
Abstract
The lung is an important open organ and the primary site of respiration. Many life-threatening diseases develop in the lung, e.g., pneumonia, asthma, chronic obstructive pulmonary diseases (COPDs), pulmonary fibrosis, and lung cancer. In the lung, innate immunity serves as the frontline in both anti-irritant response and anti-tumor defense and is also critical for mucosal homeostasis; thus, it plays an important role in containing these pulmonary diseases. Innate lymphoid cells (ILCs), characterized by their strict tissue residence and distinct function in the mucosa, are attracting increased attention in innate immunity. Upon sensing the danger signals from damaged epithelium, ILCs activate, proliferate, and release numerous cytokines with specific local functions; they also participate in mucosal immune-surveillance, immune-regulation, and homeostasis. However, when their functions become uncontrolled, ILCs can enhance pathological states and induce diseases. In this review, we discuss the physiological and pathological functions of ILC subsets 1 to 3 in the lung, and how the pathogenic environment affects the function and plasticity of ILCs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Regional immunity greatly differs from the conventional immune organ or system. Because human diseases are tightly connected with regional immunity, researchers have recently begun to focus on the regional immunity of organs such as the lung, intestine, liver, and skin. The lung is an open organ that is involved in gas conduction and exchange. Approximately 8,000 to 9,000 liters of air are breathed into the lung every day. Compared with the gut and the skin, the lung has a wider surface area, up to 90 m2. A single layer of pulmonary epithelial cells covers the alveoli (Kopf et al., 2015). Because of these characteristics, the lung is constantly exposed to environmental stressors, such as pathogens, allergens, and airborne toxins, e.g., cigarette smoke. In the battle between the mucosal immune cells and the invaders, the innate immune cells are the first line of defense, fortifying the trenches. The innate immune cells in the lung mainly comprise lung-resident macrophages, lung-resident dendritic cells (DCs) (Holt et al., 2008; Kopf et al., 2015), and emerging sets of innate lymphoid cells (ILCs) (Spits and Di Santo, 2011; Spits and Cupedo, 2012; Eberl et al., 2015).
ILCs are important tissue-resident innate immune cells. They are promptly activated by danger signals from injured mucosa and produce an array of effective cytokines to repel pathogens and tumor cells, thereby maintaining mucosal integrity. However, if they are excessively activated, they may cause pathologic tissue damage, e.g., asthma, Crohn’s disease, or psoriasis (Buonocore et al., 2010; Spits and Cupedo, 2012; Spits et al., 2013; Karta et al., 2016). Research into ILCs in the lung is only now in its infancy but it is known that there are three groups of ILCs in the lung, namely ILC1s, ILC2s, and ILC3s. Recently Lai et al. reviewed the origin, development, heterogeneity, and interaction of ILCs with other cells in the lung (Lai et al., 2016), however the roles of ILCs in lung pathologies have not been extensively reviewed, especially with respect to ILC1s and ILC3s. With this in mind, here we describe the general characteristics, functions, and phenotypic plasticity of ILCs, focusing especially on the roles of all three groups of ILCs in diseases of the lung.
CLASSIFICATION AND GENERAL CHARACTERISTICS OF ILCs
Lymphoid tissue inducer (LTi) cells and NK cells are prototypic ILCs, which require the common γ chain of the interleukin-2 receptor (IL-2Rγ) and transcriptional repressor inhibitor of DNA binding 2 (Id2) for their development (Kelly and Scollay, 1992; Held et al., 2011; Hesslein and Lanier, 2011). Over the last ten years, numerous cells have been identified whose development is also IL-2Rγ and Id2-dependent; these cells are referred to as “innate lymphoid cells” (ILCs). ILCs have three main characteristics: a lymphoid morphology, the absence of rearranged antigen-specific receptors, and a lack of myeloid dendritic cell phenotypical markers; ILCs also do not express antigen receptors or undergo clonal selection (Spits and Cupedo, 2012; Spits et al., 2013). In 2013, Spits et al. classified the ILCs into three groups according to their cytokine secretion ability, mirroring CD4+ T helper (Th) cells: group 1 ILCs (ILC1s), comprising conventional NK cells and ILCs that produce interferon-γ (IFN-γ); group 2 ILCs (ILC2s), which are ILCs that secrete type 2 cytokines, such as IL-5 and IL-13; and group 3 ILCs (ILC3s), that produce IL-17 and/or IL-22 (Spits and Cupedo, 2012; Spits et al., 2013).
ILCs mirror Th cells but also differ from them. Although ILCs and Th cells both arise from a common lymphoid progenitor (CLP), the development of ILCs is unique. It is generally believed that ILCs initially develop in fetal liver, whereas after birth they develop in the bone marrow (Vosshenrich et al., 2005; Sawa et al., 2010; Klose et al., 2014), and are then subsequently recruited into other tissues (Eberl et al., 2015; Gasteiger et al., 2015). Migration into tissues is likely mediated by the co-ordinated action of adhesion molecules and chemokines (Eberl et al., 2015). Interestingly, some researchers found that ILC progenitors seed themselves in tissues in the embryonic and adult phases, and in these tissue micro-environments they undergo development and differentiation (Montaldo et al., 2014; Bando et al., 2015). It should be noted that, in contrast to Th cells, ILCs remain tissue-resident; they are maintained locally in one organ and do not re-enter the circulation or migrate to other organs (Gasteiger et al., 2015; Fan and Rudensky, 2016). Following acute environmental challenges tissue-resident ILCs expand locally and in this way the pool of cells is renewed, although phenotypic transformation can also occur. Hematogenously derived ILC precursors or mature ILCs can also partially supply the local tissue ILC pool (Gasteiger et al., 2015). In addition, ILCs lack recombination-activating gene (RAG), meaning that unlike B and T cells, ILCs can be activated directly (Spits and Cupedo, 2012; Spits et al., 2013). When the mucosa is invaded by pathogens, allergens, or tumor cells, damaged epithelial cells secrete cytokines to directly stimulate ILCs. ILCs then become promptly activated, proliferate, and produce copious amounts of cytokines to repel the invaders and maintain mucosal homeostasis; this response, however, may also lead to pathological damage (Spits and Di Santo, 2011; Scanlon and McKenzie, 2012; Philip and Artis, 2013; Salimi and Ogg, 2014). Thus, ILCs become activated by sensing danger signals from the tissue milieu rather than by antigen presentation with antigen-presenting cells (APCs) (Drake and Kita, 2014; Eberl et al., 2015). Compared with the few days or weeks required by Th cells (Hansen et al., 1999), ILCs can therefore be activated more quickly (Spits and Cupedo, 2012; Fan and Rudensky, 2016) (Table 1).
ILC plasticity
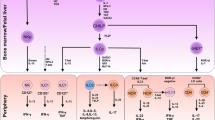
The phenotype of ILCs is not stable; these cells are highly plastic and can change phenotypes under the influence of the environment. IL-2 and IL-12 drive human natural cytotoxicity receptor (NCR)-positive ILC3s (NCR+ILC3s) to transform into ILC1s (Cella et al., 2010; Bernink et al., 2013; Bernink et al., 2015). In response to the tissue environment in vivo, ILC3s down-regulate the expression of RORγt (retinoic acid receptor-related orphan receptor γt) and produce IFN-γ (Vonarbourg et al., 2010a). IL-1β and IL-12 induce ILC2s to express T-bet and to produce IFN-γ while down-regulating ST2 and GATA3, and losing the ability to produce IL-5 and IL-13 (Bal et al., 2016; Kim et al., 2016; Lim et al., 2016). IL-23 and IL-1β cause CD127+ ILC1s to differentiate into IL-22-producing ILC3s (Bernink et al., 2015). Enhanced GATA3 expression by ILC1s results in their conversion to ILC2s with the capacity to produce greater amounts of type 2 cytokines (Mjosberg et al., 2012; KleinJan et al., 2014). Such plasticity occurs not only between ILC groups, but also between ILC subgroups. For example, mouse NKp46−RORγt+ LTi-like cells may transform into NKp46+RORγt+ cells either in vivo or in vitro (Vonarbourg et al., 2010a; Klose et al., 2013; Rankin et al., 2013; Rankin et al., 2016). Human NKp44−ILC3s undergo a profound shift toward NKp44+ ILC3s upon culture in the presence of IL-2, IL-1β, and IL-23, and they display pro-inflammatory properties (Bernink et al., 2013; Glatzer et al., 2013). Plasticity is one of the important characteristics of ILCs, and this property is especially important in the lung; the shift of ILC2s to ILC3s and the plasticity within ILC2 subgroups will be discussed below in detail (Table 2) (Fig. 1).

ILC plasticity. ILCs recruit into the lung and become resident in the mucous epithelium. When the tissue is exposed to danger signals elicited by pathogens, allergens or tumor cells, the epithelium or other innate immune cells produce many cytokines. In response to these cytokines, ILCs may alter their phenotype to respond to the environment. IL-2 and IL-12 drive the transformation of ILC3s to ILC1s. ILC1s convert to ILC3s under the influence of IL-1β and IL-23; ILC2s also transform to ILC1s when cultured with IL-12 and IL-1β. Upon increased GATA3 expression, ILC1s gain ILC2s characteristics; when cultured with TGF-β and IL-6, ILC2s become ILC3-like. Whether ILC3s convert into ILC2s is still unclear. In the ILC2 and ILC3 sub-groups, iILC2 cells give rise to cells with nILC2 phenotype when cultured in the presence of IL-2, IL-7, IL-25, and IL-33 in vitro or in vivo. Under the influence of IL-2, IL-1β, and IL-23, NCR−ILC3s express NCR+. The hypothesis that nILC2s convert to iILC2s and NCR+ILC3s convert to NCR−ILC3s should be confirmed in the future. See text for details
Identification and characterization of ILC1s
Compared with the other ILC groups, ILC1s are the least studied. Their characteristics are not yet well defined, and indeed there is even still debate concerning their classification. As of now, the ILCs nomenclature introduced by Spits et al. (2013) is widely accepted, with NK cells and ILC1s belonging to group 1 ILCs. Group 1 ILCs are defined by their ability to produce IFN-γ and inability to produce IL-4, IL-13, IL-22, and IL-17. They require transcription factor T-bet for their development (Spits et al., 2013). NK cells have been reviewed elsewhere, and therefore we will only focus on ILC1s in this review.
