
Abstract
Amyotrophic lateral sclerosis (ALS) is a debilitating neurodegenerative disorder in which the neuromuscular junction progressively degenerates, leading to movement difficulties, paralysis, and eventually death. ALS is currently being treated by only two FDA-approved drugs with modest efficacy in slowing disease progression. Often, the translation of preclinical findings to bedside terminates prematurely as the evaluation of potential therapeutic compounds focuses on a single study or a single animal model. To circumscribe these issues, we screened 3,765 novel small molecule derivatives of pimozide, a recently identified repurposed neuroleptic for ALS, in Caenorhabditis elegans, confirmed the hits in zebrafish and validated the most active compounds in mouse genetic models. Out of the 27 small molecules identified from the high-throughput screen in worms, 4 were found to recover locomotor defects in C. elegans and genetic zebrafish models of ALS. TRVA242 was identified as the most potent compound as it significantly improved efficiency in rescuing locomotor, motorneuron, and neuromuscular junction synaptic deficits in a C. elegans TDP-43 model and in multiple zebrafish genetic (TDP-43, SOD1, and C9ORF72) models of ALS. The actions of TRVA242 were also conserved in a mammalian model as it also stabilized neuromuscular junction deficits in a mouse SOD1 model of ALS. Compounds such as TRVA242 therefore represent new potential therapeutics for the treatment of ALS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by weakness and atrophy of muscles, eventually leading to paralysis and death with a mean survival window of 2–5 years post clinical diagnosis [1]. There are only two FDA-approved drugs with modest efficacy in slowing disease progression—Riluzole and Radicava. This prompts the identification and validation of new therapeutic molecules for the treatment or cure for this debilitating disease. Although much of the research in ALS has focused on developing suitable genetic models to recapitulate pathological hallmarks of the disease and generate meaningful insights of the neuropathology of this disease, the mechanism of neuronal dysfunction in ALS needs more elucidation.
The notion of ALS neuropathology as either a dying forward or “die back” phenomenon presents interesting hypotheses. The dying forward phenomenon involves glutamate-mediated excitotoxicity or abnormal neuronal excitability with consequently induced neuronal degeneration in the cortex that spreads to corticospinal projections, subsequently affecting spinal cord motor neurons [2]. On the other hand, motoneuron degeneration commencing at nerve endings in the spinal cord and eventual progression towards motoneuron soma constitute the dying back or distal axonopathy hypothesis [3, 4]. Both phenomena lead to perturbations in the neuromuscular junction structure and function, culminating in skeletal muscle denervation.
Distal axonopathy appears early in the pathological scenario of ALS and presents itself before the clinical manifestations of the symptoms. Different genetic models of ALS have consistently confirmed this observation. For example, it has been observed that muscles of mutant super oxide dismutase 1 (SOD1) mice display neuromuscular junction (NMJ) and distal axonal abnormalities before the onset of clinical symptoms [5,6,7]. Similarly, disruption of the NMJ preceding a clinical phenotype and motor neuron loss has also been observed in fused in sarcoma (FUS) transgenic mice [8]. Mutation in the transactive response DNA-binding protein-43(TDP-43) gene of mice also induces synaptic neurotransmission defects at the NMJ before the appearance of motor deficits and motor neuron loss [9]. Additionally, our laboratory has developed a new zebrafish model that highlights C9ORF72 mediated dysfunction in ALS. C9ORF72 has been identified as the most prevalent genetic cause of sporadic ALS [10]. RNA- or dipeptide-mediated toxic gain-of-function as well as haploinsufficiency mediated loss-of-function hypotheses have been proposed and expression of GR dipeptides in the motor neurons of zebrafish is sufficient to induce spinal motorneuron outgrowth and locomotor defects [11, 12]. Such compelling evidence combined with a similar observation in tissue from patients with ALS suggest NMJ defects as an early ALS pathology and provides a target for tailoring therapeutics [13].
Genetic models of ALS in Caenorhabditis elegans and zebrafish have emerged as robust tools to screen and validate active small molecules. Age-dependent paralysis and neurodegeneration in fused in sarcoma (FUS) and TDP-43 models in C. elegans serve as biomarkers to assess the potential of compounds in stabilizing motility defects and minimizing neurodegeneration [14]. Morpholino mediated transient downregulation TDP-43 and its orthologues or overexpression of mutant human TARDBP in zebrafish have been shown to induce defects in NMJ architecture and transmission, suggesting that the NMJ manifests both loss- and/or gain-of-function toxic effects and as such these phenotypes could possibly be recovered by active compounds [15,16,17].
However, one of the major impediments in translation of findings from bench to bedside of potentially effective molecules is the restriction of the evaluation to a single study or single animal model. In this light, we have shown the versatility of the repurposed neuroleptic pimozide in different animal models of ALS, thereby increasing its chances of success when tested in a sporadic population of ALS patients [17]. In a similar attempt to identify more effective molecules of therapeutic interest, here, we tested structurally similar derivatives of pimozide and related compounds in genetic models of ALS and screened 3,765 compounds from two different libraries (Treventis and Zalicus) in our drug screening platform and identified TRVA242 as our most active compound. To assess and validate the neuroleptic properties of TRVA242, we subjected this compound to subsequent analyses in our genetic models of ALS in C. elegans, zebrafish, and mice, focusing on the NMJ as a biomarker. Our observations indicate that TRVA242 restores disturbances in NMJ transmission and rescues important pathological phenotypes in all our models of ALS, suggesting its potential as a therapeutic alternative in ALS.
Methods
C. elegans Studies
Standard methods of culturing and handling C. elegans were used [18]. Worms were maintained on standard NGM plates streaked with OP50 Escherichia coli. Strain xqIs133[unc-47::TDP-43[A315T];unc-119(+)] was described previously (Vaccaro et al. 2012 PMID: 22363618) and was used for drug screening assays.
Drug Testing in Liquid Culture
Age-synchronized young adult mTDP-43 transgenic worms were distributed in 96-well plate in M9 buffer (20 μl per well, approximately 60 worms per well), containing DMSO or test compounds. The swimming activity of the nematodes was measured by a WMicroTracker machine (Phylum Tech) [19] and measured over a period of 10 h. Experiments were done in triplicate. All positively acting compounds were retested and those showing comparable results kept for further investigation. All experiments were conducted at 20 °C. Significance was determined using a two-way ANOVA with multiple comparison post hoc analysis (GraphPad Prism).
Paralysis Assays on Solid Media
Transgenic TDP-43 worms were exposed from hatching (by synchronization) to 5 μM TRVA242 incorporated into NGM solid medium or NGM solid medium only as a control. All the plates were streaked with OP50 E. coli. 30–40 mTDP-43 worms were monitored for motility from adult day 1 to adult day 12. Animals were scored as paralyzed if they failed to move after being prodded with a worm pick. Worms were scored as dead if they were unable to respond to tactile head stimulus. They were transferred to fresh plates every 2 days until the cessation of progeny production. All experiments were conducted at 20 °C and, each condition was done in triplicates with 30–40 worms/plate. Paralysis curves were generated and compared using the Log-rank (Mantel-Cox) test. All experiments were repeated at least three times.
Neurodegeneration Assay
For scoring of neuronal processes for gaps or breakages, unc-47::GFP; mTDP-43 worms collected at adult day 9 for visualization of motor neurons processes in vivo. For assessment of neuronal integrity, mean ± standard deviation of eight independent experiments was calculated and significance determined using two-tailed t tests.
Zebrafish Genetic Models
Wild-type zebrafish (Danio rerio) were bred and maintained according to standard procedures [20]. All experiments were performed in compliance with the guidelines of the Canadian Council for Animal Care and conducted at the Centre de Recherche du centre hospitalier de l’Universite de Montreal (CRCHUM) and approved by the animal care committee of the CRCHUM (N15018PMDz). All experiments were performed on sexually undifferentiated zebrafish larvae between 1 and 11 days post-fertilization (dpf). Humane endpoints were in place during the study and all larvae were monitored and assessed daily for well-being as per guidelines established by the Canadian Council of Animal Care committee at the CRCHUM. Behavioral signs of poor health in adult larvae necessitating euthanasia included an inability to feed and swim. Physical abnormalities were also monitored daily and adult larvae displaying a distended abdomen, skin ulcerations/wounds, and skeletal deformities were euthanasia immediately.
Zebrafish GOF Model
For studies using zebrafish GOF models, we pressure injected human TDP-43 WT and mutant (G348C) or human SOD1 WT and mutant (G93A) which were diluted in nuclease-free water (Ambion) with 0.05% Fast Green vital dye (MilliporeSigma) at a concentration of 30 ng/μl (TDP-43), and 100 ng/μl (SOD 1) at 1–2 cell stage embryos, using a Picospritzer III pressure ejector. Briefly, these mRNAs were transcribed from NotI-linearized pCS2+ using SP6 polymerase with the mMESSAGE Machine Kit (Ambion). This was followed by a phenol-chloroform extraction and isopropanol precipitation, and diluted in nuclease-free water (Ambion).
