
Abstract
Human prion diseases are etiologically categorized into three forms: sporadic, genetic, and infectious. Sporadic Creutzfeldt-Jakob disease (sCJD) is the most common type of human prion disease that manifests as subacute progressive dementia. No effective therapy for sCJD is currently available. Potential therapeutic compounds are frequently tested in rodents infected with mouse-adapted prions that differ from human prions. However, therapeutic effect varies depending on the prion strain, which is one of the reasons why candidate compounds have shown little effect in sCJD patients. We previously reported that intraperitoneal administration of FK506 was able to prolong the survival of mice infected with a mouse-adapted prion by suppressing the accumulation of abnormal prion protein (PrP) and inhibiting the activation of microglia. In this study, we tested oral administration of FK506 in knock-in mice expressing chimeric human prion protein (KiChM) that were infected with sCJD to determine if this compound is also effective against a clinically relevant human prion, i.e., one that has not been adapted to mice. Treatment with FK506, started either just before or just after disease onset, suppressed typical sCJD pathology (gliosis) and slightly but significantly prolonged the survival of sCJD-inoculated mice. It would be worthwhile to conduct a clinical trial using FK506, which has been safety-approved and is widely used as a mild immunosuppressant.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sporadic Creutzfeldt-Jakob disease (sCJD) accounts for about 75% of human prion diseases and leads to death through rapidly progressing dementia and akinetic mutism [1, 2]. Histologically, human prion diseases are characterized by massive brain atrophy accompanied with spongiosis, gliosis, and accumulation of abnormally aggregated prion protein (PrPSc) [3, 4]. It has been proposed that the conformational conversion of normal prion protein (PrPC) to abnormal form (PrPSc) in neurons plays a central role in sCJD pathogenesis. Some drugs, such as pentosan polysulfate (PPS) [5] and quinacrine [6], inhibit this conversion process of PrP and they have been proposed as potential therapeutic agents for prion diseases because of their effects in prion-infected mouse models. However, the corresponding clinical trials on sCJD patients did not show any improvement in either patient symptoms or survival periods [7,8,9]. Doxycycline has also been reported to prolong survival periods of PrPSc-inoculated mice [8]; however, its effect in humans appears to be limited to patients with early-stage sCJD [10].
There are several reasons that can explain the difficulty in developing therapeutics for sCJD. First, anti-prion drugs only show effects on animal prions in experimental models when the administration of drugs was started at the same time as prion infection, indicating that the drugs may inhibit the propagation of prions [11, 12]. However, the human trials of these drugs were conducted only after the disease was established because early definitive diagnosis has yet to be developed [13]. Second, drug effects will be dependent on the prion strain. Prions exhibit strain-diversity and the mechanisms for this diversity are still unknown. Notably, the Rocky Mountain Laboratory (RML) strain, a mouse-adapted scrapie prion, is often used in testing therapeutics, but its biochemical and pathological properties are distinct from those of human sCJD prions [14, 15]. Therefore, even a compound that doubles the survival period of prion-inoculated mice may not necessarily be effective for treating sCJD. This problem has been previously described [16]. To evaluate therapeutic effects against human prions, drug-evaluation can be conducted using sCJD prion-inoculated mice. Another possible reason for the failure of previous clinical trials may be more fundamental. Most anti-prion compounds inhibit PrP-conversion in prion-infected cells. However, the direct neurotoxicity of PrPSc is not clear and reduction of PrPSc may not be enough to stop neuronal loss if the pathological reactions have already started. For example, microglia have important roles in protecting neurons but over-activated microglia can induce neuronal cell death by releasing pro-inflammatory cytokines [17,18,19,20].
We have previously reported that the immunosuppressant, FK506, can prolong survival periods of mice infected with a mouse-adapted prion strain, Fukuoka-1, by regulating both glial activation and the activation of neuronal autophagy. This resulted in reduced PrPSc accumulation [21]. To elucidate the effect of FK506 on human prions, we administered the compound to sCJD prion-inoculated mice.
Materials and Methods
Reagents
3F4 antibody (BioLegend, San Diego, USA) is a mouse monoclonal antibody recognizing the amino acid residues 109–112 of human PrP. Anti-ionized calcium-binding adapter molecule 1 (IBA1) antibody to detect microglia (Wako Pure Chemical Industries, Japan) and anti-glial fibrillary acidic protein (GFAP) antibody to detect astrocytes (DAKO, Japan) are rabbit polyclonal antibodies. FK506 (Selleck Chemicals, Houston, USA) was dissolved as previously described [22]. Briefly, FK506 was mixed with Kolliphor HS15 (gifted from BASF Japan), tetraglycol (Sigma-Aldrich, Japan), and ethyl oleate (Nacalai Tesque, Japan) and was stored at 25 °C. Doxycycline hyclate (Sigma-Aldrich) was dissolved in dH2O and stored at − 20 °C.
