
Abstract
Background and Aim
Diabetes reduces the levels of circulating endothelial progenitor cells (EPCs), which contribute to vascular homeostasis. In turn, low EPCs levels predict progression of chronic complications. Several studies have shown that hyperglycaemia exerts detrimental effects on EPCs. Improvement in glucose control with glucose-lowering medications is associated with an increase of EPCs, but only after a long time of good glycaemic control. In the present study, we examined the effect of a rapid glycaemic amelioration on EPC levels in subjects hospitalized for decompensated diabetes.
Methods
We used flow cytometry to quantify EPCs (CD34+/CD133+KDR+) in patients hospitalized for/with decompensated diabetes at admission, at discharge, and 2 months after the discharge. During hospitalization, all patients received intensive insulin therapy.
Results
Thirty-nine patients with type 1 or type 2 diabetes were enrolled. Average (± SEM) fasting glucose decreased from 409.2 ± 25.9 mg/dl at admission to 190.4 ± 12.0 mg/dl at discharge and to 169.0 ± 10.3 at 2 months (both p < 0.001). EPCs (per million blood cells) significantly increased from hospital admission (13.1 ± 1.4) to discharge (16.4 ± 1.1; p = 0.022) and remained stable after 2 months (15.5 ± 1.7; p = 0.023 versus baseline). EPCs increased significantly more in participants with newly-diagnosed diabetes than in those with pre-existing diabetes. The increase in EPCs was significant in type 1 but not in type 2 diabetes and in those without chronic complications.
Conclusion
In individuals hospitalized for decompensated diabetes, insulin therapy rapidly increases EPC levels for up to 2 months. EPC defect, reflecting impaired vascular repair capacity, may be reversible in the early diabetes stages.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Endothelial progenitor cells (EPCs) reflect the endogenous regenerative capacity and are reduced in diabetes. |
Though diabetes therapy can improve circulating EPC levels, we herein examined the effect of acute glycaemic re-compensation on EPCs. |
What was learned from the study? |
Despite glycaemic control took months to increase EPC levels in prior studies on outpatients with mildly decompensated diabetes, we found that intensive insulin therapy in people hospitalized with severe hyperglycaemia elicits a rapid and significant increase in EPCs. |
The EPC increase was mostly evident in patients with newly diagnosed diabetes and in those without chronic complications. |
Introduction
Diabetes is a chronic progressive disease and episodes of acute hyperglycaemia are relatively common. Patients with decompensated diabetes are often admitted to the hospital, especially in case of severe symptomatic hyperglycaemia, or in the presence of infections or because of acute manifestations of chronic complications, such as diabetic foot ulcers [1]. Acute hyperglycaemia is known to worsen vascular function by compromising endothelial homeostasis [2]. Endothelial progenitor cells (EPCs) are a subtype of hematopoietic stem/progenitor cells (HSPCs), provided with vascular tropism, identified by surface expression of VEGF receptor 2 (or KDR) along with stem cell markers (CD34 and/or CD133) [3]. EPCs contribute to vascular repair and regeneration, though the mechanism whereby EPCs exert these actions is debated. Initial studies suggested that EPCs were recruited to sites of vascular injury, where they differentiated into functional mature endothelium and were integrated into the vasculature [4, 5]. It is now recognized that EPCs may contribute to endothelial repair and neoangiogenesis mostly by providing paracrine signals [6,7,8,9]. Evidence accumulated over the last two decades suggests that HSPCs and EPCs play a role in the pathogenesis of diabetes complications [10]. HSPCs and EPCs are significantly reduced in people with diabetes as compared to those without. Furthermore, among people with diabetes, lower levels of HSPC predict the future onset or progression of micro- and macroangiopathy [11, 12]. EPCs are particularly reduced in patients with peripheral arterial disease (PAD), one of the most typical cardiovascular disease manifestation of diabetes [13, 14]. In addition, a decrease in EPCs was associated with a higher risk of mortality and PAD-related events [15].