Drug Treatments
At 24-h post-fertilization (hpf), zebrafish embryos were dechorionated and placed in individual wells in a 24-well plate and treated overnight with compounds. For the initial screen, all 24 compounds were tested at 20 μM (dissolved in DMSO at a final concentration of 0.01%). For subsequent analyses in zebrafish, TRVA242 was tested at a concentration of 5 μM. DMSO at a concentration of 0.01% or 0.05% were tested at 24 hpf in uninjected, wt TDP-43, mut-TDP-43, wt-SOD1, mut-SOD-1, and GR100UAS:Hb9 larvae and no gross developmental or physiological effect was detected in our assays.
Touch Evoked Escape Response Assay
Touch-evoked escape response was tested at 48 dpf in control and drug-treated conditions. Larvae were place in the center of a circular aquatic arena (150-mm diameter) and touched lightly on the tails with forceps. The water temperature was maintained at 28.5 °C. Locomotor behavior was recorded digitally at 30 Hz for 10 s (Grasshopper 2 camera; Point Gray Research). Swim duration, swim distance, and maximum swim velocity were quantified off-line using the manual tracking plug-in for ImageJ.
Motor neuron outgrowth patterns in GOF models and C9ORF72 model: human TDP-43 WT and mutant (G348C) or human SOD1 WT and mutant (G93A) were injected in the zebrafish Hb9:Tg line and treated with compounds at 24 hpf (as explained in the drug treatment protocol section) and imaged at 48 hpf using a Quorum Technologies spinning disk confocal microscope with a CSU10B (Yokogawa) spinning head mounted on an Olympus BX61W1 fluorescence microscope and connected to a Hamamatsu ORCA-ER camera. Images were acquired and analyzed using Volocity software (Improvision).
Immunohistochemistry
For whole mount immunohistochemistry to elucidate NMJ structure, 48 hpf larvae were fixed in 4% paraformaldehyde overnight at 4 °C. After fixation, the larvae were rinsed several times (1 h) with phosphate-buffered saline (PBS) and then incubated in PBS containing 1 mg/ml collagenase (25 min) to remove skin. The collagenase was washed off with PBS (1 h) and the larvae were incubated in PBS with Triton X-100 (PBST) for 30 min. The larvae were then incubated with PBST containing 10 μg/ml sulforhodamine-conjugated α-bungarotoxin (αBTX; 30 min), which binds irreversibly to acetylcholine receptors (AChRs). The larvae were then rinsed several times with PBST (30 min) and then incubated in fresh block solution prepared from PBS containing goat serum, bovine serum albumin, dimethyl sulfide (DMSO), and Triton X-100, for 1 h at room temperature and then treated with a solution containing a primary antibody against presynaptic synaptotagmin 1 (ZNP-1) (1:200; Molecular Probes) overnight at 4 °C. Samples were then washed in PBST and were incubated in block solution containing a secondary antibody (Alexa Fluor 488, 1:1000; Invitrogen) for 6 h at 4 °C. Before imaging, larvae were transferred to a solution containing 70% glycerol and mounted the following day on a slide. The NMJs were visualized using a Quorum Technologies spinning disk confocal microscope with a CSU10B (Yokogawa) spinning head mounted on an Olympus BX61W1 fluorescence microscope and connected to a Hamamatsu ORCA-ER camera. Images were acquired using Volocity software (Improvision) and analyzed using Image J.
Whole-Cell Voltage-Clamp Recordings in Fast-Twitch Muscle Fibers
As previously described [21], zebrafish 48 hpf were anesthetized in 0.02% tricaine (Sigma) dissolved in modified Evans solution containing the following (in mM): 134 NaCl, 2.9 KCl, 2.1 glucose, adjusted to 290 mOsm, pH 7.8. The zebrafish were then pinned with fine (0.001 in.) tungsten wires through their notochords to a Sylgard-lined dish. The outer layer of skin was removed using a fine glass electrode and forceps, to expose the underlying musculature. The preparation was visualized by oblique illumination (Olympus BX51WI). Standard whole-cell voltage-clamp recordings were obtained from fast-twitch (embryonic white) muscle cells [21]. To isolate and record spontaneous (quantal) miniature endplate currents and block the generation of action potentials, 1 μM tetrodotoxin (TTX) was perfused over the preparation throughout the duration of the recordings (10 min). Glass electrodes (8–12 MO) were pulled from filament-containing thin-walled borosilicate glass capillary (A-M Systems) and filled with the following intracellular solution containing (in mM): 116 K-gluconate, 16 KCl, 2, and 10 EGTA adjusted to pH 7.2, 290 mOsm. Cells were held near their resting potential at − 60 mV and series resistance was between 12 and 35 MO compensated to 40–60%. All electrophysiological data were sampled at 50 kHz using an Axopatch 200B amplifier (molecular devices), digitized using a Digidata 1440A (molecular devices) and analyzed off-line using pCLAMP 10.1 software (molecular devices). In addition to recording mEPCs, we also recorded the muscle cell membrane potential (Vm), whole-cell capacitance (Cm), membrane resistance (Rm), and access resistance of the electrode (Ra) in all the drug treated and control conditions.
Zebrafish LOF Model
For LOF studies in zebrafish, we utilized an already existing zebrafish tardbp loss-of-function model containing a point mutation (c660C > T) resulting in a premature stop codon at the 220th amino acid [22]. We also generated a tardbpl loss-of-function model by CRISPR/Cas9 editing, with a 5base pair deletion encompassing the ATG start codon. Subsequently, we incrossed adult zebrafish that were heterozygous for tardbp and homozygous for tardbpl (tardbp+/−tardbpl−/−) to obtain a population of embryos that were homozygous, for both tardbp and tardbpl (tardbp−/−tardbpl−/−).
Synthesis of Cas9 mRNA and gRNA
Synthesis of zebrafish-optimized nls-zCas9-nls mRNA was done using previously described methods [23] and the pCS2-nCas9n was a gift from Wenbiao Chen (Addgene plasmid # 47929). Briefly, the nls-zCas9-nls template was linearized with NotI and synthesized using the mMESSAGE mMACHINE SP6 kit (Ambion/Invitrogen) followed by a phenol-chloroform extraction and ethanol precipitation. The pT7-gRNA was a gift from Wenbiao Chen (Addgene plasmid # 46759). The pT7-gRNA was linearized with BamHI and RNA was transcribed using the MEGAshortscript T7 kit (Ambion/Invitrogen) and extracted and precipitated using the same methods for nls-zCas9-nls mRNA.
The gRNA targets the following sequence:
tardbpl: CGCATTCGGTGTAATCATGACGG (forward strand, targeting exon 1);
Restriction Fragment Length Polymorphism Assay
Genomic DNA was extracted from individual 2 days post fertilization (2 dpf) larvae using the RED Extract-N-AMP Tissue PCR kit (Sigma-Aldrich) and used as a template for PCR using the respective primer sets. Genotyping of the tardbp and tardbpl mutation was done using the following primer sets (tardbp-forward CAAGGTATAGATGAACCAATGAGGA) and (tardbp-reverse: GTCATCTGCAAAGGTGACAAAAG), (tardbpl- forward: GCCACAACTCGTGTAGCCAAC) and (tardbpl reverse: GCAGGTCGAACCCATTATGATTG).
For genotyping tardbp mutations, an amplified PCR product of 168 bp was digested with RsaI resulting in two bands (97 and 71 bp respectively) in wild-type (WT) larvae. The RsaI restriction site is lost in zebrafish carrying the 660 (C->A) point mutation, allowing identification of larvae carrying Y220X missense mutation. For genotyping tardbpl mutations, an amplified PCR product of 486 bp in length was digested with BsphI, resulting in two bands (152 and 334 bp respectively) in wild-type (WT) larvae. The BsphI restriction site is lost in the mutant allele, allowing identification of the tardbpl mutation.
Drug Treatment for LOF Model
Adult zebrafish that are heterozygous for tardbp and homozygous for tardbpl were in-crossed to obtain a mixed population of embryos that were homozygous, heterozygous, and wild type for tardbp. Forty embryos were used for each trial and compounds were tested at 5 μM and 20 μM with 0.5% DMSO as negative control. Drugs were added at 90% epiboly stage, and at 2 dpf the number of larvae exhibiting homozygous phenotype were calculated from three separate sets of experiment.
At 5 dpf, quantitative assay was performed using the Zebrabox to evaluate the motility of the treated fish. Embryos displaying milder phenotype, presuming those are homozygotic embryos whose phenotype had been alleviated due to the presence of drug were picked up for motility assay. One embryo was placed in each well in a 24-well plate and the motility was measured using the camera fixed in the Zebrabox. Wild-type embryos were used as controls. The homozygous embryos were confirmed by amplifying the mutant section from the genomic DNA and performing a RsaI enzyme digestion assay. Small distance and large distance movements were measured for the homozygous embryos using the Zebrabox Viewpoint software and analyzed manually.
