
Abstract
Introduction
Sulfonylureas (SU) are commonly used antihyperglycemic agents. VERTIS CV was the cardiovascular outcome study for the sodium-glucose cotransporter 2 inhibitor ertugliflozin. Enrollment of patients in VERTIS CV occurred in two sequential cohorts (Cohort 1 and Cohort 2).
Methods
This substudy assessed the efficacy and safety of adding ertugliflozin to SU monotherapy. The primary endpoint was the change in HbA1c from baseline at 18 weeks.
Results
Among the 8246 patients who were randomized in VERTIS CV, 157 patients in Cohort 1 and 135 patients in Cohort 2 were on SU monotherapy at baseline. In the prespecified analysis (Cohort 1 only), the least squares (LS) mean HbA1c change from baseline for placebo, ertugliflozin 5 mg, and ertugliflozin 15 mg was − 0.56%, − 0.91%, and − 0.78%, respectively (placebo-adjusted LS mean [95% CI] change: − 0.35% [− 0.72%, 0.02%]; − 0.22% [− 0.60%, 0.16%] for ertugliflozin 5 and 15 mg, respectively; p > 0.05 for both). In a post-hoc analysis that included Cohorts 1 and 2 (N = 292), the LS mean HbA1c change from baseline at week 18 for placebo, ertugliflozin 5 mg, and ertugliflozin 15 mg was − 0.31%, − 0.77%, and − 0.68%, respectively (placebo-adjusted change: − 0.46% [− 0.73%, − 0.18%]; − 0.37% [− 0.66%, − 0.09%]; p = 0.001 and 0.01 for ertugliflozin 5 and 15 mg, respectively). In Cohort 1, adverse events were reported in 45.8%, 47.3%, and 25.9% of patients with placebo, ertugliflozin 5 mg, and ertugliflozin 15 mg. The incidence rates of symptomatic hypoglycemia were 0.0%, 5.5%, and 3.7%, respectively, with no cases of severe hypoglycemia. The safety profile was similar for Cohorts 1 and 2 combined.
Conclusion
The addition of ertugliflozin to SU monotherapy reduced HbA1c but did not result in significant placebo-adjusted reductions from baseline according to the prespecified primary analysis (n = 157); however, in a post-hoc analysis with a larger patient population (n = 292), significant and clinically relevant HbA1c reductions were observed. Ertugliflozin was generally well tolerated.
Trial Registration
ClinicalTrials.gov identifier: NCT01986881.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Sulfonylureas (SUs) are still frequently prescribed to patients with type 2 diabetes (T2DM), most commonly with metformin and less commonly as monotherapy, due to their low cost and good glycemic efficacy. However, SUs are associated with an increased risk of hypoglycemia and modest weight gain, and lack sustained efficacy over the long term. |
Addition of a sodium-glucose cotransporter 2 (SGLT2) inhibitor may be an attractive treatment option for patients with T2DM inadequately controlled by SU, as SGLT2 inhibitors are not associated with an increased risk of hypoglycemia when given as monotherapy, they reduce body weight, and they have cardiovascular and renal benefits. |
This 18-week substudy, which was conducted as part of the ertugliflozin cardiovascular outcome study VERTIS CV, assessed the efficacy and safety of the SGLT2 inhibitor ertugliflozin in patients with T2DM who were inadequately controlled on SU monotherapy. In VERTIS CV, patients were enrolled in two sequential cohorts (Cohort 1 and Cohort 2). |
What was learned from this study? |
Ertugliflozin added to SU monotherapy reduced HbA1c but did not result in significant placebo-adjusted reductions from baseline (Cohort 1). However, the power to detect a difference in response between the treatments was 80% given the actual Cohort 1 sample size. In a post-hoc analysis that included a larger patient population (Cohort 1 and Cohort 2) and provided greater power (97.8%) to detect between-group differences, significant and clinically relevant HbA1c reductions were observed. |
Ertugliflozin was generally well tolerated, with no cases of severe hypoglycemia. |
Ertugliflozin may be a useful treatment option in patients with T2DM who are receiving SU monotherapy and require additional glycemic control. |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.13664000.
Introduction
Sulfonylureas (SUs) are insulin secretagogues that work by stimulating insulin secretion from pancreatic β cells and have been widely available since the 1960s for the treatment of patients with type 2 diabetes (T2DM) [1, 2]. SUs may be prescribed in combination with other antihyperglycemic agents (AHAs), and the use of a SU as the first-line monotherapy may be one of the treatment choices for patients who cannot tolerate metformin [3,4,5]. Disadvantages associated with SUs are an increased risk of hypoglycemia, modest weight gain [6], and their lack of sustained glycemic control [7,8,9]. The cardiovascular (CV) safety of SUs has been demonstrated in outcome studies conducted with the dipeptidyl peptidase-4 inhibitor linagliptin, in which no increase in CV risk was observed with linagliptin vs. placebo (CARMELINA), and with glimepiride, in which no increase in CV risk was observed vs. linagliptin (CAROLINA) [10,11,12,13]. Nevertheless, because of the disadvantages associated with SUs and the availability of newer agents that are not inherently associated with hypoglycemia, some have recently questioned the continued use of SUs in clinical practice [14,15,16].
Despite the availability of newer oral AHAs, real-world evidence indicates that SUs remain extensively used second-line agents after metformin [17], although their use in this context may be declining [18]. Current international guidelines (from the World Health Organization and the International Diabetes Federation), consensus statements from the European Association for the Study of Diabetes and the American Diabetes Association, and many regional guidances consider SUs to be safe and effective AHAs, especially in the absence of atherosclerotic cardiovascular disease (ASCVD) or the presence of multiple risk factors for ASCVD, or when cost is a consideration [3, 4].