Brains of sCJD Patients
All three patients were female and diagnosed as having classical-type sCJD (sCJD MM1) according to Parchi’s classification [23]. Patient 1 died at 75 years old [24]. Patients 2 and 3 died at 67 and 71 years old, respectively (Fig. 1 and Table 1) [25]. Tissue samples of brains were homogenized in 10% (w/v) phosphate-buffered saline (PBS; Nacalai Tesque, Japan) using a multi-bead shocker (Yasui Kikai, Japan) [25], and supplemented with a protease inhibitor mixture (Roche, Japan). Informed consents were obtained from patients and/or patient families.

Detection of PrPSc and tPrP from brain homogenates of sCJD patients. PrPSc and total PrP (tPrP) in three sCJD patients were detected by Western blotting with the 3F4 antibody. PrPSc and total tPrP are shown in the upper and middle panels, respectively. β-Actin was used as an internal control (lower panel)
Animal Models
Five-week-old knock-in mice expressing human and mouse chimeric PrP (KiChM), which were described elsewhere [25, 26], were inoculated intracerebrally with 20 μl of brain homogenate (BH) from a sCJD patient. Mice were monitored daily until the terminal stage of the disease or sacrificed at an indicated time point. Clinical onset was defined as the weight of mice falling lower than 28 g, which is about 5 g less than that of uninoculated mice, or as the appearance of any sCJD symptom, such as priapism, hunchback, ataxic gait, and abnormal reflex with non-parallel hind limbs. Clinical scores were first graded by body weight (BW) of mice: 0, healthy; 1, 26–28 g; 2, 24–26 g; 3, 22–24 g; 4, 20–22 g; 5, less than 20 g; 6, death. The score was then added one when the mice showed mentioned symptoms (Additional file 1). Mice weighed less than 22 g with at least two symptoms were euthanized under anesthesia and their clinical score was recorded as 6 (death). Some of the mice were sacrificed at 140 d.p.i. After brains were removed, the right hemispheres were frozen and homogenized at 20% (w/v) in PBS to conduct Western blotting. Total proteins were extracted by mixing the samples with an equal volume of lysis buffer (0.5% Triton X-100, 0.5% deoxycholic acid, 150 mM NaCl, 25 mM Tris-HCl, pH 7.5). The left hemispheres were fixed in 10% neutral-buffered formalin (WAKO) to analyze histopathological changes.
Administration of FK506
In mice inoculated with sCJD prion from either patient 1 or patient 2, FK506 was orally administered at 1.0 mg/kg/day from 110 or 140 d.p.i. In the case of mice inoculated with BH from sCJD patient 3, FK506 was started from 135 d.p.i. with the lower concentration of the drug which adjusted as 0.1 mg/kg/day.
Western Blotting
Total protein concentrations were measured using a Protein Assay Bicinchoninate Kit (Nacalai). To detect PrPSc, the samples were digested with 20 μg/ml of protease K (PK) for 30 min at 37 °C. Loading buffer (50 mM Tris-HCl [pH 6.8], containing 5% (v/v) glycerol, 1.6% (w/v) sodium dodecyl sulfate [SDS], and 100 mM dithiothreitol) was added to the proteins, and the mixtures were incubated at 95 °C for 10 min. SDS polyacrylamide gel electrophoresis was performed using 15% (w/v) acrylamide gels. The proteins were transferred onto an Immobilon-P membrane (Merck, Japan) in transfer buffer containing 20% (v/v) methanol, and the membrane was blocked with 5% (w/v) nonfat dry milk in tris-buffered saline with Tween 20 (TBST, 10 mM Tris-HCl [pH 7.8], 100 mM NaCl, 0.1% [v/v] Tween 20) for 1 h before blotting with the primary antibody overnight at 4 °C. Immunoreactive bands were visualized using Clarity Western ECL substrate (Bio-Rad, Japan).