Zebrafish C9ORF72 Model
Generation of UAS-GR Transgenic Line
UAS-GR transgenic line was used for assessing locomotor and motor neuron defects as described previously [12]. Briefly, the multisite Gateway technology-based Tol2kit was used to assemble the construct to generate the transgenic zebrafish line [24]. The pDONR221 middle donor vector containing the GR construct was a gift from Prof. Aaron Gitler, Stanford University. This was used along with the 50 entry clone p5E-UAS and 30 entry clone p3E-polyA to insert these elements into the pDestTol2CG2 destination vector. For the recombineering reaction, 10 fmol of each of the entry/donor vectors and 20 fmol of the destination vector were mixed with LR Clonase II Plus enzyme mix (Invitrogen), and incubated at 25 °C for 16 h. A part of the reaction mix was transformed into chemically competent Escherichia coli following proteinase K treatment. The presence of the GR construct in plasmids extracted from selected clones was confirmed by sequencing. A mixture containing 40 ng/ll of the destination vector with the GR construct and 40 ng/ll of transposase mRNA was injected into 1 cell stage embryos (1 nl per embryo). Screening was done by performing outcrosses and checking for the presence of progeny with green heart. These embryos were grown to adulthood and then crossed with Hb9:Tg line to selectively express GR in the motorneurons and subsequently used for experimental analysis.
Motor Neuron Outgrowth Patterns in Zebrafish C9ORF72 Model
For C9ORF72 studies, UAS:GR transgenic line was crossed with Hb9:Tg to selectively express GR in motorneurons. Embryos with green heart and GFP labeled motorneurons and axons were subjected to treatment with TRVA242 (5 μM) at 24 hpf and imaged at 48 hpf using a Quorum Technologies spinning disk confocal microscope with a CSU10B (Yokogawa) spinning head mounted on an Olympus BX61W1 fluorescence microscope and connected to a Hamamatsu ORCA-ER camera. Images were acquired and analyzed using Volocity software (Improvision).
Measurement of Swimming Activity
Swimming activity was measured in 5 dpf larvae transferred individually into a 96-well plate in drug treated and control conditions and incubated in the Daniovision recording chamber (Noldus) for 1 h in the dark for habituation. Activity was then recorded over 1 h using Basler GenIcam camera. Analysis was performed using the Ethovision XT 12 software (Noldus) to quantify the distance swam.
Mouse Studies
Genetic Model
Mice used in this study were overexpressing the G37R mutant human SOD1 transgene. SOD1G37R male mice were acquired from the Jackson Laboratory and bred with females on a C57BL/6 background at our animal facility at Université de Montréal. Heterozygous SOD1G37R mutants usually present an overexpression of 2 to 5 copies of the mutant SOD1 protein in the spinal cord and the brain [25]. The SOD1G37R mouse model is characterized by a slow progression of the disease, with onset of locomotor deficits and hind-limb paralysis after 1 year of age. Mice show clear motor symptoms and phenotype similar to ALS human patients. Experiments were performed on SOD1G37R male mice and their WT littermates. To identify mutant mice from their WT counterparts, a small biopsy of the tail was obtained directly after the weaning times (3–4 weeks of age), in order to perform PCR amplification of the human SOD1 gene. To investigate the symptomatic period, 13- to 15-month-old mice (P400–450) were used to assess the early clinical period of the disease (see Tremblay et al. [7] for the characterization of disease progression in this specific model).
Nerve-Muscle Preparations
The Extensor Digitorum Longus (EDL), a fast-twitch muscle, was carefully dissected along with the deep peroneal nerve in an oxygenated Ringer’s solution (95% O2, 5% CO2) containing the following compounds (in mM): 110 NaCl, 5 KCl, 1 MgCl2, 25 NaHCO3, 2 CaCl2, 11 glucose, 0.3 glutamic acid, 0.4 glutamine, 5 BES, 0.036 choline chloride, and 4.34 × 10−7 cocarboxylase. Muscles were pinned in a sylgard-coated recording chamber containing the Ringer’s solution after dissection, and the nerve was suctioned by a stimulating electrode in the recording chamber. Muscle contractions were first assessed by stimulating the deep peroneal nerve (threshold around 1–10 mV). Then, muscles ere perfused with an oxygenated (95% O2, 5% CO2) modified Ringer’s solution with low Ca2+ (1 mM) and high Mg2+ (6.4 mM) to block muscle contractions during synaptic recordings. Temperature of the Ringer’s solution was adjusted continuously between 28 and 30 °C.
Electrophysiological Recordings of NMJ Synaptic Transmission
Electrophysiological recordings were performed as previously described [7, 17]. Briefly, we recorded EDL evoked synaptic activity by performing a suprathreshold stimulation of its nerve (5×–10×) using a suction electrode filled with the Ringer’s solution. Sharp glass microelectrodes filled with 3 M KCl (30–60 MΩ) were used to perform intracellular recordings of end plate potentials (EPPs) and miniature endplate potential (mEPPs). After localization of a given NMJ, muscle fiber associated to this NMJ was impaled around 30–50 nm from this NMJ [17]. Using an HS-2A Headstage by Axon Instruments, EPP and mEPP signals were first amplified. A second amplification was applied (1,000×) and finally filtered with a low-pass filter (2 KHz) by a Warner Instruments DC amplifier. We used a National Instruments BNC 2110 board to digitize the recordings. Acquisition of the data was performed with WinWCP software (provided by John Dempster, Strathclyde University, Strathclyde, UK).
Synaptic strength of the recorded NMJs was assessed using four parameters: mEPP frequency, mEPP amplitude, EPP amplitude, and quantal content. First, basal spontaneous activity of the muscle was assessed by recording during 5 to 10 min, without any motor nerve stimulation, to allow the measurements of mEPP amplitude (mean of 80–100 mEPPs) and mEPP frequency. The latter was obtained by calculating the number of mEPPs events occurring over a period of at least 5 min. mEPP frequency was expressed in Hz (mEPP/s). Then, stimulation protocol to elicit evoked activity and calculate synaptic strength consisted of 2 nerve stimuli (0.1-ms duration) every 5 s (0.2 Hz), with a 10-ms interval between stimuli. The method of mEPPs was used to determinate quantal content according to the following formula: mean amplitude of the first EPPs/mean amplitude of mEPPs. The quantal content calculation included also the failures.
TRVA241 and TRVA242 impact on neurotransmission. To investigate the effect of the Treventis 1441 and 1442 compounds on neuromuscular transmission at the SOD1G37R mice NMJ, we performed acutely bath applications of either one of the compounds on EDL nerve-muscle preparations, after initially recording basal and evoked transmission for 25 min. TRVA241 and TRVA242 powders were first dissolved in DMSO (20 mM) and then added to the low Ca++/high Mg++ Ringer’s solution at a concentration of 5 μM (0.02% DMSO). To investigate drug effect on neurotransmission, nerve-muscles preparations were perfused for a period of 30 min to allow the drug to exert its effect on the NMJ. Spontaneous activity and evoked activity were then recorded again following the same protocol previously described for a period of 30–60 min. Only paired experiments are presented in this study, with the same mutant NMJ being recorded before and after bath application of the drug.
Results
High-Throughput Screen in Genetic Models of ALS—C. elegans and Zebrafish
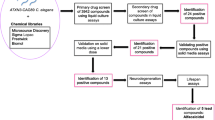
Figure 1A shows a schematic of the initial screening of compounds in zebrafish and worms. Specifically, we conducted a high-throughput screen of 3,768 compounds using transgenic C. elegans strains expressing mutant human TDP-43 (TARDBPA315T, referred to herein as mTDP-43). These worms show age-dependent motility defects leading to paralysis phenotypes in a matter of hours when grown in liquid culture [22]. Upon rescreening candidate molecules, we initially identified 11 compounds that improved motility in mTDP-43 worms when tested for 6 h at a concentration of 20 μM (Fig. 1B).

TRVA242 identified as the most potent compound from small molecule libraries (3765 compounds) in genetic models of ALS. (A) Small molecule libraries from Zalicus and Treventis were initially screened in a TDP-43 C. elegans model of ALS. Individual animals were placed in 96-well plates containing test compounds and treated for 6 h to assess improvement in motility. The 11 hits from this initial screen were tested in the GOF zebrafish model of ALS at a range of concentrations (1–20 μΜ). 10–12 mut-TDP-43 animals were placed in each well of a 24-well plate chamber containing the 24 hits and incubated overnight starting at 24 hpf and 4 compounds were found to be potent in recovering mobility impairments and TRVA242 was identified as the most potent compound, active at 5 μΜ. (Β) Chemical genetic screen identified 11 compounds that suppress motility defects in transgenic TDP-43 worms. Drug screening identified 11 molecules that suppressed motility defects in mutant TDP-43 transgenics compared to untreated controls (*p < 0.0001, †p < 0.01, p < 0.001, °p < 0.05)
Overexpression of mutant human TDP-43(TARDBP G348C, herein referred to as mut-TDP-43) or SOD-1(SOD-1G93A, herein referred to as mut-SOD-1) by mRNA injection in zebrafish leads to a toxic gain-of-function (GOF) and induces abnormalities that are reminiscent of pathological hallmarks of zebrafish models of ALS [16, 26]. At 48-h post-fertilization (hpf), larvae display a locomotor defect assayed as an impairment in generating a touch evoked escape response (TEER). Upon overnight treatment at 20 μM (starting at 24 hpf) with the hits identified from the initial high-throughput screen in worms, our primary screen in mut-TDP43 zebrafish larvae confirmed the potential of some of the active compounds (4 out of 11) (Supplementary figure S1 A-C) and TRVA 242 was identified as the most potent compound in both the worm and zebrafish screens. Therefore, all subsequent analyses were conducted with TRVA242.