Given the continued wide usage of SUs, evaluating their efficacy and safety in conjunction with newer agents is of considerable clinical importance. Sodium-glucose cotransporter 2 (SGLT2) inhibitors are not associated with an increased risk of hypoglycemia when given as monotherapy, they reduce body weight (BW), and they have CV and renal benefits [19,20,21,22]. In contrast, SU treatment can increase BW with neutral CV and renal effects [3]. Therefore, the addition of a SGLT2 inhibitor may be an attractive treatment option for patients with T2DM inadequately controlled by SU monotherapy.
VERTIS (eValuation of ERTugliflozin effIcacy and Safety) CV, the CV outcome study (CVOT) for ertugliflozin, evaluated CV and renal outcomes in patients with T2DM and established ASCVD [22]. In VERTIS CV, approximately 40% of patients were taking a SU at baseline either as part of a combination therapy or as monotherapy, with similar proportions of patients on background SU therapy in CVOTs with other SGLT2 inhibitors (EMPA-REG, CANVAS program, and DECLARE-TIMI 58) [19,20,21]. This highlights the prevalent use of SUs for the treatment of patients with T2DM and ASCVD or those at high risk for ASCVD. Although a small proportion of the patients in VERTIS CV were on background SU monotherapy, the inclusion of these patients in the study provided a unique opportunity to evaluate the addition of ertugliflozin to SU therapy in the absence of other background AHAs such as metformin.
This report evaluates the glycemic and cardiometabolic efficacy and safety of ertugliflozin in patients with T2DM inadequately controlled with SU monotherapy. Patients in VERTIS CV were enrolled in two cohorts due to a protocol amendment that doubled the size of the trial to test additional CV and renal endpoints [22, 23]. The results reported here are from an 18-week substudy of patients on background SU monotherapy who were enrolled in Cohort 1 of VERTIS CV, and a post-hoc analysis combining data from patients on background SU monotherapy who were enrolled in Cohorts 1 and 2. The assessment of cardiac endpoints and renal parameters in this subset of patients in VERTIS CV, which requires a longer duration of follow-up, was not an objective of the substudy.
Methods
Objectives
The primary objectives of this substudy were to assess the effects of 5 mg and 15 mg ertugliflozin on glycated hemoglobin (HbA1c) at week 18, and to assess the safety and tolerability of ertugliflozin in patients on background SU monotherapy. The secondary objectives were to assess the effects of 5 mg and 15 mg ertugliflozin at week 18 on fasting plasma glucose (FPG), BW, the proportion of patients with HbA1c < 7.0%, systolic blood pressure (SBP), and diastolic blood pressure (DBP).
Study Design
The VERTIS CV study (ClinicalTrials.gov identifier: NCT01986881) was a multicenter, randomized, double-blind, placebo-controlled, parallel-group, event-driven study. This substudy was conducted during the first 18 weeks of the double-blind treatment period in patients with T2DM and established ASCVD who had inadequate glycemic control on SU monotherapy. The original protocol was finalized in August 2013 and included a planned sample size of approximately 4000 patients. Based on emerging data about the SGLT2 inhibitor class, the protocol was amended in March 2016, without knowledge of any interim results, to increase the sample size to approximately 8000 patients in order to have sufficient power to evaluate cardiorenal endpoints. Patients who had been enrolled in the trial before the March 2016 amendment were designated Cohort 1, and those who were enrolled after the March 2016 amendment were designated Cohort 2 [22]. The protocol-defined treatment, assessment for efficacy and safety endpoints, and follow-up were the same for all patients regardless of cohort. The prespecified analysis included patients in Cohort 1 only, as it was expected that there would be sufficient power based on sample sizes to demonstrate the primary objective (i.e., superiority of ertugliflozin vs. placebo in reducing HbA1c from baseline at week 18). A post-hoc analysis was also conducted using data from patients enrolled in Cohort 1 and Cohort 2.
Patients were randomized 1:1:1 to oral, once-daily ertugliflozin 5 mg, ertugliflozin 15 mg, or matching placebo. A computer-generated randomization code using a random permuted blocks method was utilized to assign patients to treatment groups. The final assignment of patients included in the substudy was determined prior to database lock by a statistical analysis software (SAS) program that checked the background medication (SU) and doses at study entry.
The study was conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki, and in compliance with all International Council for Harmonisation Good Clinical Practice Guidelines. The final protocol, any amendments, and informed consent documentation were reviewed and approved by an institutional review board or independent ethics committee at each of the investigational centers participating in the study. A list of the names of all these independent ethics committees and institutional review boards is provided in Table S1 of the “Supplementary Information.” All participants provided their informed consent.
Patient Population
Patients were included in the VERTIS CV study if they were adults (aged ≥ 40 years) with T2DM according to American Diabetes Association criteria and had baseline HbA1c levels of 7.0–10.5% as well as stable, established ASCVD involving the coronary, cerebrovascular, and/or peripheral arterial systems. The specific inclusion and exclusion criteria for the overall VERTIS CV trial have been published previously [23]. A subset of patients receiving SU monotherapy was included in this substudy. According to the protocol, patients were required to have a stable SU dose for at least 8 weeks prior to the screening visit (see Table S2 of the “Supplementary Information” for the minimum SU dose allowed for patient inclusion in the study), and to continue on the same SU dose for the 18-week duration of the substudy in order to enable the glycemic effects of ertugliflozin to be assessed. During the 18-week substudy, changes to background AHA were not allowed unless patients met predefined glycemic rescue thresholds (FPG increased to > 270 mg/dL between randomization and week 6, > 240 mg/dL during weeks 6–12, or > 200 mg/dL during weeks 12–18) or experienced clinically significant hypoglycemia.