Quaking-Induced Conversion Assay
Ninety microliter of reaction buffer and 10 μ L of serially diluted BH was loaded into 96-well black plate with clear bottom (Greiner, Japan). The composition and final concentration of each in reaction buffer was 500 mM NaCl, 50 mM PIPES (pH 7.0), 1 mM EDTA, 0.001% SDS 0.01 mM thioflavin T (ThT), and 0.1 mg/mL recombinant human PrP (residues 23-231). Ten percent BHs used for seeds were serially diluted 10−2 to 10−9. The plate was sealed (plate sealer, Nalgene Nunc International) and incubated at 42 °C in a Thermomixer C (Eppendorf, Japan) with cycles of 1-min shaking (1000 rpm double orbital) and 1-min incubation. ThT fluorescence was measured with infinite F200 PRO (Tecan, Japan) (430 ± 20-nm excitation and 485 ± 20-nm emission; bottom read) at 48 h after the reaction was started. The fluorescent value larger than average values of uninoculated mice brains plus three standard deviations was determined positive. The 50% positive reactivity (50% of seeding dose: SD50) was calculated using the Spearman–Kärber method [27].
Histochemistry
The fixed hemispheres were embedded in paraffin and sectioned into 3-μm slices. Tissue sections were stained with hematoxylin and eosin. For IBA1 and GFAP staining, after deparaffinization and rehydration, the sections were treated with 0.3% (v/v) hydrogen peroxidase in methanol for 30 min to inactivate endogenous peroxidase and then incubated with 3% nonfat dry milk in TBST for 60 min at room temperature. The blocked sections were subsequently reacted with primary antibody overnight at room temperature, then reacted with the Envision polymer horseradish peroxidase (HRP)–conjugated anti-rabbit immunoglobulin G antibodies (DAKO) for 60 min at room temperature. Immunostaining was visualized using 3,3′-diaminobenzidine (DAB; Dojindo Lab, Japan). The hydrolytic autoclaving and formic acid method for PrPSc staining was performed as described previously [21].
Statistical Analysis
An unpaired t test was used for comparison between two groups. The log-rank test was used for analyzing the survival time. All statistical analyses were performed using GraphPad Prism software.
Ethical Approval
All animal experiments were approved by the Ethics Committee of Nagasaki University and were performed under the Guidelines for Animal Experimentation of Nagasaki University. These experiments also confirmed to the recommendations issued in the Guide for the Care and Use of Laboratory Animals by the National Institutes of Health.
Results
Comparison of the Therapeutic Effects of FK506 and Doxycycline in a sCJD-Inoculated Mouse Model
When wild-type mice were inoculated with BH from a sCJD patient, they did not show any symptoms. In contrast, KiChM mice were susceptible to human prions and died about 150 days post-inoculation (d.p.i.) when they are intracerebrally inoculated with sCJD prion [25, 26]. Doxycycline (Dox), which promotes the degradation of PrPSc, has been one of the most promising candidate therapeutic agents for sCJD [8]. To investigate and compare the therapeutic effects of FK506 and Dox, KiChM mice were inoculated with 10% BH from patient no. 1 (sCJD-1 in Fig. 1 and Table 1). When symptoms appeared in the mice from 130 d.p.i., they were administered 0.1 mg/kg/day FK506 or 2.0 mg/kg/day Dox or vehicle. The mice-administered Dox lived until 149.2 ± 6.7 d.p.i., which was about 10 days longer than the vehicle-treated group (p < 0.05). The mice-administered FK506 survived until 161.8 ± 18.0 d.p.i., which was about 20 days longer than the vehicle-treated group (Fig. 2 and Table 2). The mice administered with both FK506 and Dox lived until 165 ± 23.3 d.p.i. They lived significantly longer than vehicle and Dox-only groups, but for almost the same survival period as the FK506-only group.

Survival curves of sCJD-1-inoculated mice treated with FK506 and doxycycline. The survival curves of sCJD-1-inoculated mice that were administered with 0.1 mg/kg/day of FK506 (FK) and/or 2 mg/kg/day of doxycycline (Dox) from disease onset (130 d.p.i.). The survival of the groups treated with vehicle, Dox alone, FK506 alone, and combined therapy (Dox and FK506 together) are indicated by circles, square, upright triangles, and upside-down triangles, respectively
FK506 Suppresses the Progression of Symptoms and Prolongs the Survival Period of sCJD Prion-Inoculated Mice
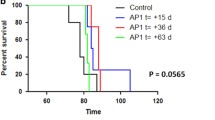
To investigate the effect of FK506 on sCJD prion-inoculated model, KiChM mice with sCJD prion were administered vehicle or FK506 (1.0 mg/kg/day) either from 110 d.p.i. (just before disease onset) or from 140 d.p.i. (after the symptoms appeared), and symptom scores were recorded (see Additional File 1). When we inoculated KiChM mice with BH from patient no. 2 (sCJD-2 in Fig. 1 and Table 1), the deterioration in symptom scores from 135 to 145 d.p.i. in mice that received FK506 from 110 d.p.i were suppressed (Fig. 3a).