The original report concerning mouse ILC1s described them as RORγt−NKR−LTi, capable of releasing IFN-γ, and as being potent inducers of experimental colitis (Vonarbourg et al., 2010b). Another group of mouse ILC1s was then described as NKp46+NKp1.1+Eomes−T-bet+, with the ability to produce IFN-γ (Klose et al., 2014). In humans, ILC1s can be divided into CD127low ILC1s and CD127high ILC1s, based on the expression of CD127. CD127low ILC1s are defined as CD3−CD56+NKp44+CD103+ and CD3−CD56+NKp44−CD103−; the former have an intraepithelial localization (Fuchs et al., 2013). CD127high ILC1s were first identified in human tonsil and intestine, and they are defined as Lineage (Lin)−CD127+CRTH2+CD117−NKp44− (Bernink et al., 2013). Although the markers that define ILC1s in mouse and human are somewhat different, the two are generally defined as lineage negative, T-bet positive, and both are capable of producing IFN-γ.
CD127high and CD127low ILC1s have also been identified in the human lung. CD127high ILC1s are identified as Lin−CD127+CD117−NKp44−; they express T-bet but do not express C-C motif chemokine receptor 6 (CCR6), CD103, or CD25. CD127low ILC1s are defined as Lin−CD127−NKp46+, and express T-bet. Similar to CD127high ILC1s in the tonsil and the intestine, most Lin−CD127−NKp46+NKp44+cells express CD103 (Carrega et al., 2015). Another group identified non-toxic ILC1s in the human lung was Lin−CD56−IL12Rβ2+ (De Grove et al., 2016). In mouse lung, ILC1s are defined by the Lin−CD90+T-bet+ phenotype, and they express IL-12Rβ2 and IL-18Rα; furthermore, IL-12 and IL-18 enhance ILC1 expansion in vivo, and they produce copious amounts of IFN-γ (Silver et al., 2016a) (Table 2).
ILC1s in the lung
ILC1s and infection
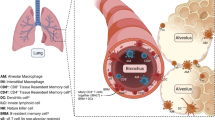
ILC1s produce large amounts of IFN-γ and protect the organism against pathogens as first reported in mouse models of intracellular infection, e.g., infections caused by the parasite Toxoplasma gondii and by Clostridium difficile in the intestine (Klose et al., 2014; Abt et al., 2015). Silver et al. (2016a, b) found that during lung infection in mice caused by either influenza A, Haemophilus influenzae, respiratory syncytial virus (RSV), or Staphylococcus aureus, GATA3 expression in the resident ILC2s was rapidly down-regulated (within two days after infection), and this was accompanied by decreased expression of ST2, CD25 (IL-2Rα), IL-7Rα, inducible costimulator (ICOS), and the stem cell factor receptor c-kit (CD117). Meanwhile, the T-bet+ ILC1 number in the lung increased and the expression of IL-12 and IL-18 receptors (IL-12Rβ2 and IL-18Rα) in ILC1s was up-regulated. The down-regulation of GATA3 expression was negatively correlated with IL-18Rα up-regulation. These results indicate that during infection of the lung, ILC2s may lose their properties and phenotypically convert into ILC1s. To confirm these findings, Silver et al. used a reporter mouse that expressed ST2 labeled with green fluorescent protein (GFP), they found that upon stimulation with IL-12, IL-18, and IL-33, the number of IL-18Rα+ ILC1 cells increased, and 50% of these cells expressed ST2-GFP. Collectively, these data suggest that lung ILC1s are derived from ILC2s that are resident in the lung rather than from ILC1 proliferation.
In the same study, GFP+ ILC2s (GFP-labeled ILC2s) were transferred to Rag2 −/−/Il2rg −/− mice with mature lymphocyte deficiency, and then the mice were infected with influenza A virus. GFP+ ILC2s infiltrated into the lungs of host mice 7 d after infection. GATA3 expression in these cells was significantly down-regulated and accompanied by a striking up-regulation of both IL-18Rα and IL-12Rβ2 expression. Double IHC (immunohistochemistry) revealed that ILCs were localized to the influenza virus-infected airways. IHC combined with hybridization in situ revealed that Il-12 and Il-18 mRNAs produced by myeloid-derived cells were present near GFP+ ILC2s in the inflamed region. GATA3highILCs were predominantly localized in uninfected tissue regions, whereas GATA3low ILCs were enriched in virus-associated areas (Silver et al., 2016a).
In summary, these data demonstrate that during infection, ILC2s migrate to the inflamed regions, where the myeloid-derived pro-inflammatory cytokines IL-12 and IL-18 drive ILC2 conversion into ILC1s, enabling their participation in the anti-pathogen response (Fig. 2).

ILC1 functions in the lung. When pathogens, such as viruses or bacteria, or tumor cells invade the airway epithelium, the myeloid cells receive danger signals from the epithelium and produce IL-12 and IL-18. These pro-inflammatory cytokines down-regulate GATA3 expression of ILC2s and then drive the conversion of ILC2s into ILC1s. IL-12 and IL-18 also enhance the activation and expansion of ILC1s. After activation, ILC1s produce copious amounts of IFN-γ. IFN-γ plays potentially important roles in clearing both pathogens and tumors, and also in the development of chronic obstructive pulmonary disease (COPD). See text for details
ILC1s and chronic obstructive pulmonary disease (COPD)
COPD is widely regarded as a heterogeneous disease associated with increased numbers of alveolar macrophages, T lymphocytes (predominantly Tc1, Th1, and Th17 cells), B lymphocytes, and neutrophils (Barnes, 2009; Kearley et al., 2015). Recently, two groups almost simultaneously reported a relationship between ILC1s and COPD (Bal et al., 2016; Silver et al., 2016a). The percentage of ILC1s is much higher in patients with COPD than in healthy controls, and is accompanied by a lower occurrence of ILC2s, either in the lung or in the circulation (Bal et al., 2016; Silver et al., 2016a). According to the classification of the Global Initiative for Chronic Obstructive Lung Disease (GOLD), ILC1s occur more frequently in severe COPD (GOLD III–IV) than in milder COPD (GOLD I–II). A strong negative correlation exists between the occurrence of ILC1s in the blood and lung function, with a higher proportion of ILC1s associated with worse lung function. The numbers of circulating ILC1s are higher in patients with two or more exacerbations of COPD per year than in patients with one exacerbation per year (Bal et al., 2016; Silver et al., 2016a).
The development and exacerbation of COPD are associated with cigarette smoke and viral and bacterial infection. Silver et al. (2016a, b) reported that the occurrence of GATA3+ ILC2s declines promptly and that the fraction of T-bet+IL-18Rα+ ILC1s is increased in response to cigarette smoke or viral and bacterial infections in mouse models (Silver et al., 2016b). When ILC2s from human fetal lung are cultured with IL-2, IL-1β, and IL-12, CRTH2 and c-kit in ILC2s are down-regulated, and the cells produce IFN-γ but not IL-5. These results indicate that ILC2s have the potential to transform into ILC1s when exposed to a type 1 inflammatory environment, such as cigarette smoke or infection, and participate in the development of COPD (Bal et al., 2016) (Fig. 2).
ILC1s and tumors
Recently, Dadi et al. (2016) discovered that unconventional type 1-like innate lymphoid cells and type 1 innate-like T cells play a role in tumor-elicited immune surveillance in murine cancer models (Dadi et al., 2016). This suggests that ILC1s may possess an anti-tumor function; however, no data on the function of ILC1s in lung tumors are available, and future studies should thus be performed to further elucidate this issue.
Identification and characterization of ILC2s
In 2001, Fort et al. were the first to report that non-T/non-B T cells in Rag2 −/−mice could produce IL-5 and IL-13, leading to a type 2 response (Fort et al., 2001). Ten years later, these cells were observed by different researchers and were differently named as nuocytes, natural helper cells, or innate helper type 2 cells (Moro et al., 2010; Neill et al., 2010; Price et al., 2010). These three groups of cells share similar molecular surface markers and function, and finally they were collectively named as group 2 innate lymphoid cells. In addition to Id2 and IL-2Rγ, the development and function of ILC2s depend on GATA3, Notch, and RORα (Halim et al., 2012b; Mjosberg et al., 2012; Gentek et al., 2013). Growth factor independence-1 (Gfi1), B cell leukemia/lymphoma 11b (Bcl11b), lysine methyltransferase G9a, and ETS1 are also essential for the development of ILC2s (Spooner et al., 2013; Walker et al., 2015; Yu et al., 2015; Zook et al., 2016).