It has been previously established that motor neuron defects due to loss of the tardbp gene can be rescued by an alternative splice variant encoded by the tardbp-like gene tardbpl [22]. Knock-out of tardbpl gene by antisense morpholino in the tardbp mutant fish leads to developmental defects and reduced survival in the zebrafish embryos [15, 22]. In this study, through a CRISPR/Cas9-mediated system, we generated a mutant zebrafish line that is a homozygotic for tardbpl and heterozygotic for tardp. When in-crossed, a population (25%) of zebrafish embryos are homozygous for both tardbp and tardbpl (tardbp−/−tardbpl−/−) and displayed a curved body, reduced motility and heart edema phenotype. These double loss-of-function (LOF) mutants had a significantly higher mortality rate when compared to the tardbp heterozygotes and wild-type siblings (p < 0.05, Log-rank test) (Supplementary figure S1 (D and E)). Based on the results from the TDP43 GOF function model, we selected the seven most active compounds and tested them on the zebrafish TDP-43 LOF model to examine whether the drugs could alleviate the phenotype and extend survival of the homozygotic larvae. Embryos were treated at 90% epiboly stage with TRVA242 at a concentration of 5 and 20 μM. After 2-day post-fertilization (dpf), treatment with 20 μM TRVA242 showed a 25% reduction in homozygotic embryos with a homozygotic phenotype at 5dpf compared to untreated controls (Supplementary figure S1 F). No rescue of homozygotic phenotype or improvement in survival was observed at 5 μM, and we attribute this to the severity of the phenotype in the double mutants.
TRVA242 Rescues Locomotor Deficits in Zebrafish and C. elegans Models of ALS
Following the initial screen in mut-TDP-43 larvae, all subsequent analyses were conducted in mut-TDP-43 and mut-SOD1 injected GOF embryos. Such an approach was undertaken to test for the neuroprotective effects of TRVA242 in two independent zebrafish models of ALS. Locomotor defects in zebrafish were characterized by monitoring the spontaneous coiling frequency (burst count/min) between 17 and 18 hpf, when the first signs of embryonic motor behavior manifest [27, 28]. We found that mut-TDP-43 and mut-SOD1 embryos displayed significantly decreased and increased coiling frequency, compared to their respective wild-type human mRNA injected controls, respectively. However, both these parameters were not found to be significantly different in uninjected zebrafish larvae compared to wild-type human mRNA injected larvae (data not shown). Upon treatment with TRVA242 (5 μM), the frequencies of coils were closer to those of wild-type human mRNA injected controls (herein referred to as wt-TDP43 and wt-SOD1) (Fig. 2A; p < 0.05 Kruskal–Wallis test, post hoc Duncan’s test). However, the mean burst duration (time taken to generate a single coil) in both mut-TDP43 and mut-SOD1 embryos was not significantly different (Fig. 2B). This is suggestive that electrically coupled spinal network activity is altered differently in SOD1 vs TDP-43 zebrafish models of ALS, leading to subsequent locomotor defects [29, 30].

TRVA242 rescues early motor behavior and locomotor deficits in zebrafish GOF models of ALS. Burst count per minute (A) and burst duration (B) was measured for the following treatment groups wt TDP-43, mut TDP-43, mut TDP-43+TRVA242 (5 μM), wt SOD-1, mut SOD-1, mut SOD-1+TRVA242 (5 μM) at 17 hpf. mut TDP-43 and mut SOD-1 displayed significantly reduced and increased burst count per minute compared to their respective controls. No significant change in burst duration was observed across all groups. Number in parentheses include sample sizes. Data are expressed as mean ± SEM. N = 3; **p < 0.01. Superimposed locomotor path traces of 48 hpf larvae from the following treatment groups: wt TDP-43, mut TDP-43, mut TDP-43+TRVA242 (5 μM), wt SOD-1, mut SOD-1, mut SOD-1+TRVA242(5 μM) (C). Swim distance (D), swim duration (E), and maximum velocity (F) were measured for wt TDP-43, mut TDP-43, mut TDP-43+TRVA242 (20 μM), mut TDP-43+TRVA242 (5 μM), mut TDP-43+TRVA242 (1 μM) treatment groups in the TEER assay. mut TDP-43 larvae displayed a significant locomotor defect in all parameters of the TEER assay compared to wt TDP-43 and TRVA242 (5 μM,20 μM) but not TRVA242(1 μM) recovered the locomotor deficits significantly. Swim distance (G), swim duration (H), and maximum velocity (I) were measured for wt SOD-1, mut SOD-1, mut SOD-1+TRVA242(5 μM), and mut SOD-1+TRVA242(1 μM) treatment groups in the TEER assay. mut SOD-1 larvae displayed a significant locomotor defect in all parameters of the TEER assay compared to wt SOD-1 and TRVA242 (5 μM) but not TRVA242 (1 μM) recovered the locomotor deficits significantly. Number in parentheses indicate sample sizes to the TEER assay in each treatment group. Data are expressed as mean ± SEM; **p < 0.01. Broken lines in the figures represent the effect of pimozide at 20 μM (positive control)
At 48 hpf, zebrafish display a robust touch evoked escape response (TEER) and axonal morphology which is altered upon overexpression of mutant human TDP-43 mRNA or mutant human SOD1 mRNA; Drapeau and Armstrong 2013; Kabashi et al., 2011). As TRVA242 was potent in rescuing coiling deficits in both mut-TDP43 and mut-SOD1 embryos, we next investigated whether TRVA242 was also potent in rescuing TEER defects in these embryos. We tested a range of concentrations (1 μM, 5 μM, and 20 μM) in mut-TDP43 zebrafish. We found that mut-TDP43 zebrafish treated with 20 μM and 5 μM but not 1 μM TRVA242 showed ameliorated TEER parameters as reflected by restoration of swim distance, swim duration, and maximum swim velocity (Fig. 2C–F; p < 0.001; Kruskal–Wallis test, post hoc Duncan’s test), with the maximal effect observed with TRVA242 at 5 μM. We subsequently analyzed the TEER response in mut-SOD1 embryos by following the same treatment protocol and found that TRVA242 at 5 μM but not 1 uM was effective in ameliorating TEER parameters (Fig. 2G–I; p < 0.001; Kruskal–Wallis test; post hoc Duncan’s test). However, no significant difference was found between uninjected WT larvae and overexpression of wild-type human TDP-43 or SOD-1 mRNA (data not shown).
As TRVA242 was able to rescue the homozygotic phenotype of tardbp double mutants in the LOF model, we next evaluated the effect of TRVA242 on the rescue of locomotor deficits at two different concentrations (5 and 20 μM). At 5 dpf, the small and large distance movements of the treated and untreated larvae were measured. We found that 20 μM but not 5 μM, TRVA242 was able to significantly rescue the locomotor defects of the tardbp−/−tardbpl−/− larvae (Fig. 3A, B; p < 0.001; Kruskal–Wallis test). To confirm the embryos rescued by TRVA242 were indeed tardbp homozygotes, a PCR-based RsaI digestion assay was performed (Dzieciolowska et al., 2017).

TRVA242 rescues locomotor defects in the zebrafish tardbpl−/−tardbp−/− LOF model of ALS. Large distance movement (A) and small distance movement (B) of larvae tardbpl−/−tardbp−/− treated and untreated with TRVA242 were measured using Zebrabox. tardbp LOF fish treated with TRVA242 showed significant increase in both small and large distance movement compared to untreated larvae. Data are expressed as mean ± SD. N = 3; n = 5; ***p < 0.001. Broken lines in the figures represent the effect of pimozide at 20 μM (positive control)
To assess whether the neuroleptic potential of TRVA242 extends to recovering spinal motor neuron outgrowth deficits in our third, C9ORF72 model, we in-crossed zebrafish homozygous for a 100 times GR repeat, or GR100 [12]. At 24 hpf, GR100 embryos were treated with TRVA242 (5uM) for 4 days. At 5 dpf, swimming activity was monitored in control and treated conditions. We found that treatment with TRVA242 significantly restored swimming distance and swimming velocity compared to untreated controls (Fig. 4A, B; p < 0.001; Mann–Whitney test).