Prior studies widely support the detrimental role of high glucose on EPC function and levels [16, 17]. An improvement in glucose control with basal insulin or sodium–glucose co-transporter 2 inhibitor (SGLT2i) has been associated with an increase of EPCs, but only after a relatively long time (3–6 months) of good glycaemic control [18, 19]. On the other hand, the effect of glucose control on EPCs in acutely decompensated patients is mostly unknown.
In the present study, we examined the effect of a rapid glycaemic amelioration with intensive insulin therapy on EPC levels in patients hospitalized for decompensated diabetes.
Methods
Study Design and Objective
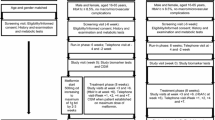
This was a longitudinal observational study performed on patients consecutively admitted to the Division of Metabolic Diseases of the University Hospital of Padova between January 2017 and July 2019. The protocol was approved by the ethics committee of the same hospital (no. 50n/AO/20) and was conducted in accordance with the Declaration of Helsinki. All patients signed informed consent for blood sampling and data collection. The aim of the study was to evaluate the change in EPC levels after glycaemic control in individuals hospitalized for/with decompensated diabetes. For this purpose, EPC quantification was assessed in the acute decompensated phase at hospital admission, at the time of discharge, and 2 months after discharge. Furthermore, to evaluate to what extent the known EPC reduction observed in diabetes is reversible with glycaemic control, we explored whether baseline clinical characteristics impacted on EPC changes.
Study Patients
Inclusion criteria were as follows: type 1 diabetes (T1D) or type 2 (T2D), age 18–85 years, hospitalization for acute glycaemic decompensation or patients hospitalized for another cause with acute glycaemic decompensation (define as plasma glucose level greater than 250 mg/dl). Exclusion criteria were ongoing sepsis, neoplasms, estimated life expectancy less than 6 months, dialysis, chronic immune-inflammatory conditions, immunosuppression or steroid therapy, organ transplantation, pregnancy or lactation, and inability to provide informed consent.
Data Collection
For all patients, we recorded the following information at baseline: age, sex, weight, height, body mass index (BMI, calculated as weight in kilograms divided by the square of height in metres), type of diabetes (T1D or T2D), known diabetes duration, HbA1c and fasting plasma glucose (FPG), lipid profile (total, HDL and LDL cholesterol, and triglycerides), serum creatinine, urinary albumin to creatinine ratio (milligrams per gram), smoking habit (defined as habitual active smoking of one or more cigarettes per day), medical history, and ongoing medications. Hypertension was defined as systolic blood pressure of at least 140 mmHg and/or diastolic blood pressure of at least 90 mmHg and/or use of antihypertensive medications. Dyslipidaemia was defined as a total cholesterol level greater than 200 mg/dl or a triglycerides level greater than 150 mg/dl confirmed after glucose control amelioration or use of lipid-lowering medication. Diabetic kidney disease was defined as urinary albumin to creatinine ratio of at least 30 mg/g (using the latest data available before the hospitalization) and/or an estimated glomerular filtration rate (eGFR according to the CKD-EPI equation [20]) equal to or lower than 60 ml/min/1.73 m2. Diabetic retinopathy was defined on the basis of digital fundus photography scored by ophthalmologists. Somatic peripheral neuropathy was defined, after exclusion of non-diabetic causes, by the presence of typical symptoms (tingling, numbness, or pain in the toes, feet, legs, fingers, hands, and arms, or wasting of the muscles of the feet or hands), confirmed by clinical examination (ankle reflexes, 10-g monofilament sensitivity, vibratory perception threshold, and pinprick) and eventually by electromyography. Autonomic neuropathy was defined using four cardiovascular autonomic function tests, as follows: deep breathing, lying-to-standing, Valsalva manoeuvre, and orthostatic hypotension. Cerebrovascular disease was defined as the presence of carotid artery plaques at routine ultrasound examination or a history of transient ischemic attack, or cerebral ischemia. Coronary artery disease was defined as a history of angina or myocardial infarction, or evidence of significant coronary artery disease at coronary angiography or history of revascularization. Peripheral arterial disease was defined as a history of claudication or rest pain, significant stenosis in leg arteries upon ultrasound examination or history of revascularization.