Efficacy Endpoints and Assessments
The primary efficacy endpoint was change from baseline in HbA1c at week 18. Secondary efficacy endpoints included change from baseline in FPG, BW, SBP, and DBP at week 18 and the proportion of patients with HbA1c < 7.0% at week 18. The proportion of patients who received glycemic rescue therapy up to week 18 was also assessed. Efficacy assessments (HbA1c, FPG, BW, SBP, and DBP) were performed at weeks 0 (baseline), 6, 12, and 18. Laboratory assessments, including HbA1c and FPG, were performed at a central laboratory. BW was measured in duplicate using a standardized digital scale. Sitting blood pressure was measured in triplicate using an automated oscillometric device.
Safety Endpoints and Assessments
Adverse events (AEs) and serious AEs (SAEs) were evaluated at each study visit. Prespecified AEs of interest (also referred to as tier 1 AEs) included symptomatic hypoglycemia and AEs associated with urinary tract infections (UTI), genital mycotic infections (GMI) by gender, and hypovolemia. AEs were classified using MedDRA version 22.1. For the tier 1 analysis, UTIs, GMIs, and hypovolemia were identified by prespecified sponsor-generated customized MedDRA queries of preferred terms. Tier 2 AEs were those that occurred in ≥ 4 patients in any treatment arm. Symptomatic hypoglycemia was defined as an event with clinical symptoms reported by the investigator as hypoglycemia (biochemical documentation not required). Documented hypoglycemia (episodes with a glucose level of ≤ 70 mg/dL with or without symptoms) and severe hypoglycemia (episodes that required medical or nonmedical assistance) were also recorded.
Statistical Methods
The sample size was estimated based on the primary outcome of reduction in HbA1c from baseline at week 18. Assuming a standard deviation (SD) of 1.0% and a lost to follow-up rate of 10%, an expected sample size of 170 subjects (~ 56 per arm) would have provided approximately 85% power to detect a between-treatment difference of 0.6% in the reduction in HbA1c from baseline at week 18 using a two-sided alpha level of 0.05. As the Cohort 1 sample size was 157, and the lost to follow-up (missing value at week 18) rate was 14.6% (combined from all three groups using the excluding rescue approach), the power was actually approximately 81%. The post-hoc analysis was conducted using data from patients enrolled in Cohort 1 and Cohort 2 to increase the sample size and thereby increase the power to detect a difference in the primary endpoint and reduce the risk of type 2 error. This post-hoc analysis was conducted using the same statistical methodology as the prespecified analysis. With Cohort 1 and Cohort 2 combined, using the same parameters as for the prespecified analysis of a standard deviation (SD) of 1.0% and a lost to follow-up rate of 10%, the sample size of 292 subjects provided a power of 97.8% to detect a between-treatment difference of 0.6% in the reduction of HbA1c from baseline to week 18 using a two-sided alpha level of 0.05. All statistical analyses were performed using SAS version 9.3 (Cary, NC, USA).
A step-down hierarchical approach was used to control the type I error rate across all key efficacy endpoints in the following order: HbA1c, FPG, BW, proportion of patients with HbA1c < 7.0%, SBP, and DBP. For each endpoint, the 15-mg dose was tested vs. placebo first, followed by the 5-mg dose vs. placebo if and only if a statistically significant result was achieved for the 15-mg dose. Each test was performed at the 0.05 level, and testing was advanced to the next endpoint if and only if a statistically significant result was obtained for both doses. Efficacy assessments used the full analysis set (FAS), which included all randomized patients who received ≥ 1 dose of study medication and had ≥ 1 baseline or postbaseline measurement of the respective endpoint. Efficacy data obtained after the initiation of glycemic rescue therapy were censored (treated as missing), with the exception of the proportion of patients receiving glycemic rescue therapy. Changes in efficacy endpoints from baseline at week 18 were evaluated using a longitudinal data analysis model that assumed a common mean baseline value across treatment groups while adjusting for treatment, visit, treatment by visit interaction, and baseline estimated glomerular filtration rate (eGFR) (continuous). Logistic regression analysis was used to evaluate the proportion of patients with HbA1c < 7.0% at week 18. The statistical model included terms for treatment, baseline HbA1c (continuous), and baseline eGFR (continuous). The proportion of subjects receiving rescue therapy in each treatment group was summarized.
Safety analyses used the all subjects as treated (ASaT) population, which comprised all randomized patients who received ≥ 1 dose of study medication. With the exception of hypoglycemia, the safety analyses used the including rescue therapy approach. For the tier 1 AEs, the incidence, risk difference, 95% confidence interval (CI), and p values (not adjusted for multiplicity) were calculated. For tier 2 AEs, the incidence, risk difference, and 95% CIs were calculated.
Results
Patient Disposition and Baseline Characteristics
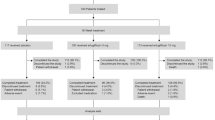
Of the 8246 patients randomized to VERTIS CV, 157 patients in Cohort 1 from across 68 centers in 15 countries were included in this substudy (Fig. 1). A high completion rate on the study medication was observed (> 90% of patients for each treatment group) at week 18, with 2.1%, 5.5%, and 9.3% of patients permanently discontinuing the study medication prior to week 18 on placebo, ertugliflozin 5 mg, and ertugliflozin 15 mg, respectively (Fig. 1). Baseline characteristics were balanced across treatment groups for most categories (Table 1). The majority of the patients (61.8%) were male, although the proportion was lower within the ertugliflozin 15 mg group (44.4% male). Most patients were White (91.7%), not Hispanic/Latino (98.1%), and from Europe (82.2%). Baseline HbA1c, FPG, and eGFR values were slightly higher with ertugliflozin 15 mg compared with placebo and ertugliflozin 5 mg. The mean body mass index (BMI) (standard deviation; SD) was 30.7 (5.5) kg/m2, and the mean duration of T2DM (SD) was 8.5 (6.5) years. A summary of SU doses at baseline is provided in Table 2.