ILC2s can be activated by IL-25, IL-33, and thymic stromal lymphopoietin (TSLP), which are produced by epithelial cells and certain immune cells. Other ILC2 activators were later identified, namely, tumor necrosis factor (TNF)-family cytokine TL1A, prostaglandin D2, cysteinyl leukotriene receptor 1 (CysLT1), arginase 1, receptor for advanced glycation end-products, and surfactant protein D (Barnig et al., 2013; Doherty et al., 2013; Meylan et al., 2014; Yu et al., 2014; Tait Wojno et al., 2015; Taniguchi et al., 2015; Monticelli et al., 2016; Thawer et al., 2016). IL-1β has been considered to be an activator of ILC3s, but a recent study demonstrated that IL-1β may also activate ILC2s (Bal et al., 2016). These cells can also produce amphiregulin to enhance the recovery of the mucosa during viral infection (Monticelli et al., 2011). ILC2 inhibitors were recently identified and include prostaglandin I2, IFN-γ, IL-27, and lipoxin A4. Because these molecules inhibit ILC2 proliferation and cytokine production (McHedlidze et al., 2016; Moro et al., 2016; Zhou et al., 2016), they may be used to control ILC2-related diseases (See below). Activated ILC2s predominantly produce type 2 cytokines such as IL-5, IL-13, and IL-4, and also IL-9 (Moro, 2010; Mjosberg et al., 2011; Wilhelm et al., 2011; Kim et al., 2013).
In the mouse lung, ILC2s are defined as Lin−CD90+ICOS+CD25+ST2+CD127+. They also express CD44 and IL-17BR, and 20% of nuocytes (ILC2s) express c-kit (CD117) (Monticelli et al., 2011; Barlow et al., 2012; Bartemes et al., 2012). Based on their killer cell lectin-like receptor G1 (KLRG1) expression, mouse lung ILC2s can be divided into two groups: nILC2s, designated as Lin−ST2+KLRG1int cells, and iILC2s, designated as Lin−ST2−KLRG1hi (Huang et al., 2015). ILC2s are also found in the fetal and adult lung, and in the bronchoalveolar lavage fluid (BLF) in human. They are defined as Lin−CD127+CD161+CRTH2+ and also express ICOS, CD25 and ST2; they also partially express CD117 (Mjosberg et al., 2011; Monticelli et al., 2011). ILC2s in mouse and human have similar markers and function, although CRHT2 is specific for human since mouse ILC2s do not express this marker (Table 2).
ILC2s in the lung
ILC2s were the first ILC group identified in lung (Monticelli et al., 2011). ILC2-related diseases in the lung involve pathogen infections (virus and helminth parasites), asthma, pulmonary fibrosis, and eosinophilic pleural effusion in primary spontaneous pneumothorax.
ILC2s and viral infection
During viral infection, ILC2s may exert a dual effect in the lung: on the one hand, ILC2s play a protective role in repelling the virus; on the other hand, if ILC2 function is not tightly controlled, these cells may induce airway hyperactivity (Fig. 3).

ILC2 functions in the lung. Following interaction with pathogens or allergens, the airway epithelium secretes IL-25, IL-33, TSLP, and TGF-β. Upon a molecular cue from the damaged epithelium, activated macrophages and DCs also release IL-33. All of these cytokines activate ILC2s. After activation, ILC2s proliferate and produce copious amounts of IL-4, IL-5, IL-9, IL-13, and amphiregulin. IL-4 stimulates B cells and also activates DCs and enhances Th2 cell maturation and activation. IL-5 and IL-13 stimulate the proliferation and recruitment of eosinophils, which are involved in parasite clearance and airway hyper-responsiveness (AHR). IL-5 also enhances B1 cell self-renewal and antibody production by B cells. ILC2s produce IL-9, and the autocrine effect of IL-9 stimulates secretion of effector cytokines by ILC2s. IL-13 induces smooth muscle contractility and airway remodeling that leads to AHR. Additionally, IL-9 plays an essential role in mast cell differentiation and also can induce mast cells to secrete IL-6; IL-9 can also promote both the proliferation of eosinophils and induce their migration into the lung. IL-13 can also induce collagen deposition and the development of pulmonary fibrosis. Furthermore, IL-13 enhances eosinophil recruitment and alternative macrophage activation. Finally, ILC2s also play an important role in airway remodeling by secreting amphiregulin. See text for details
Maintenance of epithelial integrity
During the early phase of respiratory infection caused by influenza A virus subtype H1N1, no significant differences were observed between wild-type mice and Rag −/− mice that lack adaptive immunity with respect to the decline in lung function and pathology of the lung. Because ILC2s accumulated in the lung, it was inferred that lung ILC2s play a key role in the regulation of lung innate immunity and tissue homeostasis (Monticelli et al., 2011). Depletion of ILCs led to substantial lung epithelial degeneration and necrosis, as a result of significantly impaired epithelial integrity. Adoptive transfer of ILCs to ILC-depleted mice effectively restored epithelial integrity (Monticelli et al., 2011). In the same study it was found that IL-33/IL-33R signaling was essential for the accumulation of ILC2s in H1N1 virus infected lungs whereas IL-13 and IL-22 appeared to be dispensable for tissue homeostasis (Monticelli et al., 2011). Data from genome-wide transcriptional profiling has suggested that ILC2s in the lung express wound healing-associated genes at higher levels. In vivo, lung ILC2s have the ability to produce high levels of amphiregulin, a molecule that regulates tissue remodeling and repair during acute epithelial injury and asthma (Dolinay et al., 2006; Enomoto et al., 2009; Fukumoto et al., 2010; Monticelli et al., 2011). In summary, lung ILC2s play an important role in maintaining the integrity of the respiratory epithelium and restoring lung function by producing amphiregulin during H1N1 infection.
Induction of lung inflammation and airway hyper-reactivity
Chang et al. (2011) observed that BALB/c mice infected with H3N1 virus rapidly develop AHR (airway hyper-responsiveness) that peaked on day 5 of infection. Because the sensitization of Th2 cells and enrollment of adaptive immunity during allergen-induced AHR required 7~14 d to develop (Hansen et al., 1999), based on the speed with which H3N1 induced AHR, it was hypothesized that this response was mediated by innate immune mechanisms. The authors provided evidence that indeed H3N1-induced AHR development did involve the innate immune pathway and did not require T, B, or NKT cells (Chang et al., 2011). Depletion of natural helper cells suppressed the AHR response, and furthermore only the transfer of IL-13-producing ILC2s, but no other IL-13-producing cells (such as mast cells or basophils), was sufficient for the development of H3N1-induced AHR. Furthermore, H3N1-induced AHR was tightly correlated with the presence of ILC2s, IL-33, and IL-13. IL-33 derived from alveolar macrophages likely plays an essential role in the activation of ILC2s (Chang et al., 2011). These data confirm that ILC2s are essential for the development of AHR during H3N1 infection.
During RSV infection, the number of IL-13-producing ILC2s triples compared with basal levels, and the IL-13 levels increase simultaneously. This increase contributes to airway hyper-reactivity and airway mucus accumulation in a TSLP-dependent manner (Stier et al., 2016). ILC2s have also been shown to produce IL-13 in order to recruit eosinophils and induce AHR through the IL-33/ST2 pathway during RSV infection (Liu et al., 2015b). Similarly, neonatal rhinovirus induces AHR and mucus metaplasia through IL-25 and ILC2s (Hong et al., 2014).
ILC2s and helminth parasites
Upon infection with helminthic parasites, the host undergoes a strong type 2 response to clear the pathogens (Paul and Zhu, 2010; Maizels et al., 2012). During Strongyloides venezuelensis infection of the lung, IL-33 levels increase, activating ILC2s to release the effector cytokines IL-5 and IL-13 that, in turn, can recruit eosinophils to fight the infection. IL-33−/− mice are unable to recruit eosinophils to the lungs or expel S. venezuelensis (Yasuda et al., 2012). During lung inflammation induced by Nippostrongylus brasiliensis, epithelial cells produce TSLP and IL-33, synergistically stimulating ILC2s to produce IL-5 and IL-13. Furthermore, IL-9, which is produced by ILC2s in an autocrine manner, stimulates IL-5 and IL-13 production by ILC2s. In IL-9 receptor-deficient mice infected with N. brasiliensis, ILC2s numbers are reduced and IL-5, IL-13, and amphiregulin levels are decreased, whereas the numbers of Th2 cells remain unchanged. As a result, helminth clearance is strongly impaired (Turner et al., 2013). The IL-9 signal plays an important role in the activation of ILC2s, especially in the early phase of helminth infection. This process also requires interferon regulatory factor 4 (IRF4) (Turner et al., 2013; Mohapatra et al., 2016).
During N. brasiliensis infection, the ILC2 subgroup iILC2 can convert into nILC2. N. brasiliensis-infected Il17rb −/− mice lack iILC2 cells and, interestingly, nILC2 cell numbers are significantly decreased in these mouse models. This suggests that iILC2s contribute to the nILC2 population during N. brasiliensis infection. Researchers simultaneously transferred CD45.1+ nILC2s and CD45.2+ iILC2s into Rag2 −/− Il2rg −/− mice infected with N. brasiliensis, and 14 d later found that all iILC2 cells had developed into nILC2-like cells. iILC2s may thus comprise a transient progenitor population of cells that can transform into nILC2-like cells and be involved in the clearance of N. brasiliensis (Huang et al., 2015) (Fig. 1).
Interestingly, another natural mouse parasite, Heligmosomoides polygyrus, that causes parasitic infections suppresses inflammatory responses in models of asthma, food allergy, diabetes, and colitis (McSorley and Maizels, 2012). H. polygyrus excretory/secretory (HES) products can suppress both Treg (Grainger et al., 2010) and dendritic cells (Massacand et al., 2009). McSorley et al. found that HES products inhibit the allergic reaction in the ovalbumin (OVA)-induced mouse asthma model by suppressing the release of IL-33 and inhibiting the activation of ILC2s (McSorley et al., 2014; McSorley et al., 2015).
ILC2s and asthma
Asthma is a heterogeneous disease that occurs worldwide and whose pathology involves chronic airway inflammation. The type 2 response is regarded as a central mechanism for allergic asthma (Deckers et al., 2013; Licona-Limon et al., 2013). Conventionally, Th2 cells have been regarded as the main source of type 2 cytokines, but this notion has now been challenged by the emergence of ILC2 cells.