Treatment and Study Protocol
During the hospital stay, all patients were treated with insulin therapy (continuous intravenous and/or subcutaneous) according to the international guidelines of care for the hospitalized patient with hyperglycaemic decompensation [21, 22]. After patients signed written informed consent, three fasting blood samples were collected for the quantification of circulating EPCs: at the time of hospital admission (or within 48 h), at the time of discharge and 2 months later.
EPC Quantification
EPCs were measured by flow cytometry, as previously described in detail [12], based on the surface expression of the CD34, CD133, and kinase insert domain-containing receptor (KDR). Briefly, EDTA-anticoagulated blood samples were freshly analysed within 4 h. After red blood cell lysis, cells were stained with fluorescein isothiocyanate-conjugated anti-CD34, allophycocyanin-conjugated anti-CD133, PE-conjugated anti-KDR and PerCP-Cy5.5-conjugated anti-CD45 monoclonal antibodies. CD45 diminished staining was used to confirm stem/progenitor cell identity. At least 5 × 105 events were acquired. Relative cell count was expressed as cells/106. EPCs were defined as CD34+KDR+, CD133+KDR+ or CD34+CD133+KDR+ cells. Since there are few differences among the various EPCs phenotypes, we combined cells expressing CD34 or CD133 together with KDR as EPCs (CD34+/CD133+KDR+ cells). The same trained operator performed all analyses throughout the study.
Statistical Analysis
On the basis of our previous experience in a similar setting [23], we calculated that the standard deviation of EPC is approximately 9 cells/106. We hypothesized a 25% increase in the level of EPC after a few days of glucose control compared to baseline. At least 38 patients were needed to identify this difference with α = 0.05, β = 0.20 and a dropout rate of 5%.
Data are expressed as mean ± standard deviation if normally distributed or as median (interquartile range) if not. Categorical variables are presented as numbers and percentages. Normality was checked using the Shapiro–Wilk test and variables with markedly skewed distribution were log-transformed before analysis with parametric tests. Variables collected at follow-up were compared to data at baseline using the two-tailed paired Student’s t test. Comparisons between different time points were performed using the analysis of variance for repeated measures. Comparisons between two groups were performed using the two-tailed unpaired Student’s t test for continuous variables and the χ2 test for categorical variables. Statistical significance was accepted at p < 0.05. SPSS version 22.0 was used.
Results
Patients Characteristics
Thirty-nine patients were enrolled in the study, 21 male and 18 female. Among them, 18 had T1D and 21 had T2D. The baseline characteristics of the study population are summarized in Table 1. Patients were on average 47.6 years old, and 38.5% had newly diagnosed diabetes, among them seven had T1D and eight had T2D. Patients had a baseline HbA1c value of 11.9% (106 mmol/mol), indicating severely decompensated diabetes. Almost 40% of the patients had hypertension. Regarding diabetes complications, 28% had diabetic kidney disease, 23% had neuropathy, and a quarter had retinopathy, while the prevalence of known macroangiopathy was lower.
Effect of Glycaemic Control on EPC Levels
During hospitalization, all patients were treated with insulin therapy to optimize glycaemic control. The mean duration of time spent in hospital was 6 days. On average, FPG declined from 409.2 ± 25.9 mg/dl to 190.4 ± 12.0 mg/dl at discharge (p < 0.001). At the end of observation, 2 months after hospital discharge, FPG remained stable at 169.0 ± 10.3 mg/dl (p < 0.001 vs baseline; p = 0.11 vs discharge; Fig. 1a).