Study flow diagram (Cohort 1)
Post-Hoc Analysis (Cohort 1 and Cohort 2 Combined)
The post-hoc analysis included 292 patients for Cohort 1 (n = 157) and Cohort 2 (n = 135) combined (Table S3 in the “Supplementary Information”). In Cohort 1 and Cohort 2 combined, the completion rate on the study medication was high (≥ 95% of patients for each treatment group); the study medication was permanently discontinued prior to week 18 by 5.0%, 3.6%, and 5.0% of the patients on placebo, ertugliflozin 5 mg, and ertugliflozin 15 mg, respectively. Baseline characteristics were balanced across treatment groups in Cohort 1 and Cohort 2 combined and were generally similar between Cohort 1 and Cohort 2 (Table S3 in the “Supplementary Information”). A summary of SU doses at baseline for Cohorts 1 and 2 combined is given in Table S4 of the “Supplementary Information.”
Efficacy
A reduction in HbA1c was observed early (at week 6) with both ertugliflozin 5 mg and 15 mg (Fig. 2). At week 18, the LS mean reduction from baseline in HbA1c for ertugliflozin 5 mg and 15 mg was − 0.91% and − 0.78%, respectively. However, a large HbA1c reduction (0.56%) was also observed with placebo. Thus, at week 18, the LS mean reduction from baseline in HbA1c was not significantly greater with ertugliflozin 5 mg or 15 mg relative to placebo (placebo-adjusted LS mean [95% CI] change: − 0.35% [− 0.72%, 0.02%] and − 0.22% [− 0.60%, 0.16%], respectively; p > 0.05 for both comparisons) (Fig. 3a and Table 3). Further hypothesis testing stopped after the comparison of ertugliflozin 15 mg with placebo for HbA1c.

LS mean change from baseline in HbA1c over time (Cohort 1)

Efficacy outcomes (Cohort 1): a LS mean change from baseline in HbA1c at week 18; b LS mean change from baseline in FPG at week 18. Efficacy outcomes in the post-hoc analysis (Cohort 1 and Cohort 2 combined); c LS mean change from baseline in HbA1c at week 18; d LS mean change from baseline in FPG at week 18
The proportion of patients with HbA1c < 7.0% at week 18 was not notably different across the treatment groups (Table 3). At week 18, the LS mean reductions from baseline in FPG were greater with ertugliflozin 5 mg and 15 mg relative to placebo (Fig. 3b and Table 3). At week 18, the LS mean change from baseline in BW decreased over time across all treatment groups, including placebo (Table 3). The LS mean reductions from baseline in SBP and DBP at week 18 were not notably different across the treatment groups (Table 3). The percentage of patients who received glycemic rescue therapy through week 18 was low and similar across treatment groups (placebo: 4.2%, ertugliflozin 5 mg: 3.6%, ertugliflozin 15 mg: 1.9%).
Post-Hoc Analysis (Cohort 1 and Cohort 2 Combined)
At week 18, the LS mean reduction from baseline in HbA1c was greater with ertugliflozin 5 mg and 15 mg relative to placebo (placebo-adjusted LS mean [95% CI] change: − 0.46% [− 0.73%, − 0.18%]; p = 0.001 and − 0.37% [− 0.66%, − 0.09%]; p = 0.01] respectively; Fig. 3c and Table S5 in the “Supplementary Information”). The proportion of patients with HbA1c < 7.0% at week 18 was not notably different across the treatment groups (Table S5 in the “Supplementary Information”). The LS mean reductions from baseline in FPG at week 18 were greater with ertugliflozin 5 mg and 15 mg relative to placebo (Fig. 3d and Table S5 in the “Supplementary Information”). The LS mean change from baseline in BW decreased over time in all treatment groups, including placebo (Table S5 in the “Supplementary Information”). The LS mean reductions from baseline in SBP and DBP at week 18 were not notably different across the treatment groups (Table S5 in the “Supplementary Information”).
Safety
The overall incidence rates of AEs and SAEs were similar across the treatment groups in Cohort 1 (Table 4). The incidence of AEs resulting in discontinuation from study medication was low (≤ 3.6% of patients in any group). One patient in the ertugliflozin 15 mg group in Cohort 1 died of an ischemic stroke during the substudy (Table 4).
Symptomatic hypoglycemic events (excluding rescue therapy use) occurred among 0.0%, 5.5%, and 3.7% of the patients who received placebo, ertugliflozin 5 mg, and ertugliflozin 15 mg, respectively (Table 4). There were few cases of documented hypoglycemia (symptomatic and asymptomatic) across treatment groups, and no cases of severe hypoglycemia (Table 4). One male patient (on ertugliflozin 5 mg) had a GMI AE of balanoposthitis that was nonserious, mild in severity, and did not lead to discontinuation of the study medication. The incidence of UTI AEs was low across treatment groups (Table 4). None of the UTI AEs were serious, and 1 UTI AE (in a patient on ertugliflozin 5 mg) led to discontinuation of the study medication. No hypovolemia AEs were reported.