In mouse asthma models that use ovalbumin (Kim et al., 2012), house dust mite (HDM) (Wilhelm et al., 2011; Halim et al., 2012a; Klein Wolterink et al., 2012), papain (Halim et al., 2012a), and fungal allergens, ILC2s uniformly increase in number and are the major source of IL-5 or/and IL-13, especially in the early phases of the disease. In mouse models that lack adaptive immune cells, i.e., T and B cells, allergens can also induce significant AHR, a high level of type 2 cytokines, and increased numbers of ILC2s in the lung (Bartemes et al., 2012; Halim et al., 2012a). In these allergen-induced asthma models, the pulmonary epithelium, macrophages, and DCs release IL-33; in addition, the pulmonary epithelium also produces IL-25, TSLP, and transforming growth factor-β (TGF-β). All of these cytokines can activate ILC2s (Halim et al., 2012a; Kim et al., 2012; Iijima et al., 2014; Denney et al., 2015). In vitro, IL-25 and IL-33 enhance the proliferation of ILC2s and stimulate them to produce IL-4, IL-5, IL-13, and/or IL-9 (Wilhelm et al., 2011; Barlow et al., 2012; Bartemes et al., 2012; Halim et al., 2012a; Kim et al., 2012; Klein Wolterink et al., 2012). This is important because, IL-4 stimulates B cells to produce IgE, IL-5 recruits and activates eosinophils and also enhances B cell antibody production and B1 cell self-renewal, and IL-13 enhances smooth muscle contraction, epithelial mucous production, airway remodeling, and eosinophil recruitment. IL-9 plays an essential role in mast cell differentiation and can also induce mast cells to secrete IL-6; IL-9 promotes both the proliferation of eosinophils and their migration into the lung (Renauld, 2001). Otherwise, the IL-9 production of ILC2s depends on IL-2 secreted by adaptive immune cells. IL-9 also stimulates ILC2s to produce IL-13 and IL-5 (Wilhelm et al., 2011). All of these cytokines play an important role in the development of AHR (Fig. 3).
During AHR development, ILC2s crosstalk with DCs, CD4+ Th2 cells, B cells, and Th9 cells, thereby potentiating the pathology. ILC2s produce IL-13, which directly activates DCs to express CCL17, enhancing CD4+ Th2 cell activation; ILC2s also activate Th2 cells directly by producing IL-4 and OX40L. ILC2s and CD4+ Th2 cells thereby exert a synergistic effect on the development of AHR (Drake et al., 2014; Gold et al., 2014; Halim et al., 2014; Halim et al., 2015; Liu et al., 2015a). Mouse lung ILC2s enhance the proliferation of B1- and B2-type B cells and stimulate their production of IgM, IgG1, IgA, and IgE in vitro. Specifically, ILC2-derived IL-5 is critically involved in increased IgM production (Drake et al., 2016). Polarized ILC2s and Th9 cells also stimulate each other in mouse models of asthma in the lung (Ying et al., 2016).
Researchers have recently observed that similar to Th2 cells, ILC2s gain memory-like properties upon allergen challenge. ILC2s stimulated by inhalation of either IL-33 or papain persist for a long time; even after resolution of the inflammatory response; in fact, some ILC2s persist for more than 4 weeks. Furthermore, ‘allergen-experienced’ ILC2s responded better to unrelated allergen than naïve ILC2s, mediating a more severe allergic inflammation (Martinez-Gonzalez et al., 2016). Allergen-experienced ILC2s are also more responsive and produce higher amounts of the same cytokines than unexperienced ILC2s. Compared with memory lymphocytes, ILC2s are activated by cytokines, while memory lymphocytes are activated by specific antigens, thus the memory-like ILC2s are antigen non-specific. This means that memory-like ILC2s can be activated by unrelated allergens but produce a stronger response than naïve ILC2s. Memory-like ILC2s can also enhance Th2 cell-mediated adaptive type 2 lung inflammation. These two important characteristics may explain the phenomena that some asthma patients react against multiple allergens, while some do not (Martinez-Gonzalez et al., 2016).
In comparison with mouse, far fewer studies describing the role of ILC2s in human asthma have been published. The occurrence of ILC2s is more frequent in the blood of subjects with allergic asthma than in healthy individuals and allergic donors. When stimulated with IL-25 or IL-33, peripheral blood mononuclear cells from patients with allergic asthma produced significantly greater amounts of IL-5 and IL-13 than those from patients with allergic rhinitis and those from healthy donors (Bartemes et al., 2014; Jia et al., 2016). Recently, ILC2s were found in the bronchoalvoelar lavage (BAL) and sputum of patients with asthma, and the proportion of IL-5+IL-13+ ILC2s in the sputum of patients with severe asthma was higher than that in corresponding samples from patients with mild asthma (Smith et al., 2016). ILC2 levels are also increased in the sputum from severe asthmatic children (Nagakumar et al., 2016). Other researchers have also compared the relationship between ILC2s and asthma control status, and the results indicate a positive correlation between IL-13-producing ILC2s and asthma control status (Jia et al., 2016).
Based on the above studies of examining ILC2s in human asthma, the number of ILC2s is increased in asthmatic patients’ blood, these ILC2s are activated, and the number of IL-13-producing ILC2s negatively correlates with asthma control status. These results might suggest that level of ILC2s in the blood maybe partially indicate asthma status. However, ILCs are generally tissue-resident cells and the characteristics of ILCs in tissue are very different from ILCs in the circulation since tissue-resident ILCs are affected by the tissue microenvironment, which can cause changes of biomarker profile as well as function. Therefore, the blood ILC profile and function does not absolutely represent ILC2s resident in the lungs of asthmatics; blood ILC2s therefore only partially indicates the status of the disease. In the future, it will be important to study the function of human tissue-resident ILC2s, for example ILC2s from BAL and sputum, and even from lung tissue itself.
ILC2s and pulmonary fibrosis
Pulmonary fibrosis is a heterogeneous disease that is prevalent worldwide, and that is typically regarded as a chronic progressive disease, with high morbidity and mortality (Hutchinson et al., 2015). The key pathogenic mechanism involves extracellular matrix deposition in the lung. The pro-inflammatory cytokines TGF-β, IL-13, IL-1β, and IL-17A all play an important role in the fibrotic process (Kolb et al., 2001; Wynn, 2011).
In the mouse model of pulmonary fibrosis that uses injection of S. mansoni eggs, IL-25 has been shown to be the key cytokine in the development of fibrosis. IL-25 induces a dramatic increase in both IL-13 and TGF-β in the lungs. In humans, increased levels of IL-25 and ILC2s are found in the BAL and lung tissue of patients with idiopathic pulmonary fibrosis (Hams et al., 2014). Compared with wild-type mice, pulmonary collagen deposition is impaired in ILC2-deficient mice after S. mansoni egg injection (Hams et al., 2014). Furthermore, ILC2s induce pulmonary collagen deposition in an IL-13-dependent manner (Hams et al., 2014). Li et al. (2014) have suggested that the IL-33-ST2 axis is essential for the initiation and progression of pulmonary fibrosis. In this model IL-33 activates M2 macrophages to produce IL-13 and TGF-β1, and then further induces the expansion of ILC2s to produce IL-13, ultimately resulting in the development of pulmonary fibrosis (Li et al., 2014).
Dermal and circulating ILC2 counts correlate closely with the occurrence of pulmonary fibrosis in systemic sclerosis patients. This implies that ILC2s may aggravate the pulmonary fibrosis in these patients (Wohlfahrt et al., 2016) (Fig. 3).
ILC2s and eosinophilic pleural effusion (EPE) in primary spontaneous pneumothorax (PSP)
EPE is defined as > 10% eosinophilia in the pleural fluid and is frequently associated with the presence of blood and/or air in the pleural space (Kalomenidis and Light, 2003). PSP is a common complication, with a high rate of recurrence in EPE (Kalomenidis and Light, 2003).
The levels of IL-4, IL-5, IL-13, and Eotaxin-3, as well as TSLP and IL-33, have been shown to be increased in the pleural fluid of PSP patients. These cytokines are type 2 immune response-related, and it therefore appears that Th2 cells and ILC2s might play an essential role in this pathology; however, CD4+ and CD8+ T cells are known not to be involved in the pathogenesis of PSP (Kwon et al., 2013). IL-33 directly stimulates ILC2s to produce increased amounts of IL-5, which then recruits eosinophils into the pleural space, resulting in EPE; Th2 cells are not involved in this process. This indicates that the type 2 immune response is associated with the development of EPE in PSP through an ILC2-dependent but Th2-independent manner (Kwon et al., 2013).
Identification and characterization of ILC3s
ILC3s can be classified into two main groups according to the host developmental stage when they mature: fetal LTi cells and post-natal (adult) ILC3s. Fetal LTi cells, the prototypical ILC3s, were originally reported two decades ago. In mice, LTi cells are identified as CD127+CD3−CD4+. Their development is RORγt-dependent, they originate in the fetal liver, and they are found in fetal lymphoid nodes and the intestine (Kelly and Scollay, 1992; Mebius et al., 1997; Yoshida et al., 1999; Eberl et al., 2004; Finke, 2005). In humans, fetal LTi cells were first found in 2009 and recognized as Lin−RORγt+CD127+CD4−; they play a role similar to that of mouse LTi cells (Cupedo et al., 2009).