Glucose control and change in EPCs. Levels of fasting plasma glucose (a) and EPC (b) in study patients at baseline (admission to hospital), at discharge and 2 months after discharge *p < 0.05 versus baseline. c Shows the correlation between fasting glucose and EPC levels, with superimposed average and 95% CI levels at each time point
EPCs were also measured at the time of hospital admission (baseline), at the time of discharge, and 2 months after discharge. Compared to baseline, EPC significantly increased during the hospitalization from 13.1 ± 1.4/106 to 16.4 ± 1.1/106; p = 0.022) and then remained stable at the end of the observation period (15.5 ± 1.7/106; p < 0.05 versus baseline; p = 0.023 versus discharge; Fig. 1b).
Considering all time points together, there was a significant inverse correlation between glucose concentrations and EPC levels (r = − 0.20; p = 0.039; Fig. 1c).
Change in EPCs by Patient Characteristics
We then investigated whether EPC changes at discharge compared to baseline were affected by baseline characteristics of study patients.
Patients were categorized into two groups according to the presence or absence of history of diabetes before hospital admission, type of diabetes (T1D vs T2D), and presence/absence of diabetes complications. Compared to baseline, EPCs significantly increased in patients with newly diagnosed diabetes whereas they remained stable in those with known pre-existing diabetes. The change from baseline was significantly different between the two groups (+ 7.0 ± 2.5/106 in newly diagnosed vs + 1.0 ± 1.8/106 in pre-existing; p < 0.05; Fig. 2a).

Factors affecting changes in EPC levels. Patients were divided into subgroups according to the presence or absence of history of diabetes before the hospital admission (a), type of diabetes (b) and presence or absence of diabetes complications (c) *p < 0.05 versus baseline. #p < 0.05 between groups
EPCs significantly increased in subjects with T1D while they remained stable in those with T2D. However, the change from baseline between the two groups did not reach a statistically significant difference (+ 6.6 ± 2.5/106 in T1D vs + 1.0 ± 1.6/106 in T2D; p = 0.070; Fig. 2b).
Finally, EPCs significantly increased in patients without diabetic complications, whereas they remained stable in the group of patients with chronic complications. However, the change from baseline was non-significantly different between the two groups (+ 6.2 ± 2.2/106 without vs + 0.9 ± 2.0/106 with complications; p = 0.082; Fig. 2c).
Discussion
This study demonstrates that improvement of glycaemic control in patients hospitalized with severely decompensated diabetes was followed by a rapid increase of EPC levels that persisted at least for 2 months after hospital discharge. Considering the extensive literature on the biological roles of EPCs, our data indicate that glycaemic compensation can rapidly improve endothelial function and rescue the endogenous organism’s repair capacity.
When we analysed factors associated with EPC changes after glycaemic optimization, we found that the effect was significant in the entire cohort, in those with newly diagnosed diabetes, with T1D, and without chronic complications. On the other hand, no significant effect was found for people with T2D, those with pre-existing diabetes, and with complications. The study was not powered to detect effects within some small subgroups, but results are consistent with the hypothesis that glycaemic control can acutely improve EPC levels when pursued during the early disease stages and in younger ages. Indeed, the increase in EPC levels was significantly greater in those with decompensated newly diagnosed diabetes than in those with pre-existing diabetes. Results of the analysis stratified by type of diabetes and by presence/absence of complications are consistent with this view, though not formally achieving significance in between-group comparison.
It has been demonstrated that hyperglycaemia is an important determinant of EPC levels. Indeed, the reduction of circulating EPCs is related to the degree of hyperglycaemia [16, 17]. Previous studies have shown that lowering glucose levels using different glucose-lowering medications has the potential to raise EPCs [24]. However, the rapid effect of few days of glucose control on EPC levels was partially unexpected. In prior studies conducted in outpatients with mildly decompensated T2D, months of good glycaemic control were needed to increase the levels of EPCs [18, 19]. Fadini et al. reported optimized glucose control by add-on basal insulin in patients with T2D and an average baseline HbA1c of 8.7% increased circulating EPCs only after 6 months of optimized treatment. The levels of EPCs remained unchanged after 3 months of therapy despite substantial reductions in HbA1c [18]. Similarly, 12 weeks of treatment with dapagliflozin in patients with T2D and a baseline HbA1c of 8.1% had no effect on EPC levels despite significant improvements of glucose control (− 0.9%). Only after a further observation of 74 weeks did EPCs increase significantly compared to baseline in patients who received dapagliflozin, despite HbA1c remaining stable [19]. A short-term assessment of EPC levels was not performed in those studies. We would like to underline that most prior studies recruited outpatients with T2D and inadequate glycaemic control, while those with acute glucose decompensation were excluded [24].