Clinical scores and survival curves of sCJD prion-inoculated mice. Clinical scores were recorded from 140 d.p.i until the first mouse died. Survival periods were also compared between the vehicle-treated control mice (circles) and FK506-treated mice (squares) from 110 d.p.i. a Clinical scores of FK506- or vehicle-treated mice 110 days after they were inoculated with sCJD-2 prion. b Survival curves of these mice. c Clinical scores of FK506- or vehicle-treated mice 110 days after they were inoculated with sCJD-3 prion. d Survival curves of these mice. Statistical significance of differences in clinical scores was determined using a two-tailed Student t test, and that of differences in the survival curves was determined using log-rank test. *: p < 0.001 compared with control group. Error bars indicate standard error of the mean (SEM)
After treatment with FK506 beginning at 110 d.p.i., symptom scores of mice inoculated with BH from patient no. 3 (sCJD-3 in Fig. 1) were better than those of vehicle-treated mice (Fig. 3c). In addition, both groups inoculated with sCJD-2 and sCJD-3 prion survived 12 and 30 days longer than the untreated control, respectively (Fig. 3b, d and Table 3). Moreover, mice inoculated with sCJD-3 prion survived significantly longer than vehicle-treated group even when the treatment was started from 140 d.p.i (Table 3).
FK506 Suppresses the Activation of Glial Cells and Spongiform Change
After the 30-day administration of FK506 or vehicle (140 d.p.i), we collected brains from sCJD-3 prion-inoculated mice and evaluated the degree of glial activation. IBA1 [17], which is also called allograft inflammatory factor-1 (AIF-1), was assessed as a marker of activated microglia [18]. The expression levels of IBA1 in whole brains of mice at 140 d.p.i were analyzed by Western blotting. The levels of IBA1 in whole brains of FK506-treated mice were significantly lower than those of vehicle-treated mice (Fig. 4a, b). Immunohistochemistry revealed that the areas occupied by microglia in the cortex and thalamus were significantly smaller in FK506-treated mice compared with those in vehicle-treated mice, but there was no significant difference in the hippocampus or striatum (Fig. 4c, d).

Comparison of microglia in sCJD-3 prion-inoculated mice. Some of the sCJD-3-inoculated mice that had been treated with FK506 or vehicle from 110 d.p.i were sacrificed at 140 d.p.i. a Western blot analysis of the expression level of IBA1. β-Actin (ACTB) was used as an internal control. b Band intensities of samples from FK506-treated mice are expressed as a percentage of those of the control mice. The results in the graph are the mean ± SD. c IBA-1-positive cells in the cortex (Cx), hippocampus (Hp), and thalamus (Th) were visualized by immunohistochemical staining. d The percentages of occupied by IBA-1-positive cells were calculated and compared between the FK506-treated group (FK) and the vehicle-treated group (Ve). Scale bars represent 100 μm. Statistical significance was determined using a two-tailed Student t test. *: p < 0.05, **: p < 0.01 compared with the control. Error bars indicate standard deviation (SD)
Next, we analyzed the expression levels of GFAP at 140 d.p.i. [28]. The levels of GFAP in the brains of FK506-treated mice were significantly lower than those of vehicle-treated mice (Fig. 5a, b). The areas occupied by astrocytes in the cortex, hippocampus, and striatum of the FK506-treated group were less than half those of the vehicle-treated group (Fig. 5c, d).

Comparison of astrocyte in sCJD-3 prion-inoculated mice. a Western blot analysis of the expression level of GFAP. ACTB was used as an internal control. b Band intensities of samples from FK506-treated mice are expressed as a percentage of those of the control mice. The results in the graph are the mean ± SD. c GFAP-positive cells were visualized by immunohistochemical staining. d The percentages of occupied by IBA-1-positive cells were calculated and compared between the FK506-treated group (FK) and the vehicle-treated group (Ve). Scale bars represent 100 μm. Statistical significance was determined using a two-tailed Student t test. *: p < 0.05, **: p < 0.01 compared with the control. Error bars indicate standard deviation (SD)
The spongiform areas in the cortex, hippocampus, thalamus, and striatum were analyzed by staining brain sections with hematoxylin and eosin and calculating the percentage of the vacuolated area in each brain region. In FK506-treated mice inoculated with sCJD-3 prion, the percentages of vacuolated areas were lower than those in control mice, particularly in the cortex, hippocampus, and thalamus (Fig. 6).