Post-Hoc Analysis (Cohort 1 and Cohort 2 Combined)
The overall incidence rates of AEs and SAEs were similar across the treatment groups in Cohorts 1 and 2 combined (Table S6 in the “Supplementary Information”). The incidence of AEs resulting in discontinuation from the study medication was low (≤ 2.7% of the patients in any group). There were no additional deaths.
Symptomatic hypoglycemic events (excluding rescue therapy use) occurred among 1.3%, 8.0%, and 2.0% of the patients who received placebo, ertugliflozin 5 mg, and ertugliflozin 15 mg, respectively (Table S6 in the “Supplementary Information”). There were few cases of documented hypoglycemia (symptomatic and asymptomatic) across the treatment groups, and 1 case of severe hypoglycemia in the placebo group (Table S6 in the “Supplementary Information”). Two male patients (2.6%; both receiving ertugliflozin 5 mg) had a GMI AE. The incidence of UTI AEs was low across the treatment groups (Table S6 in the “Supplementary Information”). No hypovolemia AEs were reported.
Discussion
In the prespecified analysis of this substudy of the VERTIS CV trial, reductions in HbA1c from baseline at week 18 were observed with both ertugliflozin doses when they were added to SU monotherapy in patients with T2DM and ASCVD, but these reductions were not significantly different from placebo, resulting in a failure to meet the primary objective. LS mean (95% CI) changes in HbA1c from baseline of − 0.56% (− 0.84, − 0.27), − 0.91% (− 1.17, − 0.65), and − 0.78% (− 1.06, − 0.51) were observed for placebo, ertugliflozin 5 mg, and ertugliflozin 15 mg, respectively. The atypically large reduction in HbA1c with placebo contributed to the lack of differentiation from ertugliflozin. This result precluded the testing of subsequent endpoints in the testing sequence—the comparison of changes in HbA1c with ertugliflozin 5 mg vs. placebo, and comparisons of changes in FPG, BW, proportion of patients with HbA1c < 7.0%, SBP, and DBP with ertugliflozin 15 mg and 5 mg vs. placebo.
Multiple factors may have contributed to the unusually large placebo response observed in Cohort 1 in this substudy. In this regard, it is noteworthy that the LS mean change from baseline at week 18 in the placebo group of Cohort 1 in this substudy (− 0.56%) was two- to threefold that observed for the placebo group in the other VERTIS CV substudies (− 0.23% in the metformin + SU substudy and − 0.19% in the insulin substudy) [24, 25]. Demographic characteristics were similar for Cohort 1 and Cohort 2, although compared with the overall VERTIS CV population (56.2%), Cohorts 1 and 2 had greater percentages of patients from Europe (82.2% and 88.1%, respectively) [22]. A potential explanation for this is that there was a large effect of study participation after randomization for those patients with T2DM inadequately controlled by SU monotherapy. It is possible that enrollment in the VERTIS CV trial led to improved adherence and compliance with prescribed SU treatment regimens compared with everyday clinical practice due to the greater access to caregiver support, more frequent monitoring, and greater motivation. While the protocol required patients to have a stable SU dose for at least 8 weeks prior to the screening visit, this may not have been sufficient to stabilize HbA1c levels prior to initiation of the study medication if adherence to the prescribed SU monotherapy was also improving over this time period. Another potential explanation is that, due to the relatively small numbers of patients in the individual treatment groups in Cohort 1 (48, 55, and 54 patients in the placebo, ertugliflozin 5 mg, and ertugliflozin 15 mg groups, respectively), the precision of the endpoint estimates was predisposed to chance variability to a greater extent than when the larger number of patients from the combined Cohort 1 and Cohort 2 populations were utilized. The precision of the observed treatment response estimates increased with the larger sample size in the post-hoc analysis of patients from Cohorts 1 and 2 combined. The subsequent differentiation between the effects of ertugliflozin and placebo observed in this post-hoc analysis was consistent with prior ertugliflozin phase 3 studies [26,27,28,29], and suggests that the factor(s) that contributed to the primary result were mitigated by the addition of Cohort 2.
In a similarly designed substudy of patients with T2DM inadequately controlled by SU monotherapy that was conducted as part of the canagliflozin CVOT, a large change from baseline with placebo was not observed despite the similarly small sample size (45, 45, and 40 patients in the placebo, canagliflozin 100 mg, and canagliflozin 300 mg groups, respectively) [30]. In that substudy, LS mean changes in HbA1c from baseline at week 18 of 0.04%, − 0.70%, and − 0.79% were observed for placebo, canagliflozin 100 mg, and canagliflozin 300 mg, respectively. In a standalone study of the SGLT2 inhibitor dapagliflozin in patients with T2DM and inadequate glycemic control on glimepiride, the LS mean change in HbA1c from baseline at week 24 with placebo was − 0.13% [31]. The dapagliflozin study was conducted with a greater number of patients in the respective treatment groups (145, 154, 142, and 151 patients in the placebo and the dapagliflozin 2.5 mg, 5 mg, and 10 mg groups, respectively). In that study, LS mean changes in HbA1c from baseline at week 24 of − 0.13%, − 0.58%, − 0.63%, and − 0.82% were observed with the placebo and the 2.5 mg, 5 mg, and 10 mg doses of dapagliflozin, respectively. Both the canagliflozin substudy [30] and the dapagliflozin study [31] used a last observation carried forward statistical approach to address missing data, whereas the VERTIS CV SU substudy used a longitudinal data analysis method for handling missing data. Acknowledging the limitations of cross-study comparisons, the placebo-adjusted changes in HbA1c from baseline at week 18 of − 0.46% (− 0.73%, − 0.18%) with ertugliflozin 5 mg and − 0.37% (− 0.66%, − 0.09%) with ertugliflozin 15 mg, as observed in the post-hoc analysis of the VERTIS CV substudy described here, are consistent with those observed in the dapagliflozin study [31].