In mice, adult ILC3s are defined as CD45+Lin−Thy1+RORγt+, and they partially express CCR6 and NKp46 (Sanos et al., 2009; Takatori et al., 2009; Vonarbourg et al., 2010b; Song et al., 2015). RORγt, AHR, GATA3, and T-bet are required for their development (Luci et al., 2009; Klose et al., 2013; Hughes et al., 2014; Serafini et al., 2014). In humans, adult ILC3s are defined as Lin−CD127+CRTH2−CD117+; they also heterogeneously express NKp44, NKp46, CD56, and NKp30 (Crellin et al., 2010; Hoorweg et al., 2012; Glatzer et al., 2013). Upon stimulation with IL-1β and IL-23, ILC3s express IL-17, IL-22, and granulocyte-macrophage colony-stimulating factor (GM-CSF) (Takatori et al., 2009; Crellin et al., 2010; Hoorweg et al., 2012; Song et al., 2015).
Adult ILC3s are the most heterogeneous ILCs. Depending on the level of the expressed NK receptor, e.g., NKp44, NKp46, or NKp30, ILC3s can be divided into NCR−ILC3s and NCR+ILC3s. ILC3s can also be divided according to the level of expression of CCR6 into CCR6− ILC3s and CCR6+ ILC3s. Recently, it was shown that NCR engagement enables ILC3s to play a pro-inflammatory role (Glatzer et al., 2013); based on this an increasing numbers of researchers use the NCR status to classify the function of adult ILC3s. Similar markers and functions are found in both mouse and human ILC3s with the exception that mice do not express NKp44 (Killig et al., 2014).
In the mouse lung, ILC3s were initially reported by Centre et al. (2015), who found that 30% of ILCs were ILC3s, defined as Lin−CD90+CD127+RORγt+; almost 70% of ILC3s also co-express CCR6. IL-1β and IL-23 have been shown to activate these cells. ILC3s were also identified as a major source of IL-22 produced in response to IL-23 stimulation (Van Maele et al., 2014). In the human lung, ILC3s are identified as Lin−CD127+CRTH2−CD117+, and are NCR− or NCR+. NCR+ILC3s produce IL-22, TNF-α, IL-8, IL-2, and GM-CSF upon stimulation (Carrega et al., 2015; De Grove et al., 2016) (Table 2).
ILC3s in the lung
ILC3s and infection
ILC3s play an essential role in the maintenance of mucosal barrier function because they produce effector cytokines, especially IL-22 and IL-17. IL-22 and IL-17 activate epidermal cells to produce antimicrobial molecules and protect the host from extracellular bacteria and fungi (Liang et al., 2006). IL-22 can also enhance the epithelial production of mucus-associated molecules (McAleer and Kolls, 2014). A protective role of ILC3s against infections in the digestive system has also been reported. During an intestinal infection with Citrobacter rodentium, the number of IL-22-producing ILC3s increased in the intestinal lamina propria (Cella et al., 2009; Sanos et al., 2009). Gladiator et al. found that ILC3s can also produce IL-17 that helps eliminate pathogens during fungal infection in mice, especially in the early phase of infection. (Gladiator et al., 2013). In 2014, Van Maele et al. reported the function of ILC3s in the lung. During Streptococcus pneumoniae infection, ILC3s rapidly accumulate in the lung tissue to produce IL-22 in a DC- and MyD88-dependent manner (Van Maele et al., 2014). Boosting lung ILC3 numbers might therefore represent an interesting strategy for fighting respiratory bacterial infections.
One study suggested that Toll-like receptor 5 (TLR5) signaling could activate ILCs (CD3−CD127+) and enhance the production of IL-17 and IL-22, which are crucial for anti-pathogen defenses in the mucous membrane of the intestine and the lung (Van Maele et al., 2010). This raises the interesting notion that TLR5 may activate ILC3s to reject the pathogen in the lung. Later, Van Maele et al. found that during S. pneumoniae infection, the TLR5 agonist flagellin accelerates and over-stimulates lung ILC3s to produce more IL-22 (Van Maele et al., 2014).
Klebsiella pneumoniae is a gram-negative bacterium that is highly resistant to antibiotics and is a common pathogen in pneumonia (Doorduijn et al., 2016). In 2014, Xu et al. found that IL-22-producing NK cells are required for optimal host defense in mouse models of K. pneumoniae infection (Xu et al., 2014). Recently, another group reported that following infection with K. pneumoniae in mice, inflammatory monocytes are immediately recruited to the lungs, where they produce TNF, which then increases the number of IL-17-producing ILC3s. IL-17A-dependent clearance of K. pneumoniae is impaired in monocyte- or TNF-depleted mouse models, whereas IL-17-producing ILC3s enhance monocyte-mediated bacterial clearance. These results indicate that ILC3s and monocytes participate in a positive feedback cycle that promotes the clearance of highly antibiotic-resistant bacterial pathogens from the lung (Xiong et al., 2016) (Fig. 4).

ILC3 functions in the lung. After receiving the danger signals from the airway epithelium upon pathogen and tumor cell invasion, DCs and macrophages release IL-1β and IL-23, which activate ILC3s. Activated ILC3s release IL-17, IL-22, IL-8, IL-2, TNF, and LTα1β2. IL-17 and IL-22 play a dual role: on the one hand, they stimulate the epithelium to produce antimicrobial peptides and proteins, and mucus-associated molecules to clear the pathogen and tumor cells to maintain homeostasis of the mucosa; on the other hand, they may aggravate the development of obesity-related asthma and COPD. IL-22-producing-ILC3s may have anti-asthma effects, whereas IL-17-producing ILC3s may participate in the pathology of asthma, especially in obesity-related asthma. IL-2 and IL-8 recruit neutrophils to the lung. LTα1β2 stimulates mesenchymal stem cells (MSCs) to express ICAM-1 and VCAM-1. These molecules participate in the formation of a tertiary lymphoid organ in the tumor. See text for details
iILC2-derived ILC3-like cells also play a role in Candida albicans infections. iILC2s express an intermediate amount of RORγt, i.e., one that is significantly different from nILC2s but lower than that in ILC3 cells. A small proportion of freshly isolated iILC2s produces IL-17 upon stimulation with PMA and ionomycin (Huang et al., 2015). When cultured with TGF-β and IL-6, iILC2s become ILC3-like, produce IL-17, and lose the ability to produce IL-13. During C. albicans infection, iILC2 cells aid in the clearance of this pathogen. In the lungs of mice infected with C. albicans, transferred iILC2 cells become ILC3-like cells after 5 d; these ILC3-like cells produce IL-17 but not IL-13 (Huang et al., 2015). Thus, iILC2 cells can transform into ILC3-like cells in vitro and in vivo, and gain the ability to protect the host against C. albicans (Huang et al., 2015) (Fig. 1).
ILC3s and asthma
In addition to ILC2s, ILC3s are also involved in asthma. In an OVA-induced asthma murine model, Taube et al. found that IL-22 expression increased, and the IL-22 was mainly produced by innate lymphoid cells in the lungs, rather than by TH cells. OVA challenged IL-22-deficient mice suffered from much higher AHR. In contrast mice treated with IL-22 before OVA challenge displayed significantly reduced allergic airway inflammation. Based on these data, IL-22-producing ILC3s may participate in reducing allergic asthma pathology (Taube et al., 2011).
Obesity is a risk factor associated with asthma, and obese asthma patients respond poorly to typical anti-asthma medications, including corticosteroids (Sutherland et al., 2009); therefore, a distinct immune mechanism must be at play in obese asthmatics. Mice fed a high-fat diet become obese and exhibit AHR through an IL-17A and NKRP3-dependent pathway, and this AHR also occurs in obese Rag1 −/− mice (Kim et al., 2014). In this model, the number of CCR6+ IL-17A-producing ILC3s is elevated in the lung and macrophage-derived IL-1 directly causes AHR by stimulating this IL-17-producing ILC3 population (Kim et al., 2014).
Everaere et al. found a similar result where, compared with lean mice, the number of ILCs was increased in the lung of obese mice, and this effect was accompanied by eosinophil infiltration. Following an HDM challenge, the counts of ILC2s and ILC3s in the lung further increased, as did IL-33 and IL-1β levels, whereas ILC markers in visceral adipose tissue decreased. In an obese mouse model with ILC depletion, the HDM-induced inflammatory profile of the airway was profoundly decreased, including reduced Th2 and Th17 infiltration (Everaere et al., 2016) (Fig. 4).
In humans, IL-17-producing ILCs are found in BAL fluid samples from asthma patients; their levels are increased in severe asthma patients compared with those in patients with mild asthma or control donors (Kim et al., 2014).
These results indicate that IL-22-producing-ILC3s may have an anti-asthma effect, whereas IL-17-producing ILC3s participate in the pathology of asthma, possibly providing a link between obesity and asthma. Further studies should be performed to help illuminate these potential connections.
ILC3s and COPD
IL-17A plays an essential role in the development of COPD. Since ILC3s have the ability to produce IL-17A it is therefore possible that ILC3s are linked with COPD. De Grove et al. (2016) found that in patients with COPD, the population of NCR−ILC3s comprises the largest subset of ILCs (De Grove et al., 2016). Bal et al. (2016) also found that NKp44−ILC3 levels were increased, whereas the numbers of ILC2s and NKp44+ ILC3s were significantly diminished in lung tissue from patients with severe COPD (Bal et al., 2016). This supports our hypothesis that IL-17-producing ILC3s may play a role in COPD; however, the authors suggested that the accumulation of NCR−ILC3s in COPD could be associated with the protective immunity of the host in response to bacterial respiratory tract infections that occur frequently in patients with COPD. The involvement of ILC3s in COPD therefore requires further study (Fig. 4).