Comparison of the degree of vacuolation in sCJD-3 prion-inoculated mice. a The vacuole areas in the brains of mice treated with FK506 or vehicle 110 d.p.i in sCJD-3-inoculated groups were visualized by staining with hematoxylin and eosin (HE staining) at 140 d.p.i. Ve and FK indicate the mice they were treated with vehicle or FK506 respectively. b The percentages of the vacuolated areas were calculated and compared between the FK506-treated group and the vehicle-treated group. Statistical significance was determined using a two-tailed Student t test. *: p < 0.05, **: p < 0.01, ***: p < 0.001 compared with the control. Error bars indicate SD
Comparison of the Accumulation of PrPSc and Total PrP at 140 d.p.i and Each Terminal Stage
In mice inoculated with sCJD-3 prion, PrPSc was not detected in two of the four FK506-treated mice, whereas it was detected in the brains of all mice in the vehicle-treated group by Western blotting at 140 d.p.i. (Fig. 7a, c). In the two PrPSc-negative cases, we also evaluated the amount of seeding activity in the brains by using the endpoint quaking-induced conversion (QUIC) method. Their SD50 was about 1/1000 of the control group (Additional file 2). At each terminal stage, the FK506-treated mice tended to accumulate more PrPSc than the control group, although there was no significant difference (Fig. 7b, d).

Comparison of the amount of PrPSc in the brains of sCJD-3 prion-inoculated mice. a, b PrPSc and tPrP in the brains of mice treated with FK506 or vehicle from 110 d.p.i in sCJD-3-inoculated groups were detected at 140 d.p.i (a) and each terminal stage (b). c, d Band intensities of samples from FK506-treated mice are expressed as a percentage of those of the control mice. The results in the graph are the mean ± SD. β-Actin (ACTB) was used as an internal control for evaluating the amount of tPrP
Discussion
Orally administered FK506 prolonged the survival of sCJD prion-inoculated mice expressing humanized PrP. The treatment suppressed glial cell activation and spongiform changes. These results indicate that FK506 administration possibly delays the progression of pathological damage, resulting in prolonged survival. To our knowledge, this is the first report of successfully treating sCJD in a humanized mouse model.
FK506 has already been used in humans as an immunosuppressant to treat autoimmune diseases and to prevent rejection of organ transplantation and graft versus host disease (GVHD) [29,30,31,32]; furthermore, its side effects and pharmacokinetics are predictable. Administering FK506 at 1.0 mg/kg/day to a mouse is equivalent to 0.081 mg/kg/day for a human, based on a “human equivalent dose” (HED) estimation [33]. FK506 for kidney transplantation patients typically starts at 0.3 mg/kg/day and gradually reduces to 0.12 mg/kg/day, a dose that is taken for years. Therefore, the doses of FK506 used in this study are acceptable for long-term administration to sCJD patients.
Doxycycline treatment also prolonged the survival of mice inoculated with sCJD-1 prion but its effect on survival was less than that of FK506. The survival period of mice receiving FK506 and Dox combination therapy was almost the same as that of the mice receiving FK506 only. Although further experiments are needed, these results indicate that FK506 has potential to improve the symptoms and survival of sCJD patients even after onset of the disease.
In the case of sCJD-3 prion-inoculated mice, accumulation of PrPSc was suppressed in two of four treated mice, whereas it was not inhibited in the other mice at 140 d.p.i. This result is very surprising but should be carefully considered. We have previously reported that FK506 inhibits prion disease progression by promoting PrPSc degradation and inhibiting the proliferation and/or activation of microglia when treated from an early stage of prion infection [21]. On the other hand, other groups reported that administration of FK506 from after symptomatic stage did not affect the amount of PrPSc [34, 35]. These results suggest that FK506 can suppress the accumulation of PrPSc when its amount is relatively low but cannot suppress the accumulation of large amount of PrPSc. In our experiments, the rate of PrPSc accumulation might be more likely to differ between individuals when KiChM mice were inoculated with human BHs at relatively low concentrations comparing to wild-type mice inoculated with mouse adapt-prions (Table 2). In particular, the period just before disease onset, such as 110 d.p.i. in this experiment, might be a time when PrPSc have begun to accumulate explosively, and individual differences in the accumulation of PrPSc are likely to occur. For these reasons, administration of FK506 could suppress the accumulation of PrPSc only in the mice with relatively low amount of PrPSc at 110 d.p.i. It is preferable to start treatment after estimating the amount of PrPSc using the QUIC method in further examination. It will be helpful to solve this problem if we can collect enough volume of central spinal fluid from living mice for QUIC.