The numerically greater reduction in HbA1c observed with ertugliflozin 5 mg compared with ertugliflozin 15 mg observed in this substudy is likely explained by the relatively small number of patients who were studied and the influence of chance. Dose–response modeling of efficacy endpoints observed in ertugliflozin clinical studies, including the changes from baseline in HbA1c, FPG, BW, and the mechanistic biomarker 24-h UGE, indicates that the ertugliflozin 5 mg and 15 mg doses are associated with > 80% and > 90%, respectively, of the maximum response that is attainable with ertugliflozin. Although the ertugliflozin 5 mg and 15 mg doses are on a relatively flat portion of the dose–response curve, in six randomized placebo- and active-controlled clinical studies in the VERTIS phase 3 development program, ertugliflozin 15 mg consistently showed a numerically greater reduction in HbA1c from baseline than the 5 mg ertugliflozin dose [27,28,29, 32,33,34]. Additionally, a pooled analysis of three placebo-controlled VERTIS phase 3 ertugliflozin studies demonstrated an incremental HbA1c lowering of 0.15% with the ertugliflozin 15 mg dose relative to ertugliflozin 5 mg [35].
One of the major concerns with using SU as an AHA is its association with hypoglycemia. Insulin secretagogues such as SUs stimulate the release of insulin independent of the glucose level, increasing the risk of hypoglycemia [2]. Hypoglycemia increases the risk of CV events and mortality in patients with T2DM [36]. Although this substudy was not designed to compare the incidence of hypoglycemia events associated with ertugliflozin with those of other AHAs, low rates of hypoglycemia were observed in this substudy when ertugliflozin was added to SU monotherapy in patients with T2DM. Although this substudy required a stable SU dose for at least 8 weeks prior to screening along with continuation of the same SU dose for the 18-week study duration, the addition of a SGLT2 inhibitor to SU therapy has the potential to enable the SU dose to be lowered, which could minimize the risk of hypoglycemia [37, 38].
A recent network meta-analysis of different AHA classes added to SU monotherapy concluded that SGLT2 inhibitors have distinct advantages over other AHA classes, as they not only significantly improve glycemic control but they may also reduce the risk of hypoglycemia and BW [39]. In both the primary and post-hoc analyses of the VERTIS CV substudy reported here, the mean reductions in BW from baseline with both doses of ertugliflozin were commensurate with the reduction predicted in that meta-analysis. However, the mean reduction in BW observed in the current substudy was slightly less than that observed in both the overall VERTIS CV trial [22] and other ertugliflozin phase 3 studies [26,27,28, 32, 40]. In this substudy, the mean changes in SBP and DBP from baseline at week 18 with ertugliflozin were not notably different from those in the placebo group in either the primary or post-hoc analyses, which is consistent with the BP findings in the canagliflozin SU substudy [30], but not the dapagliflozin SU study [31].
Both doses of ertugliflozin were generally well tolerated in the substudy. The incidence rates of AEs were consistent with the known safety profile of ertugliflozin.
The major limitation of the present report is that the interpretation of the findings relies on both prespecified and post-hoc analyses. With the actual sample size of 157 patients and a lost to follow-up rate of 14.6%, the power to detect a difference in HbA1c change from baseline between the ertugliflozin and placebo groups was approximately 81%. This power was lower than typical phase 3 studies, which are often powered at approximately 90% to ensure more robust estimates of treatment effect. Patients enrolled in Cohort 2 were not included in the prespecified substudy because it was expected that there would be sufficient power to detect a treatment difference based on the sample size from Cohort 1. The post-hoc analysis was conducted with a larger sample size that included patients enrolled in both cohorts in order to increase the power of the study and thus address the primary objective. Given the 80% power to detect a difference between the treatment groups in Cohort 1 and the similar inclusion criteria for Cohort 1 and Cohort 2, a post-hoc analysis was justified, and confidence in the results is validated by the similar baseline demographics of the two cohorts. Another potential limitation is the relatively short duration of the substudy. However, the HbA1c response observed within the 18-week time period is sufficient to assist clinicians and their patients with T2DM in determining the glycemic response that can be expected when they initiate ertugliflozin treatment on a background of SU monotherapy. An assessment of the longer-term glycemic efficacy would have been confounded by changes in background AHAs that were permissible after week 18. Longer-term safety assessments were also not conducted in this substudy since changes to doses and background AHAs and the addition of agents that were allowed after the 18-week placebo-controlled period could have confounded results, especially with regard to the assessment of hypoglycemia. It is well established that ertugliflozin improves glycemic control as a monotherapy [26, 29] or when combined with other AHAs [27, 28, 32,33,34, 40]. These studies included patients with or without ASCVD. Although the VERTIS CV study and hence this substudy were conducted in patients with established ASCVD, the results of this substudy are likely to be relevant to the general population of T2DM patients without established CVD.
Conclusions
In this substudy of the VERTIS CV trial, the addition of ertugliflozin to SU monotherapy reduced HbA1c from baseline at week 18 vs. placebo in patients with T2DM, but the difference was nonsignificant based on the prespecified primary analysis that was restricted to patients from Cohort 1. However, a post-hoc analysis that included a larger patient population (Cohort 1 and Cohort 2) and provided greater power to detect between-group differences demonstrated a significant difference, and is possibly more reflective of results that might occur when adding a SGLT2 inhibitor to SU monotherapy. In this substudy, ertugliflozin was generally well tolerated and the associated incidence of AEs was consistent with the mechanism of action of SGLT2 inhibitors. While recognizing the limitations of this study, these data suggest a favorable benefit–risk profile when ertugliflozin is combined with SUs in patients with T2DM.