TRVA242 rescues locomotor defects in the zebrafish C9ORF72 model of ALS. Swimming distance (A) and velocity (B) was measured in WT (without treatment), GR100UAS:Hb9 larvae without and with TRVA242 (5 μM) treatment at 5 dpf. GR100 UAS:Hb9 larvae treated with TRVA242 showed significantly increased swimming distance and velocity (p < 0.001; Mann–Whitney test). Number in parentheses indicate sample size. **p < 0.001
Next, we investigated whether TRVA242 could suppress motor neuron phenotypes in our ALS models. Returning to our C. elegans model, we observed that TRVA242 improved the swimming activity of mTDP-43 transgenics when tested in liquid culture (Fig. 5A), suppressed age-dependent paralysis of mTDP-43 animals when cultured over a period of 12 days on Petri plates (Fig. 5B), and suppressed motor neuron degeneration of mTDP-43 transgenics compared to untreated controls (Fig. 5C).

TRVA242 rescues motility defects and neurodegeneration in a C. elegans TDP-43 model. (A) Movement scores of mTDP-43 transgenics treated with TRV1442 or vehicle. mTDP-43 animals treated with TRV1442 showed a significantly higher swimming movement score (p < 0.001) compared to controls. Data are expressed as mean ± SD, N = 6. (B) mTDP-43 transgenics treated with TRV1442 show decreased age-dependent paralysis compared to untreated transgenics (p < 0.001). (C) mTDP-43 transgenics treated with TRV1442 show less motor neuron degeneration compared to untreated controls (P < 0.001). Broken lines in the figures represent the effect of pimozide at 20 μM (positive control)
TRVA242 Rescues Aberrant Spinal Motor Neuron Outgrowth and NMJ Structural Abnormalities in Zebrafish Models of ALS
Previous studies have indicated that overexpression of mutant human TDP-43 mRNA or SOD-1 mRNA induce aberrant spinal motor neuronal outgrowth [26]. Therefore, we investigated whether TRVA242 could restore disturbances in spinal motoneuronal outgrowth patterns. To do so, mutant human TDP-43 mRNA or mutant human SOD-1 mRNA was injected into the Hb9:Tg line expressing GFP mostly in motoneurons. Although no significant difference was found between uninjected WT larvae and overexpression of wild-type human TDP-43 or SOD-1 mRNA (data not shown) at 48 hpf post injection, we observed a significant reduction of spinal motor neuron axonal length in both mut-TDP43 and mut-SOD1 larvae. Following overnight treatment with TRVA242 (5 uM) starting at 24 hpf, we observed that the reduction in axonal length recovered to WT levels, confirming that TRVA242 was effective in rescuing the motor neuron phenotype observed in our zebrafish models of ALS (Fig. 5A–D; p < 0.05, Kruskal–Wallis test, post hoc Duncan’s test). We also tested for recovery of a similar phenotype in our C9ORF72 zebrafish model, wherein homozygous Hb9:GR100 embryos show a significant reduction in spinal motor neuron axonal length. Upon treatment with TRVA242 (5uM), the reduction in axonal length recovered significantly (Fig. 5E–F; p < 0.05, Kruskal–Wallis test, post hoc Duncan’s test).
We next sought to investigate if TRVA242 was efficient in preventing the NMJ structural abnormalities that we described previously in our mut-TDP-43 zebrafish larvae [16, 17]. As reported previously, overexpression of wt TDP-43 did not induce significant changes in terms of orphaned synapses (both ZNP1 and α-BTX puncta, data not shown) (Armstrong & Drapeau, 2013). Upon immunohistochemical analysis, mut-TDP-43 zebrafish larvae displayed an increase in the number of presynaptic (ZNP-1 marker) orphaned synapses, orphaned postsynaptic (α-BTX puncta), and aberrant motor axon branching, suggesting NMJ structural impairment. Overnight treatment with TRVA242 could prevent the NMJ structural abnormalities (Fig. 6A–C; p < 0.05, Kruskal–Wallis test, post hoc Duncan’s test). Similar immunohistochemical analyses revealed a slightly increased but not significantly different ratio of orphaned ZNP1 or αΒΤX synapses compared to wt-SOD1larvae (data not shown). This indicates that overexpression of human mutant TDP-43 and SOD1 variants induced distinct ALS phenotypes possibly mediated by diverse mechanisms. The possibility of distinct phenotypes is supported by the changes in coiling frequency (see above Fig. 2A–B) in mut-TDP-43 vs mut-SOD-1 larvae, wherein the former displayed a significantly reduced coiling frequency although the latter exhibited the reverse.

TRVA242 rescues aberrations in spinal motor neuron outgrowth in zebrafish genetic models of ALS. Motor neuron axonal outgrowth phenotype was assessed in wt TDP-43, mut-TDP-43 and mut TDP-43+TRVA242 (5 μM) at 48 hpf (A) and quantified (B). mut TDP-43 larvae displayed a significantly reduced axonal length compared to wt TDP-43 and overnight treatment with TRVA242 (5 μM) displayed a significant recovery. Motor neuron axonal outgrowth phenotype was assessed in wt SOD-1, mut SOD-1, and mut SOD-1+TRVA242 (5 μM) at 48 hpf (C) and quantified (D). mut SOD-1 larvae displayed a significantly reduced axonal length compared to wt SOD-1 and overnight treatment with TRVA242 (5 μM) displayed a significant recovery. Motor neuron axonal outgrowth phenotype was assessed in non-transgenic WT, GR100 UAS:Hb9 and GR100 UAS:Hb9 + TRVA242 (5 μM) at 48 hpf (E) and quantified (F). Number in parentheses indicate sample sizes. Scale bar represents 40 μm. Data are expressed as mean ± SEM; *p < 0.05. Broken lines in the figures represent the effect of pimozide at 20 μM (positive control)
Restoration of NMJ Synaptic Deficits by TRVA242 in Zebrafish and Mouse Models of ALS
As reported previously and in our current observation (data not shown), overexpression of wild-type human TDP-43 does not induce NMJ deficits leading to alterations in quantal neurotransmission. However, overexpression of mutant human TDP-43 in zebrafish induce NMJ deficits characterized by a reduction in quantal neurotransmission [16, 17]. Upon assessing passive membrane properties of fast-twitch muscle cells, we observed no significant difference in our treatment groups (Supplementary Fig. S3 A, B, C). However, mut-TDP-43 injected larvae displayed a significantly reduced mEPC frequency and amplitude. Overnight treatment with TRVA242 (5 μM) significantly maintained mEPC amplitude and frequency (Fig. 7A–C; p < 0.001; Kruskal–Wallis test, post hoc Duncan’s test). The kinetics of mEPCs were however unaffected by treatment in mut-TDP-43 larvae and wt TDP-43 larvae, suggesting that the observed aberrations in quantal transmission could have resulted from altered acetylcholine receptor density at the NMJ (Fig. 7D, E).

TRVA242 rescues NMJ structural abnormalities in a zebrafish genetic model of ALS. (A) Representative images of one ventral root projection labeled with ZNP1 (presynaptic marker 1) and sulphorhodamine conjugated αBTX (post synaptic marker 2). wt TDP-43 larvae demonstrate tight colocalization of both ZNP1 and αBTX (merged 3). Scale bar represents 25 μm for all images. Quantification of orphaned ZNP1 puncta over total ZNP1 puncta (B). All other treatment groups display significantly greater orphaned ZNP1 puncta compared to WT larvae (one-way ANOVA; post hoc Tukey test; p < 0.01). Quantification of orphaned αBTX puncta over total αBTX puncta (C). mut TDP-43 larvae mutants display significantly higher number of orphaned puncta compared to wtTDP-43 and overnight treatment with TRVA242 significantly restores number of orphaned puncta to WT levels (one-way ANOVA; post hoc Tukey test; p < 0.01). Data are expressed as ± SEM; **p < 0.01; number in parentheses indicate sample size. Broken lines in the figures represent the effect of pimozide at 20 μM (positive control)
Upon assessing quantal neurotransmission at the NMJ of mut-SOD-1 larvae, we found that the passive membrane properties of fast-twitch muscle cells from wt SOD-1, mut-SOD-1, and mut-SOD-1 larvae remained unchanged (Supplementary Fig. S3 D, E, F). Overexpression of wt SOD-1 also does not induce significant changes in quantal neurotransmission when compared to uninjected WT larvae (not shown). However, mEPCs from mut-SOD-1 displayed a significantly increased frequency compared to wt SOD-1 (Fig. 7F–J; p < 0.001, Kruskal–Wallis test, post hoc Duncan’s test). Overnight treatment with TRVA242 (5 μM) significantly reduced the mEPC frequency to wt SOD-1levels. This reassures that TRVA242 stabilizes NMJ deficits in two separate genetic models of zebrafish ALS, possibly through distinct mechanisms.