The amount of PrPSc in the brain of sCJD patient 2 was about twice as that of sCJD patient 3 (Fig. 1). Then, clinical score of vehicle group of mice inoculated with sCJD-2 prion was significantly worse than that of mice inoculated with sCJD-3 prion (2.33 ± 0.91 vs 1.1 ± 1.91 p value < 0.05). For this reason, the administration of FK506, beginning after obvious symptoms were observed, prolonged the survival of sCJD-3 prion-inoculated, but not of sCJD-2 prion-inoculated mice. Therefore, we predict that administration of FK506 from the early stage of the disease suppresses not only gliosis and vacuolation but also the accumulation of PrPSc. In other words, the early diagnosis of sCJD using QUIC method [36], molecular probes of abnormal proteins [37, 38], and MRI [39] is critical.
In recent decades, the main strategy for developing drugs against prion diseases focused on reducing abnormal PrP accumulation using either small compounds, antibodies against PrP, or siRNA to stop PrP expression. These treatments showed certain effects on prion-inoculated mice [40,41,42]. These findings indicate that the conversion of PrP has an important role in the initial pathogenesis, and that anti-PrP compounds prolong the incubation time of the disease. The preventive use of anti-PrP treatments will be especially beneficial for genetic human prion diseases. However, inhibiting the conversion may be insufficient for patients who have already shown symptoms, because the therapeutic effects of pentosan polysulfate and quinacrine for the patients have been restricted [5, 6].
Glial cells protect neurons by removing aggregated proteins and releasing anti-inflammatory cytokines [20, 43], but they can also exacerbate the disease state by inducing neuroinflammation in several neurodegenerative models including prion diseases [44, 45]. In the sCJD MM1-inoculated mice model, the expression levels of activated glial markers have been upregulated from early clinical stage [46]. Therefore, when treatment is initiated after onset, we predict that modulation of microglia and astrocytes is very important for prolonging the survival of mice with sCJD prion.
Several biological mechanisms have recently been reported as potential therapeutic targets for prion diseases. Stimulation of innate immunity [47,48,49] and autophagy [21, 50, 51] can reduce the amount of PrPSc in infected neurons or brains. The unfolded protein response (UPR) is also over-activated in prion diseases and promotes disease progression, and UPR inhibitors can restore memory loss and extend the survival of prion-inoculated mice [52, 53]. Recently, sCJD prion-infected cells were developed [54, 55]. Using these cells and humanized mice will enable the effects of these new candidates against human prions to be assessed. Although further work is needed to elucidate the details of the mechanism of FK506 action, data indicate that the effect will not depend on prion strain and oral administration of FK506 has good potential as a therapeutic for sCJD. It is worth considering the establishment of an appropriate combination therapy with other drugs that act through various anti-prion mechanisms.
Availability of Data and Materials
All data generated or analyzed during this study are included in this published article.
References
Geschwind MD, Shu H, Haman A, Sejvar JJ, Miller BL. Rapidly progressive dementia. Ann Neurol 2008;64:97-108.
Nozaki I, Hamaguchi T, Sanjo N, et al. Prospective 10-year surveillance of human prion diseases in Japan. Brain 2010, 133:3043-3057.
Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998;95:13363-13383.
Aguzzi A, Heikenwalder M. Pathogenesis of prion diseases: current status and future outlook. Nat Rev Microbiol 2006;4:765-775.
Caughey B, Raymond GJ. Sulfated polyanion inhibition of scrapie-associated PrP accumulation in cultured cells. J Virol 1993;67:643-650.
Doh-Ura K, Iwaki T, Caughey B. Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J Virol 2000;74:4894-4897.
Mead S, Ranopa M, Gopalakrishnan GS, et al. PRION-1 scales analysis supports use of functional outcome measures in prion disease. Neurology 2011;77:1674-1683.
Haik S, Marcon G, Mallet A, et al. Doxycycline in Creutzfeldt-Jakob disease: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol 2014;13:150-158.
Teruya K, Oguma A, Nishizawa K, Kamitakahara H, Doh-Ura K. Pyrene conjugation and spectroscopic analysis of hydroxypropyl methylcellulose compounds successfully demonstrated a local dielectric difference associated with in vivo anti-prion activity. PLoS One 2017;12:e0185357.
Varges D, Manthey H, Heinemann U, et al. Doxycycline in early CJD: a double-blinded randomised phase II and observational study. J Neurol Neurosurg Psychiatry 2017;88:119-125.
Ishibashi D, Nakagaki T, Ishikawa T, , et al. Structure-Based Drug Discovery for Prion Disease Using a Novel Binding Simulation. EBioMedicine 2016;9:238-249.
Miyazaki Y, Ishikawa T, Kamatari YO, et al. Identification of Alprenolol Hydrochloride as an Anti-prion Compound Using Surface Plasmon Resonance Imaging. Mol Neurobiol 2019;56:367-377.
Atarashi R, Satoh K, Sano K, et al. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat Med 2011;17:175-178.
Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol 1994;68:7859-7868.
Sano K, Atarashi R, Nishida N. Structural conservation of prion strain specificities in recombinant prion protein fibrils in real-time quaking-induced conversion. Prion 2015;9:237-243.
Giles K, Berry DB, Condello C, et al. Optimization of Aryl Amides that Extend Survival in Prion-Infected Mice. J Pharmacol Exp Ther 2016;358:537-547.
Sasaki Y, Ohsawa K, Kanazawa H, Kohsaka S, Imai Y. Iba1 is an actin-cross-linking protein in macrophages/microglia. Biochem Biophys Res Commun 2001;286:292-297.
Deininger MH, Meyermann R, Schluesener HJ. The allograft inflammatory factor-1 family of proteins. In Book The allograft inflammatory factor-1 family of proteins. (Editor ed.^eds.), vol. 514. pp. 115-121. City; 2002:115-121.
Rock RB, Gekker G, Hu S, , et al. Role of microglia in central nervous system infections. Clin Microbiol Rev 2004;17:942-964, table of contents.
Sakai K, Hasebe R, Takahashi Y, et al. Absence of CD14 delays progression of prion diseases accompanied by increased microglial activation. J Virol 2013;87:13433-13445.
Nakagaki T, Satoh K, Ishibashi D, et al. FK506 reduces abnormal prion protein through the activation of autolysosomal degradation and prolongs survival in prion-infected mice. Autophagy 2013;9:1386-1394.
von Suesskind-Schwendi M, Gruber M, Touraud D, et al. Pharmacokinetics of a self-microemulsifying drug delivery system of tacrolimus. Biomed Pharmacother 2013;67:469-473.
Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999;46:224-233.
Takatsuki H, Satoh K, Sano K, et al. Rapid and Quantitative Assay of Amyloid-Seeding Activity in Human Brains Affected with Prion Diseases. PLoS One 2015;10:e0126930.
Takatsuki H, Fuse T, Nakagaki T, et al. Prion-Seeding Activity Is widely Distributed in Tissues of Sporadic Creutzfeldt-Jakob Disease Patients. EBioMedicine 2016;12:150-155.
Taguchi Y, Mohri S, Ironside JW, Muramoto T, Kitamoto T. Humanized knock-in mice expressing chimeric prion protein showed varied susceptibility to different human prions. Am J Pathol 2003;163:2585-2593.
Wilham JM, Orru CD, Bessen RA, et al. Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog 2010;6:e1001217.
Liberski PP, Brown P. Astrocytes in transmissible spongiform encephalopathies (prion diseases). Folia Neuropathol 2004;42:71-88.
Messina C, Faraci M, de Fazio V, et al. Prevention and treatment of acute GvHD. Bone Marrow Transplant 2008;41:S65-70.
Shrestha BM. Two Decades of Tacrolimus in Renal Transplant: Basic Science and Clinical Evidences. Exp Clin Transplant 2017;15:1-9.
Cruz JL, Wolff ML, Vanderman AJ, Brown JN. The emerging role of tacrolimus in myasthenia gravis. Ther Adv Neurol Disord 2015;8:92-103.
Kraaij T, Bredewold OW, Trompet S, et al. TAC-TIC use of tacrolimus-based regimens in lupus nephritis. Lupus Sci Med 2016;3:e000169.
Bae JW, Kim DH, Lee WW, Kim HY, Son CG. Characterizing the human equivalent dose of herbal medicines in animal toxicity studies. J Ethnopharmacol 2015;162:1-6.
Mukherjee A, Morales-Scheihing D, Gonzalez-Romero D, Green K, Taglialatela G, Soto C. Calcineurin inhibition at the clinical phase of prion disease reduces neurodegeneration, improves behavioral alterations and increases animal survival. PLoS Pathog 2010;6:e1001138.
Shah SZA, Zhao D, Taglialatela G, et al. Early Minocycline and Late FK506 Treatment Improves Survival and Alleviates Neuroinflammation, Neurodegeneration, and Behavioral Deficits in Prion-Infected Hamsters. Neurotherapeutics 2017;14:463-483.
Atarashi R, Wilham JM, Christensen L, et al. Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat Methods 2008;5:211-212.
Fuchigami T, Yamashita Y, Kawasaki M, et al. Characterisation of radioiodinated flavonoid derivatives for SPECT imaging of cerebral prion deposits. Sci Rep 2015;5:18440.
Kawasaki M, Fuchigami T, Kobashi N, et al. Development of radioiodinated acridine derivatives for in vivo imaging of prion deposits in the brain. Bioorg Med Chem 2017;25:1085-1093.