References
Proks P, Reimann F, Green N, Gribble F, Ashcroft F. Sulfonylurea stimulation of insulin secretion. Diabetes. 2002;51(Suppl 3):S368–76.
Sola D, Rossi L, Schianca GP, et al. Sulfonylureas and their use in clinical practice. Arch Med Sci. 2015;11:840–8.
American Diabetes Association. 9. Pharmacologic approaches to glycemic treatment: standards of medical care in diabetes—2020. Diabetes Care. 2020;43:S98-S110.
Amod A. The place of sulfonylureas in guidelines: why are there differences? Diabetes Ther. 2020;11:5–14.
Roglic G, Norris SL. Medicines for treatment intensification in type 2 diabetes and type of insulin in type 1 and type 2 diabetes in low-resource settings: synopsis of the World Health Organization Guidelines on second- and third-line medicines and type of insulin for the control of blood glucose levels in nonpregnant adults with diabetes mellitus. Ann Intern Med. 2018;169:394–7.
Belsey J, Krishnarajah G. Glycaemic control and adverse events in patients with type 2 diabetes treated with metformin + sulphonylurea: a meta-analysis. Diabetes Obes Metab. 2008;10(Suppl 1):1–7.
Kahn SE, Haffner SM, Heise MA, et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med. 2006;355:2427–43.
Nauck MA, Del Prato S, Duran-Garcia S, et al. Durability of glycaemic efficacy over 2 years with dapagliflozin versus glipizide as add-on therapies in patients whose type 2 diabetes mellitus is inadequately controlled with metformin. Diabetes Obes Metab. 2014;16:1111–20.
Del Prato S, Camisasca R, Wilson C, Fleck P. Durability of the efficacy and safety of alogliptin compared with glipizide in type 2 diabetes mellitus: a 2-year study. Diabetes Obes Metab. 2014;16:1239–46.
Bain S, Druyts E, Balijepalli C, et al. Cardiovascular events and all-cause mortality associated with sulphonylureas compared with other antihyperglycaemic drugs: a Bayesian meta-analysis of survival data. Diabetes Obes Metab. 2017;19:329–35.
Rosenstock J, Kahn SE, Johansen OE, et al. Effect of linagliptin vs glimepiride on major adverse cardiovascular outcomes in patients with type 2 diabetes: the CAROLINA randomized clinical trial. JAMA. 2019;322:1155–66.
Rosenstock J, Perkovic V, Johansen OE, et al. Effect of linagliptin vs placebo on major cardiovascular events in adults with type 2 diabetes and high cardiovascular and renal risk: the CARMELINA randomized clinical trial. JAMA. 2019;321:69–79.
Riddle MC. A verdict for glimepiride: effective and not guilty of cardiovascular harm. Diabetes Care. 2019;42:2161–3.
Abrahamson MJ. Should sulfonylureas remain an acceptable first-line add-on to metformin therapy in patients with type 2 diabetes? Yes, they continue to serve us well! Diabetes Care. 2015;38:166–9.
Genuth S. Should sulfonylureas remain an acceptable first-line add-on to metformin therapy in patients with type 2 diabetes? No, it’s time to move on! Diabetes Care. 2015;38:170–5.
Riddle MC. Modern sulfonylureas: dangerous or wrongly accused? Diabetes Care. 2017;40:629–31.
Montvida O, Shaw J, Atherton JJ, Stringer F, Paul SK. Long-term trends in antidiabetes drug usage in the U.S.: real-world evidence in patients newly diagnosed with type 2 diabetes. Diabetes Care. 2018;41:69–78.
Kitten AK, Kamath M, Ryan L, Reveles KR. National ambulatory care non-insulin antidiabetic medication prescribing trends in the United States from 2009 to 2015. PLoS ONE. 2019;14:e0221174.
Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117–28.
Neal B, Perkovic V, Mahaffey KW, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377:644–57.
Wiviott SD, Raz I, Bonaca MP, et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2019;380:347–57.
Cannon CP, Pratley R, Dagogo-Jack S, et al. Cardiovascular outcomes with ertugliflozin in type 2 diabetes. N Engl J Med. 2020;383:1425–35.
Cannon CP, McGuire DK, Pratley R, et al. Design and baseline characteristics of the eValuation of ERTugliflozin effIcacy and Safety CardioVascular outcomes trial (VERTIS-CV). Am Heart J. 2018;206:11–23.
Budoff MJ, Davis TME, Palmer AG, et al. Efficacy and safety of ertugliflozin in patients With type 2 diabetes and established cardiovascular disease treated with metformin and sulfonylurea. Diabetes Ther. 2020; In press.
Lingvay I, Greenberg M, Gallo S, Shi H, Liu J, Gantz I. Efficacy and safety of ertugliflozin in patients with type 2 diabetes mellitus and established cardiovascular disease using insulin—a VERTIS CV sub-study. Diabetes Obes Metab. 2020;Submitted for publication.
Aronson R, Frias J, Goldman A, Darekar A, Lauring B, Terra SG. Long-term efficacy and safety of ertugliflozin monotherapy in patients with inadequately controlled T2DM despite diet and exercise: VERTIS MONO extension study. Diabetes Obes Metab. 2018;20:1453–60.