In the current study, the beneficial effect of the glycaemic improvement on EPC levels was statistically significant only in newly diagnosed diabetes and not in patients with a known history of diabetes. It is consistent with the hypothesis that long-term hyperglycaemia is responsible for progressive structural, morphological and functional damage to the bone marrow (BM), resulting in an impaired release of stem/progenitor cells to the circulation [25, 26]. Reversing such morpho-functional alterations is likely to require a much longer period of sustained good glycaemic control. Indeed, EPCs did not increase in patients with chronic complications which can accompany BM damage. This observation is not surprising since in previous studies, EPCs were found to be significantly lower in patients with a long history of T2D and especially in the presence of complications [27]. It has been widely recognized that low progenitor cell levels predict the development and the worsening of micro- and macrovascular complications in individuals with T2D [11, 12, 28, 29]. Finally, we found that levels of EPC increased in subjects with T1D but they remained stable in those with T2D. A plausible explanation for this observation is that most enrolled individuals with T1D were young and had newly diagnosed diabetes without complications. Therefore, before the onset of end-organ complications, glycaemic improvements may be able to act on reversible mechanisms of EPC reduction, which are eventually lost in the long run. Interestingly, in agreement with this hypothesis, some in vitro and in vivo studies have demonstrated the role of a poor glycaemic legacy in epigenetic changes that also involve EPCs. These changes exert a deleterious impact on proliferation activity, migration capacity, and tube formation ability and increased susceptibility to senescence and apoptosis [30, 31].
Opposite results to ours were obtained in an observational study performed by Zhang et al. [32]. They reported that 2 weeks of intensive insulin therapy reduced the levels of CD34+CD133+KDR+ cells in 36 subjects with newly diagnosed T2D. Surprisingly, they also found that patients with T2D have significantly higher levels of circulating EPC compared to controls without diabetes. The authors indicated these results as an attempt by the BM to compensate for early vascular damage. However, such data are in contrast to the existing literature which shows a reduction of progenitor cells in subjects with T2D and already in individuals with prediabetes [33]. Plausible explanations for such opposing results include differences in the study population characteristics, in the study setting, and in the method used to enumerate progenitor cells.
Limitations of our study include the relatively small simple size that might have affected the statistical power in some subgroup analyses. We also acknowledge that we do not have information on EPC traffic and function. A further limitation is that, though we assessed EPC levels after hospital discharge, we have no information on whether the increased EPC levels persist beyond 2 months.
Notwithstanding these limitations, our study supports a view whereby an acute decompensation of diabetes reduces EPCs similarly to chronic hyperglycaemia. However, the levels of EPC can be rapidly restored by an intensive insulin treatment, especially in the early disease stage. Therefore, our data highlight once more the importance of improving glycaemic control as early as possible in the natural history of diabetes.
Conclusions
This study demonstrates that in subjects hospitalized with severely decompensated diabetes, an improvement of glycaemic control by insulin therapy rapidly increases EPC levels for up to 2 months after hospital discharge. Given the role of EPCs in vascular homeostasis, glycaemic improvement may be able to revert the endogenous organism’s repair capacity, at least in the early diabetes stages.
References
Karslioglu French E, Donihi AC, Korytkowski MT. Diabetic ketoacidosis and hyperosmolar hyperglycemic syndrome: review of acute decompensated diabetes in adult patients. BMJ. 2019;365:l1114.
Avogaro A, Albiero M, Menegazzo L, de Kreutzenberg S, Fadini GP. Endothelial dysfunction in diabetes: the role of reparatory mechanisms. Diabetes Care. 2011;34(Suppl 2):S285–90.