ILC3s and tumors
A dual effect of ILC3s on tumor immunity was suggested for intestinal tumors. On the one hand, IL-22 produced by ILC3s maintains mucosal integrity and clearance of pathogens and transformed cells. On the other hand, IL-22 activates the STAT3 cascade to enhance tumor generation (Kirchberger et al., 2013). Carrega et al. (2015) observed that NCR+ILC3s were enriched in non-small cell lung cancer (NSCLC) and that the proportion of NCR+ILC3s was positively associated with tumor stage. NCR+ILC3s were present in significantly higher amounts in stage I/II NSCLC tumors than in more tumors from more advanced stages. When stimulated, NCR+ILC3s, which had been freshly isolated from NSCLC tissues, produced IL-22, TNF-α, IL-8, and IL-2, but did not secrete IL-17. IL-22 maintains the integrity of epithelial cells (Dudakov et al., 2015); TNF-α is a pro-inflammatory cytokine that exerts anti-tumor and anti-pathogen effects, and IL-8 and IL-2 enhance leukocyte recruitment and proliferation (Waugh and Wilson, 2008; Boyman and Sprent, 2012). Based on the characteristics of the cytokines produced by NCR+ILC3s, it was concluded that these cells might play a role in anti-tumor defenses (Carrega et al., 2015).
As stated above, LTi is the prototype for ILC3s. LTi cells produce lymphotoxin α, lymphotoxin β, and TNF-α. They also stimulate stromal cells to produce vascular cell adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule 1 (ICAM-1), which recruit the immune cells and form the fetal lymphoid node and Peyer’s patches (Kelly and Scollay, 1992; Mebius et al., 1997; Yoshida et al., 1999; Eberl et al., 2004; Finke, 2005; Cupedo et al., 2009). Lung cancer-derived NCR+ILC3s express more lymphotoxin mRNA than their tonsil counterparts. Furthermore, lung cancer-derived NCR+ILC3s elicit significant up-regulation of ICAM-1 and VCAM-1 in mesenchymal stem cells in a lymphotoxin α, lymphotoxin β, and TNF-α dependent manner. Additionally, NCR+ILC3s preferentially reside at the edge of lymphoid structures associated with NSCLC, and the percentage of NCR+ILC3s is correlated with the density of tertiary lymphoid structures in the tumor region. These observations suggest that NCR+ILC3s might play a role in the formation and maintenance of these structures as well as lymphoid aggregates in tumor tissue (Carrega et al., 2015). Otherwise, NCR−ILC3s can gain pro-inflammatory properties by engaging NKp44, as the number of NCR+ILC3s is increased in the lung tumor region (Fig. 4). Whether the conversion of NCR−ILC3 to NCR+ILC3 cells contributes to this increased number and how the tumor microenvironment influences this conversion are questions that should be answered in future studies. Additionally, the question of whether IL-22-producing ILC3s enhance or inhibit tumors in the lung remains unresolved and should be studied further.
Conclusions
ILCs are attracting increasing attention on account of their distinct tissue-resident properties. Although it is known that ILCs are involved in pulmonary infection, asthma, COPD, fibrosis, and tumors in the lung, in-depth studies are still only in their infancy. The mechanisms of ILC activation, proliferation, and regulation in the lung are not clear; how exactly the pathological environment affects ILC function and how ILCs respond to the environment also remain unknown. Elucidation of these mechanisms is therefore an urgent matter, especially in pulmonary diseases. Answers to these questions will hopefully provide new clues for the treatment of these serious human diseases.
Change history
06 April 2017
An erratum to this article has been published.
REFERENCES
Abt MC, Lewis BB, Caballero S, Xiong H, Carter RA, Susac B, Ling L, Leiner I, Pamer EG (2015) Innate Immune Defenses Mediated by Two ILC Subsets Are Critical for Protection against Acute Clostridium difficile Infection. Cell Host Microbe 18:27–37
Bal SM, Bernink JH, Nagasawa M, Groot J, Shikhagaie MM, Golebski K, van Drunen CM, Lutter R, Jonkers RE, Hombrink P et al (2016) IL-1beta, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat Immunol 17:636–645
Bando JK, Liang HE, Locksley RM (2015) Identification and distribution of developing innate lymphoid cells in the fetal mouse intestine. Nat Immunol 16:153–160
Barlow JL, Bellosi A, Hardman CS, Drynan LF, Wong SH, Cruickshank JP, McKenzie AN (2012) Innate IL-13-producing nuocytes arise during allergic lung inflammation and contribute to airways hyperreactivity. J Allergy Clin Immunol 129(191–198):e191–e194
Barnes PJ (2009) The cytokine network in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 41:631–638
Barnig C, Cernadas M, Dutile S, Liu X, Perrella MA, Kazani S, Wechsler ME, Israel E, Levy BD (2013) Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci Transl Med 5:174ra126
Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H (2012) IL-33-responsive lineage- CD25 + CD44(hi) lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol 188:1503–1513
Bartemes KR, Kephart GM, Fox SJ, Kita H (2014) Enhanced innate type 2 immune response in peripheral blood from patients with asthma. J Allergy Clin Immunol 134(671–678):e674
Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, Hreggvidsdottir HS, Heinsbroek SE, Legrand N, Buskens CJ et al (2013) Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol 14:221–229
Bernink JH, Krabbendam L, Germar K, de Jong E, Gronke K, Kofoed-Nielsen M, Munneke JM, Hazenberg MD, Villaudy J, Buskens CJ et al (2015) Interleukin-12 and -23 CONTROL PLASTICITY of CD127(+) Group 1 and Group 3 Innate lymphoid cells in the intestinal lamina propria. Immunity 43:146–160
Boyman O, Sprent J (2012) The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol 12:180–190
Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, Powrie F (2010) Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464:1371–1375
Carrega P, Loiacono F, Di Carlo E, Scaramuccia A, Mora M, Conte R, Benelli R, Spaggiari GM, Cantoni C, Campana S et al (2015) NCR(+)ILC3 concentrate in human lung cancer and associate with intratumoral lymphoid structures. Nat Commun 6:8280
Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, Doherty JM, Mills JC, Colonna M (2009) A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 457:722–725
Cella M, Otero K, Colonna M (2010) Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc Natl Acad Sci USA 107:10961–10966
Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, Dekruyff RH, Umetsu DT (2011) Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol 12:631–638
Crellin NK, Trifari S, Kaplan CD, Cupedo T, Spits H (2010) Human NKp44+IL-22+ cells and LTi-like cells constitute a stable RORC+ lineage distinct from conventional natural killer cells. J Exp Med 207:281–290
Cupedo T, Crellin NK, Papazian N, Rombouts EJ, Weijer K, Grogan JL, Fibbe WE, Cornelissen JJ, Spits H (2009) Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat Immunol 10:66–74
Dadi S, Chhangawala S, Whitlock BM, Franklin RA, Luo CT, Oh SA, Toure A, Pritykin Y, Huse M, Leslie CS et al (2016) Cancer immunosurveillance by tissue-resident innate lymphoid cells and innate-like T cells. Cell 164:365–377
De Grove KC, Provoost S, Verhamme FM, Bracke KR, Joos GF, Maes T, Brusselle GG (2016) Characterization and quantification of innate lymphoid cell subsets in human lung. PLoS ONE 11:e0145961
Deckers J, Branco Madeira F, Hammad H (2013) Innate immune cells in asthma. Trends Immunol 34:540–547
Denney L, Byrne AJ, Shea TJ, Buckley JS, Pease JE, Herledan GM, Walker SA, Gregory LG, Lloyd CM (2015) Pulmonary epithelial cell-derived cytokine TGF-beta1 is a critical cofactor for enhanced innate lymphoid cell function. Immunity 43:945–958
Doherty TA, Khorram N, Lund S, Mehta AK, Croft M, Broide DH (2013) Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates TH2 cytokine production. J Allergy Clin Immunol 132:205–213
Dolinay T, Kaminski N, Felgendreher M, Kim HP, Reynolds P, Watkins SC, Karp D, Uhlig S, Choi AM (2006) Gene expression profiling of target genes in ventilator-induced lung injury. Physiol Genom 26:68–75
Doorduijn DJ, Rooijakkers SH, van Schaik W, Bardoel BW (2016) Complement resistance mechanisms of Klebsiella pneumoniae. Immunobiology 221:1102–1109
Drake LY, Kita H (2014) Group 2 innate lymphoid cells in the lung. Adv Immunol 124:1–16
Drake LY, Iijima K, Kita H (2014) Group 2 innate lymphoid cells and CD4+ T cells cooperate to mediate type 2 immune response in mice. Allergy 69:1300–1307
Drake LY, Iijima K, Bartemes K, Kita H (2016) Group 2 innate lymphoid cells promote an early antibody response to a respiratory antigen in mice. J Immunol 197:1335–1342
Dudakov JA, Hanash AM, van den Brink MR (2015) Interleukin-22: immunobiology and pathology. Annu Rev Immunol 33:747–785
Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, Littman DR (2004) An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol 5:64–73
Eberl G, Colonna M, Di Santo JP, McKenzie AN (2015) Innate lymphoid cells: a new paradigm in immunology. Science 348:aaa6566