Dagogo-Jack S, Liu J, Eldor R, et al. Efficacy and safety of the addition of ertugliflozin in patients with type 2 diabetes mellitus inadequately controlled with metformin and sitagliptin: the VERTIS SITA2 placebo-controlled randomized study. Diabetes Obes Metab. 2018;20:530–40.
Rosenstock J, Frias J, Pall D, et al. Effect of ertugliflozin on glucose control, body weight, blood pressure and bone density in type 2 diabetes mellitus inadequately controlled on metformin monotherapy (VERTIS MET). Diabetes Obes Metab. 2018;20:520–9.
Terra SG, Focht K, Davies M, et al. Phase III, efficacy and safety study of ertugliflozin monotherapy in people with type 2 diabetes mellitus inadequately controlled with diet and exercise alone. Diabetes Obes Metab. 2017;19:721–8.
Fulcher G, Matthews DR, Perkovic V, et al. Efficacy and safety of canagliflozin used in conjunction with sulfonylurea in patients with type 2 diabetes mellitus: a randomized, controlled trial. Diabetes Ther. 2015;6:289–302.
Strojek K, Yoon KH, Hruba V, Elze M, Langkilde AM, Parikh S. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with glimepiride: a randomized, 24-week, double-blind, placebo-controlled trial. Diabetes Obes Metab. 2011;13:928–38.
Grunberger G, Camp S, Johnson J, et al. Ertugliflozin in patients with stage 3 chronic kidney disease and type 2 diabetes mellitus: the VERTIS RENAL randomized study. Diabetes Ther. 2018;9:49–66.
Hollander P, Liu J, Hill J, et al. Ertugliflozin compared with glimepiride in patients with type 2 diabetes mellitus inadequately controlled on metformin: the VERTIS SU randomized study. Diabetes Ther. 2018;9:193–207.
Pratley RE, Eldor R, Raji A, et al. Ertugliflozin plus sitagliptin versus either individual agent over 52 weeks in patients with type 2 diabetes mellitus inadequately controlled with metformin: the VERTIS FACTORIAL randomized trial. Diabetes Obes Metab. 2018;20:1111–20.
Liu J, Tarasenko L, Terra SG, et al. Efficacy of ertugliflozin in monotherapy or combination therapy in patients with type 2 diabetes: a pooled analysis of placebo-controlled studies. Diab Vasc Dis Res. 2019;16:415–23.
International Hypoglycaemia Study Group. Hypoglycaemia, cardiovascular disease, and mortality in diabetes: epidemiology, pathogenesis, and management. Lancet Diabetes Endocrinol. 2019;7:385–96.
European Medicines Agency. SteglatroTM: summary of product characteristics, 2018. https://www.ema.europa.eu/en/documents/product-information/steglatro-epar-product-information_en.pdf. Accessed 15 Dec 2020.
Food and Drug Administration. SteglatroTM: prescribing information, 2017. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/209803s000lbl.pdf. Accessed 15 Dec 2020.
Qian D, Zhang T, Tan X, et al. Comparison of antidiabetic drugs added to sulfonylurea monotherapy in patients with type 2 diabetes mellitus: a network meta-analysis. PLoS ONE. 2018;13:e0202563.
Miller S, Krumins T, Zhou H, et al. Ertugliflozin and sitagliptin co-initiation in patients with type 2 diabetes: the VERTIS SITA randomized study. Diabetes Ther. 2018;9:253–68.
Acknowledgements
The authors wish to thank the investigators, staff, and participants of the VERTIS CV study (protocol B1521021), as well as Philip Jones and Robert Frederich for their critical review of this manuscript.
Funding
This study and the Rapid Service Fee were funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, in collaboration with Pfizer Inc., New York, NY, USA.
Editorial Assistance
Editorial support was provided by Marion James, PhD, CMPP, of Engage Scientific Solutions (Horsham, UK) and Anil Sindhurakar, PhD, of Engage Scientific Solutions (Fairfield, USA), and was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, in collaboration with Pfizer Inc., New York, NY, USA.
Authorship
All the authors meet the International Committee of Medical Journal Editors criteria for authorship, take full responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Vanessa Dell, Melanie Sisson, and Shuai Wang are employees and shareholders of Pfizer Inc. Jie Liu, Susan Huyck, and Ira Gantz are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and own stock in Merck & Co., Inc., Kenilworth, NJ, USA. Krzysztof Strojek received honoraria for speaking engagements and research support from Boehringer Ingelheim, Novo Nordisk, Eli Lilly, Merck, Sanofi, Servier, AstraZeneca, MSD, Pfizer, Berlin Chemie, and Polpharma. A. Shekhar Pandey received honoraria for speaking engagements and research support from Boehringer Ingelheim, Novo Nordisk, Eli Lilly, Merck, Sanofi, Servier, AstraZeneca, MSD, and Pfizer.
Compliance with Ethics Guidelines
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964, as revised in 2013. The study was conducted in accordance with the Principles of Good Clinical Practice and approved by the appropriate institutional review boards and regulatory agencies. Informed consent was obtained from all individual participants included in the study. The protocol and statistical analysis plan were developed by the sponsors in consultation with an external scientific advisory committee.
Data Availability
Upon request, and subject to certain criteria, conditions, and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual deidentified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices (1) for indications that have been approved in the US and/or EU or (2) in programs that have been terminated (i.e., development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The deidentified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Strojek, K., Pandey, A.S., Dell, V. et al. Efficacy and Safety of Ertugliflozin in Patients With Diabetes Mellitus Inadequately Controlled by Sulfonylurea Monotherapy: a Substudy of VERTIS CV. Diabetes Ther 12, 1175–1192 (2021). https://doi.org/10.1007/s13300-021-01018-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-021-01018-w