We next sought to determine if the rescue of NMJ neurotransmission deficits by TRVA242 observed in zebrafish was also present at the mammalian NMJ, in which several alterations in synaptic strength have been previously reported in various mouse models [7, 9, 31]. To this purpose, we used the SOD1G37R mouse model of ALS, which successfully recapitulates the ALS human motor phenotype, with a slow progression of the disease and a late-onset of motor deficits and hind-limb paralysis [25]. To assess synaptic transmission, we performed intracellular recordings at the NMJ of the Extensor Digitorum Longus (EDL) muscle, a fast-twitch muscle composed of a vast majority of fast-fatigable motor units, highly susceptible to NMJ degeneration throughout disease progression in SOD1 mouse models (Fig. 8) [7, 32].

TRVA242 restores synaptic defects at the NMJ in zebrafish models of ALS. Recordings of mEPCs, which result from spontaneous release of quanta, were done on wt TDP-43, mut-TDP-43, and mut TDP-43 + TRVA242 (5 μM), wt SOD-1, mut SOD-1, mut SOD-1 + TRVA242 (5 μM) at 48 hpf. mut-TDP-43 larvae showed a significantly reduced amplitude (A, B) and frequency (C) compared to wt TDP-43 larvae. Treatment with TRVA242 significantly restored the amplitude and frequency. D decay constant and E rise time were measured across all treatment groups and no significant differences were found. mut-SOD-1 larvae showed no significant differences in amplitude (F), decay constant (G), and rise time (H). mEPC frequency(I) of mut SOD-1 larvae were significantly increased compared to wt TDP-43 larvae. Treatment with TRVA242 significantly restored values to wt TDP-43 levels. Representative mEPC recordings (J) from wt SOD-1, mut SOD-1, mut SOD-1 + TRVA242 (5 μM) larvae for 3 min. Number in parentheses indicates sample sizes. Data are expressed as mean ± SEM, **p < 0.01. Broken lines in the figures represent the effect of pimozide at 20 μM (positive control)
Alterations in neurotransmission after onset of the disease (P425-P450) were striking in this mouse model. Indeed, evoked neurotransmission recordings showed that SOD1G37R NMJs presented a significant decrease in EPP amplitude and quantal content compared to age-matched WT NMJs (Fig. 9A–C; respectively p ˂ 0.01 and p ˂ 0.05, one-way ANOVA, post hoc Tukey’s test), similar to what has been previously reported [7, 17]. These deficits in neurotransmitter release were abolished when TRVA242 (5 μM) was bath-applied for 30 min (Fig. 9A–C; paired experiments). Indeed, after addition of the drug, there was no longer any difference between mutant treated NMJs and WT controls for either EPP amplitude or quantal content (Fig. 9A–C; both p ˃ 0.05, one-way ANOVA, post hoc Tukey’s test). Values obtained in TRVA242 versus the positive control Pimozide reference level (Patten et al., 2017) were similar for EPP amplitude (TRVA242: 0.94 vs Pimozide: 0.8) or quantal content (TRVA242 = 3.94, Pimozide reference level: 3.52), demonstrating a preserved capacity to rescue synaptic deficits by the new compound TRVA242. The effect was however obtained faster and was obtained more reliably for a concentration of 5 μM with TRVA242 (data not shown).

TRVA242 rescues synaptic transmission alterations at the symptomatic SOD1G37R mouse NMJ. Intracellular recordings of evoked and basal neurotransmission from WT, SOD1G37R, and SOD1G37R + TRVA242 (5 μM) NMJs were performed on the EDL muscle at P450. A) Examples of nerve-evoked EPP traces from a paired-pulse facilitation protocol at 0.2 Hz from WT (left), SOD1G37R (middle, SOD1 - Ctrl), and SOD1G37R treated NMJs (right, SOD1 + TRVA242). Scatter plots showing mean ± SEM of EPP amplitude (B), quantal content (C), mEPP frequency (D), and mEPP amplitude (E) of the WT NMJs (square, blue) as well as SOD1G37R NMJs before (triangle, black) and after treatment with TRVA242 (circle, green). Quantal content was obtained by dividing the mean EPP amplitude of the first EPP, including failures, divided by the mean mEPP amplitude. Data are expressed as mean ± SEM, WT: N = 5, n = 15, SOD1 paired experiments: N = 5, n = 5;*p < 0.05, **p < 0.01. Broken lines in each panel represent values obtained with the positive control pimozide at 5 μM, from Patten et al. (2017)
We also reported alterations in basal neurotransmission at mutant NMJs, with a significant decrease in mEPP frequency for symptomatic SOD1G37R NMJs compared to WT NMJs (Fig. 9D; p ˂ 0.05, one-way ANOVA, post hoc Tukey’s test). Interestingly, after application of TRVA242, the mEPP frequency was no longer different from WT (Fig. 9D; p ˃ 0.05, one-way ANOVA, post hoc Tukey’s test). However, no change was observed for mEPP amplitude in any condition in the mutant treated and untreated NMJs compared to WT (Fig. 9E; p ˃ 0.05, one-way ANOVA, post hoc Tukey’s test). TRVA242 therefore appears to be efficient in acutely restoring neurotransmitter release and synaptic strength at the SOD1 mouse NMJ, without however altering quantal size.
We intended to replicate the same synaptic analysis with the compound TRVA241 to assess its efficacy in rescuing the defects in neurotransmission previously reported at mutant SOD1G37R NMJs. Bath application of the drug (5 μM) showed comparable results: 30 min after the drug was added to the Ringer’s solution, EPP amplitude, quantal content as well as the frequency of miniature events were all significantly increased, with the most significant effect being observed on evoked neurotransmission (Fig. S4A–C, respectively, p ˂ 0.001, p ˂ 0.0001, and p ˂ 0.01, paired t test). The mEPP amplitude was still unchanged in treated NMJs compared to their baseline amplitude (Fig. S4D, p ˃ 0.05, paired t test).
Discussion
The utility of small animal models to screen for active compounds against ALS has gained impetus following the identification of pimozide’s repurposed activity in stabilizing the NMJ in a variety of genetic models and potentially in sporadic patients [17]. However, the neuroleptic pimozide has important side-effects when treating patients with a motoneuron disease, due to the well-known diverse molecular targets of the drugs, leaving a need for more potent or effective molecules for treating ALS. In an attempt to discover and validate structurally similar derivatives of pimozide and other beneficial molecules, we implemented a high-throughput chemical screen in a C. elegans genetic model of ALS. This initial screen of 3,765 novel compounds identified 11 active compounds, which when analyzed in our genetic GOF and LOF models in zebrafish confirmed 4 active compounds. TRVA242 was found to be the most potent molecule and prevented locomotor, spinal motorneuron outgrowth and NMJ defects in both mut-TDP43 and mut-SOD1 zebrafish larvae. The compliance of TRVA242 in rescuing defects in both LOF and GOF genetic zebrafish models of ALS highlights its potential of ameliorating neuronal dysfunction irrespective of the genotype. Finally, the enhancement of synaptic strength by TRVA242 in SOD1G37R mice confirmed its role in stabilizing NMJs in a mammalian model of ALS, indicating the potential of this compound as a potential therapeutic for ALS.
The first signs of embryonic motor behavior in zebrafish manifests in the form of spontaneous tail coiling between 17 and 20 hpf and we found that although mut-TDP-43 embryos displayed a reduced burst count, mut-SOD-1 embryos displayed an increased frequency of spontaneous coiling. To our knowledge, this is the first study that reports an embryonic motor behavior abnormality associated with overexpression of mutant human TDP-43 in zebrafish. This suggests that the motor phenotype elicited in mut-TDP-43 embryos possibly stems from an impairment in the electrical properties of spinal neurons and upon maturing to 48 hpf results in an impaired TEER and a concomitant reduction in the spinal motor neuron axon length. Overexpression of mutant human TDP-43 in zebrafish larvae has been shown to induce such phenotypes [16, 26]. We found that TRVA242 exerted a neuroprotective role and prevented defects in coiling frequency, swim duration, swim distance, and maximal velocity. Upon retesting TRVA242 in the C. elegans model, it again showed potent rescue of the impaired swimming activity of mTDP-43 transgenics. Furthermore, when tested in assays examining age-dependent phenotypes, TRVA242 rescued progressive paralysis and motor neuron degeneration in mTDP-43 transgenics. These data suggest that TRVA242 has neuroprotective capacity against both acute and chronic phenotypes linked to mTDP-43.
Additionally, TRVA242 was also potent in suppressing spinal motorneuron outgrowth defects and perturbations in NMJ architecture. This led us to examine whether TRVA242 would also be potent in rescuing functional neurotransmission defects at the NMJ. Indeed, TRVA242 significantly recovered neurotransmission defects at the NMJ in mut-TDP43 embryos which displayed a robust decrease in frequency of spontaneous neurotransmission at the NMJ and a significantly reduced amplitude of unitary events. L-type Ca2+ channel agonists, Bay K 8644 and FPL 64176 have been shown to exert a stabilizing effect at the NMJ in mut-TDP-43 zebrafish larvae. These agonists possibly act by enhancing Ca2+ entry at presynaptic terminals, compensating for the loss in quantal transmission [16]. Alternatively, our laboratory reported the role of pimozide in enhancing NMJ activity by blocking low voltage activated Ca2+ current mediated by T-type calcium channels [17]. Because TRVA242 was also efficient in recovering functional NMJ defects in our mut-TDP-43 larvae, it might be possible that TRVA242 might have performed through a similar mode of recovery by engaging Ca2+ channels, although further studies are needed to validate this speculation.