Enomoto Y, Orihara K, Takamasu T, Matsuda A, Gon Y, Saito H, Ra C, Okayama Y (2009) Tissue remodeling induced by hypersecreted epidermal growth factor and amphiregulin in the airway after an acute asthma attack. J Allergy Clin Immunol 124(913–920):e911–e917
Everaere L, Ait-Yahia S, Molendi-Coste O, Vorng H, Quemener S, LeVu P, Fleury S, Bouchaert E, Fan Y, Duez C et al (2016) Innate lymphoid cells contribute to allergic airway disease exacerbation by obesity. J Allergy Clin Immunol 138(5):1309–1318
Fan X, Rudensky AY (2016) Hallmarks of tissue-resident lymphocytes. Cell 164:1198–1211
Finke D (2005) Fate and function of lymphoid tissue inducer cells. Curr Opin Immunol 17:144–150
Fort MM, Cheung J, Yen D, Li J, Zurawski SM, Lo S, Menon S, Clifford T, Hunte B, Lesley R et al (2001) IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity 15:985–995
Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, Cella M, Colonna M (2013) Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity 38:769–781
Fukumoto J, Harada C, Kawaguchi T, Suetsugu S, Maeyama T, Inoshima I, Hamada N, Kuwano K, Nakanishi Y (2010) Amphiregulin attenuates bleomycin-induced pneumopathy in mice. Am J Physiol Lung Cell Mol Physiol 298:L131–L138
Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY (2015) Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 350:981–985
Gentek R, Munneke JM, Helbig C, Blom B, Hazenberg MD, Spits H, Amsen D (2013) Modulation of signal strength switches notch from an inducer of T cells to an inducer of ILC2. Front Immunol 4:334
Gladiator A, Wangler N, Trautwein-Weidner K, LeibundGut-Landmann S (2013) Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection. J Immunol 190:521–525
Glatzer T, Killig M, Meisig J, Ommert I, Luetke-Eversloh M, Babic M, Paclik D, Bluthgen N, Seidl R, Seifarth C et al (2013) RORgammat(+) innate lymphoid cells acquire a proinflammatory program upon engagement of the activating receptor NKp44. Immunity 38:1223–1235
Gold MJ, Antignano F, Halim TY, Hirota JA, Blanchet MR, Zaph C, Takei F, McNagny KM (2014) Group 2 innate lymphoid cells facilitate sensitization to local, but not systemic, TH2-inducing allergen exposures. J Allergy Clin Immunol 133:1142–1148
Grainger JR, Smith KA, Hewitson JP, Mcsorley HJ, Harcus Y, Filbey KJ, Finney CA, Greenwood EJ, Knox DP, Wilson MS (2010) Helminth secretions induce de novo T cell Foxp3 expression and regulatory function through the TGF-β pathway. J Exp Med 207:2331–2341
Halim TY, Krauss RH, Sun AC, Takei F (2012a) Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity 36:451–463
Halim TY, MacLaren A, Romanish MT, Gold MJ, McNagny KM, Takei F (2012b) Retinoic-acid-receptor-related orphan nuclear receptor alpha is required for natural helper cell development and allergic inflammation. Immunity 37:463–474
Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, McKenzie AN, Takei F (2014) Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 40:425–435
Halim TY, Hwang YY, Scanlon ST, Zaghouani H, Garbi N, Fallon PG, McKenzie AN (2015) Group 2 innate lymphoid cells license dendritic cells to potentiate memory T2 cell responses. Nat Immunol 17:57–64
Hams E, Armstrong ME, Barlow JL, Saunders SP, Schwartz C, Cooke G, Fahy RJ, Crotty TB, Hirani N, Flynn RJ et al (2014) IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc Natl Acad Sci USA 111:367–372
Hansen G, Berry G, DeKruyff RH, Umetsu DT (1999) Allergen-specific Th1 cells fail to counterbalance Th2 cell-induced airway hyperreactivity but cause severe airway inflammation. J Clin Invest 103:175–183
Held W, Kijima M, Angelov G, Bessoles S (2011) The function of natural killer cells: education, reminders and some good memories. Curr Opin Immunol 23:228–233
Hesslein DG, Lanier LL (2011) Transcriptional control of natural killer cell development and function. Adv Immunol 109:45–85
Holt PG, Strickland DH, Wikstrom ME, Jahnsen FL (2008) Regulation of immunological homeostasis in the respiratory tract. Nat Rev Immunol 8:142–152
Hong JY, Bentley JK, Chung Y, Lei J, Steenrod JM, Chen Q, Sajjan US, Hershenson MB (2014) Neonatal rhinovirus induces mucous metaplasia and airways hyperresponsiveness through IL-25 and type 2 innate lymphoid cells. J Allergy Clin Immunol 134:429–439
Hoorweg K, Peters CP, Cornelissen F, Aparicio-Domingo P, Papazian N, Kazemier G, Mjosberg JM, Spits H, Cupedo T (2012) Functional differences between human NKp44(-) and NKp44(+) RORC(+) innate lymphoid cells. Front Immunol 3:72
Huang Y, Guo L, Qiu J, Chen X, Hu-Li J, Siebenlist U, Williamson PR, Urban JF Jr, Paul WE (2015) IL-25-responsive, lineage-negative KLRG1(hi) cells are multipotential ‘inflammatory’ type 2 innate lymphoid cells. Nat Immunol 16:161–169
Hughes T, Briercheck EL, Freud AG, Trotta R, McClory S, Scoville SD, Keller K, Deng Y, Cole J, Harrison N et al (2014) The transcription Factor AHR prevents the differentiation of a stage 3 innate lymphoid cell subset to natural killer cells. Cell Rep 8:150–162
Hutchinson J, Fogarty A, Hubbard R, McKeever T (2015) Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 46:795–806
Iijima K, Kobayashi T, Hara K, Kephart GM, Ziegler SF, McKenzie AN, Kita H (2014) IL-33 and thymic stromal lymphopoietin mediate immune pathology in response to chronic airborne allergen exposure. J Immunol 193:1549–1559
Jia Y, Fang X, Zhu X, Bai C, Zhu L, Jin M, Wang X, Hu M, Tang R, Chen Z (2016) IL-13+ Type 2 innate lymphoid cells correlate with asthma control status and treatment response. Am J Respir Cell Mol Biol 55:675–683
Kalomenidis I, Light RW (2003) Eosinophilic pleural effusions. Curr Opin Pulm Med 9:254–260
Karta MR, Broide DH, Doherty TA (2016) Insights into Group 2 innate lymphoid cells in human airway disease. Curr Allergy Asthma Rep 16:8
Kearley J, Silver JS, Sanden C, Liu Z, Berlin AA, White N, Mori M, Pham TH, Ward CK, Criner GJ et al (2015) Cigarette smoke silences innate lymphoid cell function and facilitates an exacerbated type I interleukin-33-dependent response to infection. Immunity 42:566–579
Kelly KA, Scollay R (1992) Seeding of neonatal lymph nodes by T cells and identification of a novel population of CD3-CD4+ cells. Eur J Immunol 22:329–334
Killig M, Glatzer T, Romagnani C (2014) Recognition strategies of group 3 innate lymphoid cells. Front Immunol 5:142
Kim HY, Chang YJ, Subramanian S, Lee HH, Albacker LA, Matangkasombut P, Savage PB, McKenzie AN, Smith DE, Rottman JB et al (2012) Innate lymphoid cells responding to IL-33 mediate airway hyperreactivity independently of adaptive immunity. J Allergy Clin Immunol 129(216–227):e211–e216
Kim BS, Siracusa MC, Saenz SA, Noti M, Monticelli LA, Sonnenberg GF, Hepworth MR, Van Voorhees AS, Comeau MR, Artis D (2013) TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med 5:170ra116
Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, Iwakura Y, Israel E, Bolger K, Faul J et al (2014) Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med 20:54–61
Kim J, Kwon J, Kim M, Do J, Lee D, Han H (2016) IL-1 is a critical regulator of group 2 innate lymphoid cell function and plasticity. Polym J 17:646–655
Kirchberger S, Royston DJ, Boulard O, Thornton E, Franchini F, Szabady RL, Harrison O, Powrie F (2013) Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med 210:917–931
Klein Wolterink RG, Kleinjan A, van Nimwegen M, Bergen I, de Bruijn M, Levani Y, Hendriks RW (2012) Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in murine models of allergic asthma. Eur J Immunol 42:1106–1116
KleinJan A, Klein Wolterink RG, Levani Y, de Bruijn MJ, Hoogsteden HC, van Nimwegen M, Hendriks RW (2014) Enforced expression of Gata3 in T cells and group 2 innate lymphoid cells increases susceptibility to allergic airway inflammation in mice. J Immunol 192:1385–1394
Klose CS, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d’Hargues Y, Goppert N, Croxford AL, Waisman A, Tanriver Y et al (2013) A T-bet gradient controls the fate and function of CCR6-RORgammat+innate lymphoid cells. Nature 494:261–265
Klose CS, Flach M, Mohle L, Rogell L, Hoyler T, Ebert K, Fabiunke C, Pfeifer D, Sexl V, Fonseca-Pereira D et al (2014) Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 157:340–356
Kolb M, Margetts PJ, Anthony DC, Pitossi F, Gauldie J (2001) Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J Clin Invest 107:1529–1536
Kopf M, Schneider C, Nobs SP (2015) The development and function of lung-resident macrophages and dendritic cells. Nat Immunol 16:36–44
Kwon BI, Hong S, Shin K, Choi EH, Hwang JJ, Lee SH (2013) Innate type 2 immunity is associated with eosinophilic pleural effusion in primary spontaneous pneumothorax. Am J Respir Crit Care Med 188:577–585
Lai DM, Shu Q, Fan J (2016) The origin and role of innate lymphoid cells in the lung. Mil Med Res 3:25
Li D, Guabiraba R, Besnard AG, Komai-Koma M, Jabir MS, Zhang L, Graham GJ, Kurowska-Stolarska M, Liew FY, McSharry C et al (2014) IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol 134(1422–1432):e1411
Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA (2006) Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 203:2271–2279
Licona-Limon P, Kim LK, Palm NW, Flavell RA (2013) TH2, allergy and group 2 innate lymphoid cells. Nat Immunol 14:536–542