Warden DRt, Dennison JV, Limback J, Shroff SM, Messina SA. Imaging Manifestations of Creutzfeldt-Jakob Disease and Case Series. Cureus 2018;10:e3725.
White MD, Farmer M, Mirabile I, Brandner S, Collinge J, Mallucci GR. Single treatment with RNAi against prion protein rescues early neuronal dysfunction and prolongs survival in mice with prion disease. Proc Natl Acad Sci U S A 2008;105:10238-10243.
Song CH, Furuoka H, Kim CL, et al. Effect of intraventricular infusion of anti-prion protein monoclonal antibodies on disease progression in prion-infected mice. J Gen Virol 2008;89:1533-1544.
Ohsawa N, Song CH, Suzuki A, Furuoka H, Hasebe R, Horiuchi M. Therapeutic effect of peripheral administration of an anti-prion protein antibody on mice infected with prions. Microbiol Immunol 2013;57:288-297.
Aguzzi A, Zhu C. Microglia in prion diseases. J Clin Invest 2017;127:3230-3239.
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell 2010;140:918-934.
Tang Y, Le W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol Neurobiol 2016;53:1181-1194.
Llorens F, Lopez-Gonzalez I, Thune K, et al. Subtype and regional-specific neuroinflammation in sporadic creutzfeldt-jakob disease. Front Aging Neurosci 2014;6:198.
Spinner DS, Cho IS, Park SY, et al. Accelerated prion disease pathogenesis in Toll-like receptor 4 signaling-mutant mice. J Virol 2008;82:10701-10708.
Kang SG, Kim C, Cortez LM, et al. Toll-like receptor-mediated immune response inhibits prion propagation. Glia 2016;64:937-951.
Ishibashi D, Homma T, Nakagaki T, et al. Type I interferon protects neurons from prions in in vivo models. Brain 2019;142:1035-1050.
Aguib Y, Heiseke A, Gilch S, et al. Autophagy induction by trehalose counteracts cellular prion infection. Autophagy 2009;5:361-369.
Ishibashi D, Homma T, Nakagaki T, et al. Strain-Dependent Effect of Macroautophagy on Abnormally Folded Prion Protein Degradation in Infected Neuronal Cells. PLoS One 2015;10:e0137958.
Halliday M, Radford H, Zents KAM, et al. Repurposed drugs targeting eIF2α-P-mediated translational repression prevent neurodegeneration in mice. Brain 2017;140:1768-1783.
Moreno JA, Halliday M, Molloy C, et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med 2013;5:206ra138.
Krejciova Z, Alibhai J, Zhao C, et al. Human stem cell-derived astrocytes replicate human prions in a PRNP genotype-dependent manner. J Exp Med 2017;214:3481-3495.
Groveman BR, Foliaki ST, Orru CD, et al. Sporadic Creutzfeldt-Jakob disease prion infection of human cerebral organoids. Acta Neuropathol Commun 2019;7:90.
Acknowledgments
We thank Takayuki Fuse, Atsuko Matsuo, Hanako Nakayama, Ayako Nakazaki, Megumi Tanaka, and Marie Yamaguchi for their technical assistance. We also thank Katie Oakley, Ph.D., from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript. This work was supported by a grant-in-aid for scientific research from YOKOYAMA Foundation for Clinical Pharmacology (grant no. YRY-1602) and Takeda Science Foundation; a grant-in-aid of Young Scientists (B) (17 K17126), Scientific Research (C) (16K07042) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan; and a Grant-in-Aid of the Research Committee of Prion Disease and Slow Virus Infection from the Ministry of Health, Labour and Welfare of Japan (H29-036).
Author information
Authors and Affiliations
Contributions
TN and RA designed the experiments. TM, YM, HT, and HT provided animals. TN performed the experiments. KS provided tissues of patients. TN, DI, YT, RA, and NN analyzed data. TN and NN wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
Additional file 1: Criteria of clinical scores. Criteria for assigning clinical scores are shown. These scores were determined by animal body weight and the existence of symptoms, such as priapism, hunchback, ataxic gait, and non-parallel hind limbs. Additional file 2: Comparison of Intensity of PrPSc and SD50 in the each brains of mice inoculated with sCJD-3 prion. The intensity and SD50 at 140d.p.i in the brains of mice inoculated with sCJD-3 prion were measured by Western blotting and QUIC, respectively. (PDF 171 kb).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nakagaki, T., Ishibashi, D., Mori, T. et al. Administration of FK506 from Late Stage of Disease Prolongs Survival of Human Prion-Inoculated Mice. Neurotherapeutics 17, 1850–1860 (2020). https://doi.org/10.1007/s13311-020-00870-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-020-00870-1