Tardbp double mutants displayed a severe phenotype when compared to the TDP43 GOF model. We found that TRVA242 not only increased the survivability of the mutant fish but also was able to partially rescue the more severe phenotype of tardbp LOF model. TRVA242 also significantly improved the small and large distance movements of the tardbp LOF embryos when compared to untreated controls. Thus, TRVA242, at least in this LOF model, may avoid developmental defects such as those observed in TDP-43 knock-out mice [33].
On the other hand, mut-SOD-1 embryos exhibited an increase in the frequency of spontaneous coiling although the duration of a single coil remained unaltered. This is in line with recent observations of increased spontaneous coiling in zebrafish embryos overexpressing mutant human SOD1 (G93R) and indicates that altering of electrical properties of spinal motor neurons lead to such phenotypes [28, 34,35,36]. It has also been shown that in transgenic SOD1 mouse (G93A), electrical properties of spinal cord neurons are altered and present a hyperexcitable state at early embryonic stages [29, 30, 37]. We have previously shown that overexpression of mutant human SOD-1 mRNA (G93A) in zebrafish induces an impairment in TEER and motorneuron axonal defects. Similar axonal outgrowth defects were observed in this study and significantly ameliorated by TRVA242. Although we did not observe a significant perturbation in NMJ structural assembly, we did find a significant increase in the frequency of neurotransmission at the NMJ in mut-SOD1 larvae compared to wt SOD-1 larvae. Previous studies report a similar hyperexcitability in SOD1 mice models leading to an increase in the tendency of motorneuron firing (Jiang MC et al., 2017). In fact, in a transgenic SOD1 zebrafish model, selective inhibition of the persistent pacemaker sodium current iNaP with riluzole, one of the two compounds used to treat ALS, tends to revert spinal motoneuron and interneuron hyperexcitability, consequently enhancing motor unit functioning (Benedetti L et al., 2016). The observed increase in the frequency of neurotransmission of mut-SOD1 larvae in our study could possibly be attributed to a similar hyperexcitable state which commences as early as the first signs of embryonic motor behavior in the form of spontaneous coiling. TRVA242 treatment significantly restored the frequency of neurotransmission to wild-type levels in mut-SOD-1 larvae, suggesting that TRVA242 could regulate sodium channels, consequently restoring NMJ activity. It is important to note that overexpression of mutant human TDP-43 or SOD-1 mRNA leads to distinct motor phenotypes, which are recovered by TRVA242 at equimolar concentrations. This further reaffirms our hypothesis that TRVA242 may engage a common target that involves both SOD1 and TDP-43-mediated pathology in zebrafish models of ALS or engages multiple cellular targets in these pathogenic pathways.
Using a slow-onset mouse model of ALS (SOD1G37R), several alterations of the synaptic strength were reported after disease onset. Indeed, EPP amplitude, quantal content, and frequency of mEPP were all decreased as opposed to WT NMJs, which is consistent with previous reports of deficits in fast-fatigable fibers of the EDL [7, 17]. Similar, to the impact of pimozide on synaptic properties of NMJs, TRVA242 treatment successfully restored SOD1 synaptic transmission deficits, with quantal content and EPP amplitude and mEPP frequency back to the level of their aged-matched WT littermates. The fact that the acute treatment did not cause any change in mEPP amplitude suggests that no alteration of the postsynaptic endplate or fiber diameter occurred [38], which was expected considering the acute short-term treatment administered. This implies that TRVA242 potentially has a presynaptic mechanism of action at the SOD1 mouse NMJ. Our results could be consistent with TRVA242 altering Ca2+ dynamics in the nerve terminal that would result in an enhanced release of neurotransmitters. Interaction with K+ channels cannot be ruled out as well, because acute application of a K+ channel blockers such as tetraethyl ammonium (TEA) can increase EPP amplitude more than twofold at the mouse NMJ [6]. Furthermore, TRVA241 appears to be as efficient in rescuing the deficits in synaptic strength observed at the NMJ, which suggest that the two derivative compounds probably share similar mechanisms of action, at least in the SOD1 mouse. However, future experiments are needed to highlight if NMJ structure and integrity can be improved by treatment and drug ability to slow disease progression in vivo in SOD1 mice and other mammalian disease models.
Finally, in line with the preceding study by [17], this study demonstrates the beneficial impact of acute and chronic treatment with the newly synthesized compound TRVA242 in C. elegans, as well as in zebrafish and a mouse model of ALS. Indeed, it is important to note that despite the fact that overexpression of mutant human TDP-43, SOD-1 mRNA or GR100 dipeptides in zebrafish leads to distinct motor phenotypes, all of which are recovered by TRVA242 at equimolar concentrations. This further reaffirms our hypothesis, that TRVA242 possibly engages a target that is central to the neuropathological mechanism of ALS or one that is common to SOD1, TDP-43, or C9ORF72 mediated pathology, at least in zebrafish models of ALS, or engages to multiple cellular targets in these pathogenic pathways. Owing to the similar facilitating effect of TRVA242 treatment on synaptic transmission at SOD1 mouse NMJs, the target of this compound might be conserved across species or instead several distinct mechanisms of action could be involved, as TRVA242 is a derivative of pimozide, compounds presenting numerous cellular targets. Such drugs with reduced selectivity possibly are better compounds as therapeutics due to their ability to target varied cellular substrates and increase chances of success in a more sporadic population presenting multiple forms of the disease. This type of compound could therefore represent suitable compounds for an NMJ-targeted treatment for ALS in the future. Although conclusions about the effectiveness of pimozide await completion of a large on-going clinical trial (Clinicaltrials.gov NCT03272503), our study provides alternative molecules for therapeutic consideration. These of course would need to be evaluated for pharmacokinetics, pharmacodynamics, behavioral effects, and other measures before advancing to the clinic, approaches that are beyond the goal of this initial study but potentially meritorious of further evaluation. For an untreatable, lethal disease, this is no small consideration.
Change history
07 June 2021
A Correction to this paper has been published: https://doi.org/10.1007/s13311-021-01061-2
References
Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol 2011;7(11):603–15.
Braak H, Brettschneider J, Ludolph AC, Lee VM, Trojanowski JQ, Del Tredici K. Amyotrophic lateral sclerosis--a model of corticofugal axonal spread. Nat Rev Neurol 2013;9(12):708–14.
Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, et al. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol 2004;185(2):232–40.
Moloney EB, de Winter F, Verhaagen J. ALS as a distal axonopathy: molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front Neurosci 2014;8:252.
Clark JA, Southam KA, Blizzard CA, King AE, Dickson TC. Axonal degeneration, distal collateral branching and neuromuscular junction architecture alterations occur prior to symptom onset in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. J Chem Neuroanat. 2016;76(Pt A):35–47.
Arbour D, Tremblay E, Martineau E, Julien JP, Robitaille R. Early and persistent abnormal decoding by glial cells at the neuromuscular junction in an ALS model. J Neurosci 2015;35(2):688–706.
Tremblay E, Martineau E, Robitaille R. Opposite Synaptic Alterations at the Neuromuscular Junction in an ALS Mouse Model: When Motor Units Matter. J Neurosci 2017;37(37):8901–18.
So E, Mitchell JC, Memmi C, Chennell G, Vizcay-Barrena G, Allison L, et al. Mitochondrial abnormalities and disruption of the neuromuscular junction precede the clinical phenotype and motor neuron loss in hFUSWT transgenic mice. Hum Mol Genet 2018;27(3):463–74.
Chand KK, Lee KM, Lee JD, Qiu H, Willis EF, Lavidis NA, et al. Defects in synaptic transmission at the neuromuscular junction precedes motor deficits in a TDP-43(Q331K) transgenic mouse model of amyotrophic lateral sclerosis. FASEB J 2018.
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–68.
Gitler AD, Tsuiji H. There has been an awakening: Emerging mechanisms of C9orf72 mutations in FTD/ALS. Brain Res 2016;1647:19–29.
Swaminathan A, Bouffard M, Liao M, Ryan S, Callister JB, Pickering-Brown SM, et al. Expression of C9orf72-related dipeptides impairs motor function in a vertebrate model. Hum Mol Genet 2018;27(10):1754–62.
Maselli RA, Wollman RL, Leung C, Distad B, Palombi S, Richman DP, et al. Neuromuscular transmission in amyotrophic lateral sclerosis. Muscle Nerve 1993;16(11):1193–203.
Vaccaro A, Patten SA, Ciura S, Maios C, Therrien M, Drapeau P, et al. Methylene blue protects against TDP-43 and FUS neuronal toxicity in C. elegans and D. rerio. PLoS One. 2012;7(7):e42117.
