;46(10):1052–60.
Yacoubian TA, Standaert DG. Targets for neuroprotection in Parkinson’s disease. Biochim Biophys Acta - Mol Basis Dis [Internet]. 2009;1792(7):676–87. Available from: https://doi.org/10.1016/j.bbadis.2008.09.009
Thorogood M, Armstrong B, Nichols T, Hollowell J, Nichols T, Armstrong B. Mortality in people taking selegiline: observational study. 1998;(December 1994):1989–90.
Churchyard A, Head J, Hurwitz B, Overstall P, Ockelford J, Lees AJ. Investigation by Parkinson’s Disease Research Group of combined levodopa and selegiline treatment in patients, 1995;311;1602
Stern MB, Marek KL, Friedman J, Hauser RA, LeWitt PA, Tarsy D, et al. Double-blind, randomized, controlled trial of rasagiline as monotherapy in early Parkinson’s disease patients. Mov Disord. 2004;19(8):916–23.
Viallet F, Pitel S, Lancrenon S, Blin O. Evaluation of the safety and tolerability of rasagiline in the treatment of the early stages of Parkinson’s disease. Curr Med Res Opin. 2013;29(1):23–31.
Olanow CW, Rascol O, Hauser R, Feigin PD, Jankovic J, Lang A, et al. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med. 2009;361(13):1268–78.
Study TT. A Controlled Trial of Rasagiline in Early Parkinson Disease. The TEMPO Study. 2012;59:1937–43.
Feigin PD, Ph D, Jankovic J, Lang A, Langston W, Melamed E, et al. A Double-Blind, Delayed-Start Trial of Rasagiline in Parkinson’s Disease. 2009;361:1268-1278
Bette S. Safinamide in the management of patients with Parkinson’s disease not stabilized on levodopa: a review of the current clinical evidence. 2018;1737–45.
Stocchi F, Borgohain R, Onofrj M, Schapira AH V, Bhatt M, Lucini V, et al. ARTICLE A Randomized, Double-Blind, Placebo-Controlled Trial of Safinamide as Add-on Therapy in Early Parkinson’s Disease Patients. 2012;27(1):106–12.
Schapira AH V, Stocchi F, Borgohain R, Onofrj M, Bhatt M, Lorenzana P. Long-term efficacy and safety of safinamide as add-on therapy in early Parkinson’s disease. 2012;1–10.
Ellis JM, Fell MJ. Current approaches to the treatment of Parkinson’s Disease. Bioorganic Med Chem Lett. 2017;27(18):4247–55.
Kim A, Kim YE, Yun JY, Kim H-J, Yang H-J, Lee W-W, et al. Amantadine and the Risk of Dyskinesia in Patients with Early Parkinson’s Disease: An Open-Label, Pragmatic Trial. J Mov Disord. 2018;11(2):65–71.
Laurencin C, Danaila T, Broussolle E, Thobois S, Bron L, Lyon D, et al. Movement disorders Initial treatment of Parkinson’s disease in 2016: The 2000 consensus conference revisited. Rev Neurol (Paris) [Internet]. 2016;1–12. Available from: https://doi.org/10.1016/j.neurol.2016.07.007
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(PDF 1224 kb)
Rights and permissions
About this article

Cite this article
Sy, M.A.C., Fernandez, H.H. Pharmacological Treatment of Early Motor Manifestations of Parkinson Disease (PD). Neurotherapeutics 17, 1331–1338 (2020). https://doi.org/10.1007/s13311-020-00924-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-020-00924-4