Fadini GP, Avogaro A. It is all in the blood: the multifaceted contribution of circulating progenitor cells in diabetic complications. Exp Diabetes Res. 2012;2012:742976.
Asahara T, Masuda H, Takahashi T, et al. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res. 1999;85:221–8.
Hill JM, Zalos G, Halcox JPJ, et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593–600.
Urbich C, Dimmeler S. Endothelial progenitor cells: characterization and role in vascular biology. Circ Res. 2004;95:343–53.
Barcelos LS, Duplaa C, Kränkel N, et al. Human CD133+ progenitor cells promote the healing of diabetic ischemic ulcers by paracrine stimulation of angiogenesis and activation of Wnt signaling. Circ Res. 2009;104:1095–102.
Tse H-F, Siu C-W, Zhu S-G, et al. Paracrine effects of direct intramyocardial implantation of bone marrow derived cells to enhance neovascularization in chronic ischaemic myocardium. Eur J Heart Fail. 2007;9:747–53.
Burchfield JS, Dimmeler S. Role of paracrine factors in stem and progenitor cell mediated cardiac repair and tissue fibrosis. Fibrogen Tissue Repair. 2008;1:4. https://doi.org/10.1186/1755-1536-1-4.
Fadini GP. A reappraisal of the role of circulating (progenitor) cells in the pathobiology of diabetic complications. Diabetologia. 2014;57:4–15.
Rigato M, Bittante C, Albiero M, Avogaro A, Fadini GP. Circulating progenitor cell count predicts microvascular outcomes in type 2 diabetic patients. J Clin Endocrinol Metab. 2015;100:2666–72.
Fadini GP, Rigato M, Cappellari R, Bonora BM, Avogaro A. Long-term prediction of cardiovascular outcomes by circulating CD34+ and CD34+CD133+ stem cells in patients with type 2 diabetes. Diabetes Care. 2017;40:125–31.
Fadini GP, Sartore S, Albiero M, et al. Number and function of endothelial progenitor cells as a marker of severity for diabetic vasculopathy. Arterioscler Thromb Vasc Biol. 2006;26:2140–6.
Fadini GP, Miorin M, Facco M, et al. Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. J Am Coll Cardiol. 2005;45:1449–57.
Hayek SS, MacNamara J, Tahhan AS, et al. Circulating progenitor cells identify peripheral arterial disease in patients with coronary artery disease. Circ Res. 2016;119:564–71.
Loomans CJM, de Koning EJP, Staal FJT, et al. Endothelial progenitor cell dysfunction: a novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes. 2004;53:195–9.
Tepper OM, Galiano RD, Capla JM, et al. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation. 2002;106:2781–6.
Fadini GP, de Kreutzenberg SV, Mariano V, et al. Optimized glycaemic control achieved with add-on basal insulin therapy improves indexes of endothelial damage and regeneration in type 2 diabetic patients with macroangiopathy: a randomized crossover trial comparing detemir versus glargine. Diabetes Obes Metab. 2011;13:718–25.
Bonora BM, Cappellari R, Albiero M, Avogaro A, Fadini GP. Effects of SGLT2 inhibitors on circulating stem and progenitor cells in patients with type 2 diabetes. J Clin Endocrinol Metab. 2018;103:3773–82.
Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150:604–12.
Kitabchi AE, Umpierrez GE, Miles JM, Fisher JN. Hyperglycemic crises in adult patients with diabetes. Diabetes Care. 2009;32:1335–43.
American Diabetes Association (ADA). 2. Classification and diagnosis of diabetes: standards of medical care in diabetes-2021. Diabetes Care. 2021;44:15–33.
Fadini GP, Boscari F, Cappellari R, et al. Effects of hypoglycemia on circulating stem and progenitor cells in diabetic patients. J Clin Endocrinol Metab. 2018;103:1048–55.
Albiero M, Bonora BM, Fadini GP. Diabetes pharmacotherapy and circulating stem/progenitor cells. State of the art and evidence gaps. Curr Opin Pharmacol. 2020;55:151–6.