Dzieciolowska S, Drapeau P, Armstrong GAB. Augmented quantal release of acetylcholine at the vertebrate neuromuscular junction following tdp-43 depletion. PLoS One 2017;12(5):e0177005.
Armstrong GA, Drapeau P. Calcium channel agonists protect against neuromuscular dysfunction in a genetic model of TDP-43 mutation in ALS. J Neurosci 2013;33(4):1741–52.
Patten SA, Aggad D, Martinez J, Tremblay E, Petrillo J, Armstrong GA, et al. Neuroleptics as therapeutic compounds stabilizing neuromuscular transmission in amyotrophic lateral sclerosis. JCI Insight. 2017;2(22).
Stiernagle T. Maintenance of C. elegans. WormBook. 2006:1–11.
Simonetta SH, Golombek DA. An automated tracking system for Caenorhabditis elegans locomotor behavior and circadian studies application. J Neurosci Methods 2007;161(2):273–80.
Westerfield M. The zebrafish book: a guide for the laboratory use of zebrafish(Danio rerio)1995.
Buss RR, Drapeau P. Activation of embryonic red and white muscle fibers during fictive swimming in the developing zebrafish. J Neurophysiol 2002;87(3):1244–51.
Hewamadduma CA, Grierson AJ, Ma TP, Pan L, Moens CB, Ingham PW, et al. Tardbpl splicing rescues motor neuron and axonal development in a mutant tardbp zebrafish. Hum Mol Genet 2013;22(12):2376–86.
Jao L-E, Wente SR, Chen W. Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc Natl Acad Sci 2013;110(34):13904–9.
Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, Campbell DS, et al. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn 2007;236(11):3088–99.
Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, et al. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14(6):1105–16.
Kabashi E, Bercier V, Lissouba A, Liao M, Brustein E, Rouleau GA, et al. FUS and TARDBP but not SOD1 interact in genetic models of amyotrophic lateral sclerosis. PLoS Genet 2011;7(8):e1002214.
Brustein E, Saint-Amant L, Buss RR, Chong M, McDearmid JR, Drapeau P. Steps during the development of the zebrafish locomotor network. J Physiol Paris 2003;97(1):77–86.
Saint-Amant L, Drapeau P. Time course of the development of motor behaviors in the zebrafish embryo. J Neurobiol 1998;37(4):622–32.
Kuo JJ, Schonewille M, Siddique T, Schults AN, Fu R, Bar PR, et al. Hyperexcitability of cultured spinal motoneurons from presymptomatic ALS mice. J Neurophysiol 2004;91(1):571–5.
Martin E, Cazenave W, Cattaert D, Branchereau P. Embryonic alteration of motoneuronal morphology induces hyperexcitability in the mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 2013;54:116–26.
Rocha MC, Pousinha PA, Correia AM, Sebastiao AM, Ribeiro JA. Early changes of neuromuscular transmission in the SOD1(G93A) mice model of ALS start long before motor symptoms onset. PLoS One 2013;8(9):e73846.
Valdez G, Tapia JC, Lichtman JW, Fox MA, Sanes JR. Shared resistance to aging and ALS in neuromuscular junctions of specific muscles. PLoS One 2012;7(4):e34640.
Kraemer BC, Schuck T, Wheeler JM, Robinson LC, Trojanowski JQ, Lee VM, et al. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol 2010;119(4):409–19.
Drapeau P, Saint-Amant L, Buss RR, Chong M, McDearmid JR, Brustein E. Development of the locomotor network in zebrafish. Prog Neurobiol 2002;68(2):85–111.
Tong H, McDearmid JR. Pacemaker and plateau potentials shape output of a developing locomotor network. Curr Biol 2012;22(24):2285–93.
Benedetti L, Ghilardi A, Rottoli E, De Maglie M, Prosperi L, Perego C, et al. INaP selective inhibition reverts precocious inter- and motorneurons hyperexcitability in the Sod1-G93R zebrafish ALS model. Sci Rep 2016;6:24515.
van Zundert B, Peuscher MH, Hynynen M, Chen A, Neve RL, Brown RH, Jr., et al. Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J Neurosci 2008;28(43):10864–74.
Sieck GC, Prakash YS. Morphological adaptations of neuromuscular junctions depend on fiber type. Can J Appl Physiol. 1997;22(3):197–230.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised to update an author’s name. The correct spelling is Claudia Maios.
Summary statement
This study is unique as it describes the potential efficacy of a novel small molecule TRVA242 in simple genetic models (zebrafish and C. elegans) as well as a mouse model of amyotrophic lateral sclerosis. The efficacy of TRVA242 in stabilizing neuromuscular junction deficits through diverse neuronal mechanisms and across species suggests it may have promising effects for the treatment of amyotrophic lateral sclerosis.
Electronic Supplementary Material
Fig S1
Initial screening of the four most potent compounds from the library in zebrafish GOF and LOF model. S1: Swim distance(A), swim duration(B) and maximum velocity(C) were measured for wt TDP-43, mut TDP-43 and mut TDP-43 treated with the active compounds at 20 μM (35 F06, 35 H09, TRVA241, TRVA242) from the high-throughput screen in C.elegans . 35 F06, 35 H09, TRVA241, TRVA242 significantly restored locomotor deficits and TRVA242 was identified as the most active compound. Number in parentheses indicate sample size. Data are expressed as mean ± SEM; *p < 0.05; **p < 0.01. Representative images of 2 dpf larvae of tardbpl−/−tardbp+/+, tardbpl−/−tardbp± and tardbpl−/−tardbp−/− (D). Scale bar represents 1 mm. Percentage survival curves of all three mutant lines tracked over 10 days. Tardbpl−/−tardbp−/− embryos had a reduced survival and all animals dying by 8 dpf (E), N = 3, n = 40, *p < 0.05. TRVA242 rescues the homozygotic phenotype of tardbp LOF mutants, the graph shows the reduction in the number of mutant larvae with homozygotic phenotype after treatment with 20 μM TRVA242 compared to untreated larvae (negative control) (F). Broken line in the figures represent the effect of pimozide at 20 μM (positive control). (PNG 872 kb)
Fig S2
Initial screen of the 5 most potent compounds in the zebrafish LOF model. S2: Small distance movement (A) and large distance movement (B) of larvae tardbpl−/−tardbp−/−treated and untreated with seven compounds were measured using Zebrabox. Double mutant fish treated with TRVA242 showed significant increase in both small and large distance movement compared to untreated larvae. Data are expressed as mean ± SD. N = 3; n = 5 ***p < 0.0001; **p < 0.001. (PNG 411 kb)
Fig S3
Passive membrane properties of fast-twitch muscle cells in zebrafish GOF models. S3: Passive membrane properties of wt TDP-43, mut-TDP-43 and mut TDP-43 + TRVA242 (5 μM), wt SOD-1, mut SOD-1, mut SOD-1 + TRVA242(5 μM) at 48 hpf. Membrane potential (A), capacitance(B) and membrane resistance (C) of fast-twitch muscle cells at 48 hpf from mut-TDP-43 and mut TDP-43 + TRVA242 (5 μM) larvae did not display significant difference compared to wt TDP-43 larvae. Membrane potential (D), capacitance(E) and membrane resistance (F) of fast-twitch muscle cells at 48 hpf from mut SOD-1, mut SOD-1 + TRVA242(5 μM) larvae also did not display significant difference compared to wt SOD-1 larvae. Number in parentheses indicate sample size. Broken line in the figures represent the effect of pimozide at 20 μM (positive control) (PNG 243 kb)
Fig S4
Restoration of NMJ neurotransmission deficits by TRVA241 in the SOD1G37R mouse model. We performed intracellular recordings of EPP and mEPP signals from SOD1G37R NMJs before and after bath application of TRVA241 (5 μM) for 30 min. Recordings were obtained in the EDL muscle, after disease onset (P425–450). Scatter plots showing mean ± SEM of EPP amplitude (A), Quantal content (B), mEPP frequency (C) and mEPP amplitude (D) of the SOD1G37R NMJs before (square, black) and after treatment with TRVA241 (triangle, blue). All experiments were paired, with a given NMJ being recorded during a 30 min baseline period, followed by bath-application of TRVA241 on the same NMJ. SOD1 paired experiments: N = 6, n = 6 **p < 0.01, ***p < 0.01. Broken line in each panel represent the effect of pimozide at 5 μM used as a positive control), from Patten et al. (2017) (PNG 131 kb)
ESM 1
(PDF 1661 kb)
Rights and permissions
About this article

Cite this article
Bose, P., Tremblay, E., Maios, C. et al. The Novel Small Molecule TRVA242 Stabilizes Neuromuscular Junction Defects in Multiple Animal Models of Amyotrophic Lateral Sclerosis. Neurotherapeutics 16, 1149–1166 (2019). https://doi.org/10.1007/s13311-019-00765-w
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-019-00765-w