Fadini GP. Is bone marrow another target of diabetic complications? Eur J Clin Invest Engl. 2011;41:457–63.
Fadini GP, Ferraro F, Quaini F, Asahara T, Madeddu P. Concise review: diabetes, the bone marrow niche, and impaired vascular regeneration. Stem Cells Transl Med. 2014;3:949–57.
Fadini GP, Ciciliot S, Albiero M. Concise review: perspectives and clinical implications of bone marrow and circulating stem cell defects in diabetes. Stem Cells. 2017;35:106–16.
Rigato M, Avogaro A, Fadini GP. Levels of circulating progenitor cells, cardiovascular outcomes and death. Circ Res Am Heart Assoc. 2016;118:1930–9.
Rigato M, Fadini GP. Circulating stem/progenitor cells as prognostic biomarkers in macro- and microvascular disease: a narrative review of prospective observational studies. Curr Med Chem. 2018;25(35):4507–17.
Yu C-G, Zhang N, Yuan S-S, et al. Endothelial progenitor cells in diabetic microvascular complications: friends or foes? Stem Cells Int. 2016;2016:1803989.
Rajasekar P, O’Neill CL, Eeles L, Stitt AW, Medina RJ. Epigenetic changes in endothelial progenitors as a possible cellular basis for glycemic memory in diabetic vascular complications. J Diabetes Res. 2015;2015:436879.
Zhang W, Wang H, Liu F, et al. Effects of early intensive insulin therapy on endothelial progenitor cells in patients with newly diagnosed type 2 diabetes. Diabetes Ther. 2022;13(4):679–90.
Fadini GP, Boscaro E, de Kreutzenberg S, et al. Time course and mechanisms of circulating progenitor cell reduction in the natural history of type 2 diabetes. Diabetes Care. 2010;33:1097–102.
Acknowledgements
Funding
No funding or sponsorship was received for this study or publication of this article. The Rapid Service Fee was funded by the authors.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authors’ Contributions
Benedetta Maria Bonora, study conduction, data analysis and interpretation, manuscript writing. Roberta Cappellari, study conduction, data analysis and interpretation, manuscript revision. Marco Grasso, study conduction, data analysis and interpretation, manuscript revision. Marta Mazzucato, study conduction, data analysis and interpretation, manuscript revision. Marianna D’Anna, study conduction, data analysis and interpretation, manuscript revision. Angelo Avogaro, study design, data analysis and interpretation, manuscript revision. Gian Paolo Fadini, study design and conduction, data analysis and interpretation, manuscript writing.
Disclosures
Benedetta Maria Bonora received lecture or advisory board fees from AstraZeneca, Boehringer Ingelheim, Eli Lilly, Merck Sharp & Dome, Mundipharma, Novo Nordisk, Sanofi, Takeda. Angelo Avogaro received research grants, lecture fees, or advisory board fees from Merck Sharp & Dome, AstraZeneca, Novartis, Boehringer Ingelheim, Sanofi, Mediolanum, Janssen, Novo Nordisk, Eli Lilly, Servier, and Takeda. Gian Paolo Fadini received grant support, lecture fees, or advisory board fees from AstraZeneca, Boehringer Ingelheim, Eli Lilly, Novo Nordisk, Sanofi, Genzyme, Abbott, Novartis, and Merck Sharp & Dohme. Roberta Cappellari, Marco Grasso, Marta Mazzucato and Marianna D’Anna have nothing to disclose.
Compliance with Ethics Guidelines
Prior to enrolment, the protocol will be reviewed and approved by the Padova University Hospital ethics committee. All patients signed informed consent. All procedures were conducted in accordance with the Declaration of Helsinki.
Data Availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Bonora, B.M., Cappellari, R., Grasso, M. et al. Glycaemic Control Achieves Sustained Increases of Circulating Endothelial Progenitor Cells in Patients Hospitalized for Decompensated Diabetes: An Observational Study. Diabetes Ther 13, 1327–1337 (2022). https://doi.org/10.1007/s13300-022-01273-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-022-01273-5