Lim AI, Menegatti S, Bustamante J, Le Bourhis L, Allez M, Rogge L, Casanova JL, Yssel H, Di Santo JP (2016) IL-12 drives functional plasticity of human group 2 innate lymphoid cells. J Exp Med 213:569–583
Liu B, Lee JB, Chen CY, Hershey GK, Wang YH (2015a) Collaborative interactions between type 2 innate lymphoid cells and antigen-specific CD4+ Th2 cells exacerbate murine allergic airway diseases with prominent eosinophilia. J Immunol 194:3583–3593
Liu J, Wu J, Qi F, Zeng S, Xu L, Hu H, Wang D, Liu B (2015b) Natural helper cells contribute to pulmonary eosinophilia by producing IL-13 via IL-33/ST2 pathway in a murine model of respiratory syncytial virus infection. Int Immunopharmacol 28:337–343
Luci C, Reynders A, Ivanov II, Cognet C, Chiche L, Chasson L, Hardwigsen J, Anguiano E, Banchereau J, Chaussabel D et al (2009) Influence of the transcription factor RORgammat on the development of NKp46+ cell populations in gut and skin. Nat Immunol 10:75–82
Maizels RM, Hewitson JP, Smith KA (2012) Susceptibility and immunity to helminth parasites. Curr Opin Immunol 24:459–466
Martinez-Gonzalez I, Matha L, Steer CA, Ghaedi M, Poon GF, Takei F (2016) Allergen-experienced Group 2 innate lymphoid cells acquire memory-like properties and enhance allergic lung inflammation. Immunity 45:198–208
Massacand JC, Stettler RC, Meier R, Humphreys NE, Grencis RK, Marsland BJ, Harris NL (2009) Helminth products bypass the need for TSLP in Th2 immune responses by directly modulating dendritic cell function. Proc Natl Acad Sci USA 106:13968–13973
McAleer JP, Kolls JK (2014) Directing traffic: IL-17 and IL-22 coordinate pulmonary immune defense. Immunol Rev 260:129–144
McHedlidze T, Kindermann M, Neves AT, Voehringer D, Neurath MF, Wirtz S (2016) IL-27 suppresses type 2 immune responses in vivo via direct effects on group 2 innate lymphoid cells. Mucosal Immunol 3:1384–1394
McSorley HJ, Maizels RM (2012) Helminth infections and host immune regulation. Clin Microbiol Rev 25:585–608
McSorley HJ, Blair NF, Smith KA, McKenzie AN, Maizels RM (2014) Blockade of IL-33 release and suppression of type 2 innate lymphoid cell responses by helminth secreted products in airway allergy. Mucosal Immunol 7:1068–1078
McSorley HJ, Blair NF, Robertson E, Maizels RM (2015) Suppression of OVA-alum induced allergy by Heligmosomoides polygyrus products is MyD88-, TRIF-, regulatory T- and B cell-independent, but is associated with reduced innate lymphoid cell activation. Exp Parasitol 158:8–17
Mebius RE, Rennert P, Weissman IL (1997) Developing lymph nodes collect CD4+CD3- LTbeta+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity 7:493–504
Meylan F, Hawley ET, Barron L, Barlow JL, Penumetcha P, Pelletier M, Sciume G, Richard AC, Hayes ET, Gomez-Rodriguez J et al (2014) The TNF-family cytokine TL1A promotes allergic immunopathology through group 2 innate lymphoid cells. Mucosal Immunol 7:958–968
Mjosberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, Fokkens WJ, Cupedo T, Spits H (2011) Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol 12:1055–1062
Mjosberg J, Bernink J, Golebski K, Karrich JJ, Peters CP, Blom B, te Velde AA, Fokkens WJ, van Drunen CM, Spits H (2012) The transcription factor GATA3 is essential for the function of human type 2 innate lymphoid cells. Immunity 37:649–659
Mohapatra A, Van Dyken SJ, Schneider C, Nussbaum JC, Liang HE, Locksley RM (2016) Group 2 innate lymphoid cells utilize the IRF4-IL-9 module to coordinate epithelial cell maintenance of lung homeostasis. Mucosal Immunol 9:275–286
Montaldo E, Teixeira-Alves LG, Glatzer T, Durek P, Stervbo U, Hamann W, Babic M, Paclik D, Stölzel K, Gröne J (2014) Human RORγt(+)CD34(+) cells are lineage-specified progenitors of group 3 RORγt(+) innate lymphoid cells. Immunity 41:988–1000
Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, Angelosanto JM, Laidlaw BJ, Yang CY, Sathaliyawala T et al (2011) Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol 12:1045–1054
Monticelli LA, Buck MD, Flamar AL, Saenz SA, Tait Wojno ED, Yudanin NA, Osborne LC, Hepworth MR, Tran SV, Rodewald HR et al (2016) Arginase 1 is an innate lymphoid-cell-intrinsic metabolic checkpoint controlling type 2 inflammation. Nat Immunol 17:656–665
Moro K (2010) Innate production of TH2 cytokines by adipose tissue-associated c-kit+Sca-1+ lymphoid cells. Nature 463:540–544
Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, Furusawa J, Ohtani M, Fujii H, Koyasu S (2010) Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463:540–544
Moro K, Kabata H, Tanabe M, Koga S, Takeno N, Mochizuki M, Fukunaga K, Asano K, Betsuyaku T, Koyasu S (2016) Interferon and IL-27 antagonize the function of group 2 innate lymphoid cells and type 2 innate immune responses. Nat Immunol 17:76–86
Nagakumar P, Denney L, Fleming L, Bush A, Lloyd CM, Saglani S (2016) Type 2 innate lymphoid cells in induced sputum from children with severe asthma. J Allergy Clin Immunol 137(624–626):e626
Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, Bucks C, Kane CM, Fallon PG, Pannell R et al (2010) Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464:1367–1370
Paul WE, Zhu J (2010) How are T(H)2-type immune responses initiated and amplified? Nat Rev Immunol 10:225–235
Philip NH, Artis D (2013) New friendships and old feuds: relationships between innate lymphoid cells and microbial communities. Immunol Cell Biol 91:225–231
Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, Locksley RM (2010) Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci USA 107:11489–11494
Rankin LC, Groom JR, Chopin M, Herold MJ, Walker JA, Mielke LA, McKenzie AN, Carotta S, Nutt SL, Belz GT (2013) The transcription factor T-bet is essential for the development of NKp46+ innate lymphocytes via the Notch pathway. Nat Immunol 14:389–395
Rankin LC, Girard-Madoux MJ, Seillet C, Mielke LA, Kerdiles Y, Fenis A, Wieduwild E, Putoczki T, Mondot S, Lantz O et al (2016) Complementarity and redundancy of IL-22-producing innate lymphoid cells. Nat Immunol 17:179–186
Renauld JC (2001) New insights into the role of cytokines in asthma. J Clin Pathol 54:577–589
Salimi M, Ogg G (2014) Innate lymphoid cells and the skin. BMC Dermatol 14:18
Sanos SL, Bui VL, Mortha A, Oberle K, Heners C, Johner C, Diefenbach A (2009) RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat Immunol 10:83–91
Sawa S, Cherrier M, Lochner M, Satohtakayama N, Fehling HJ, Langa F, Santo JPD, Eberl G (2010) Lineage relationship analysis of RORγt+ innate lymphoid cells. Science 330:665–669
Scanlon ST, McKenzie AN (2012) Type 2 innate lymphoid cells: new players in asthma and allergy. Curr Opin Immunol 24:707–712
Serafini N, Klein Wolterink RG, Satoh-Takayama N, Xu W, Vosshenrich CA, Hendriks RW, Di Santo JP (2014) Gata3 drives development of RORgammat+ group 3 innate lymphoid cells. J Exp Med 211:199–208
Silver JS, Kearley J, Copenhaver AM, Sanden C, Mori M, Yu L, Pritchard GH, Berlin AA, Hunter CA, Bowler R et al (2016a) Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat Immunol 17:626–635
Silver JS, Kearley J, Copenhaver AM, Sanden C, Mori M, Yu L, Pritchard GH, Berlin AA, Hunter CA, Bowler R et al (2016b) Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat Immunol 17:626–635
Smith SG, Chen R, Kjarsgaard M, Huang C, Oliveria JP, O’Byrne PM, Gauvreau GM, Boulet LP, Lemiere C, Martin J et al (2016) Increased numbers of activated group 2 innate lymphoid cells in the airways of patients with severe asthma and persistent airway eosinophilia. J Allergy Clin Immunol 137(75–86):e78
Song C, Lee JS, Gilfillan S, Robinette ML, Newberry RD, Stappenbeck TS, Mack M, Cella M, Colonna M (2015) Unique and redundant functions of NKp46 + ILC3s in models of intestinal inflammation. J Exp Med 212:1869–1882
Spits H, Cupedo T (2012) Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu Rev Immunol 30:647–675
Spits H, Di Santo JP (2011) The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol 12:21–27
Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE et al (2013) Innate lymphoid cells–a proposal for uniform nomenclature. Nat Rev Immunol 13:145–149
Spooner CJ, Lesch J, Yan D, Khan AA, Abbas A, Ramirez-Carrozzi V, Zhou M, Soriano R, Eastham-Anderson J, Diehl L et al (2013) Specification of type 2 innate lymphocytes by the transcriptional determinant Gfi1. Nat Immunol 14:1229–1236
Stier MT, Bloodworth MH, Toki S, Newcomb DC, Goleniewska K, Boyd KL, Quitalig M, Hotard AL, Moore ML, Hartert TV et al (2016) Respiratory syncytial virus infection activates IL-13-producing group 2 innate lymphoid cells through thymic stromal lymphopoietin. J Allergy Clin Immunol 138:814–824
Sutherland ER, Lehman EB, Teodorescu M, Wechsler ME, National Heart, Lung and Blood Institute’s Asthma Clinical Research Network (2009) Body mass index and phenotype in subjects with mild-to-moderate persistent asthma. J Allergy Clin Immunol 123:1328–1334
Tait Wojno ED, Monticelli LA, Tran SV, Alenghat T, Osborne LC, Thome JJ, Willis C, Budelsky A, Farber DL, Artis D (2015) The prostaglandin D2 receptor CRTH2 regulates accumulation of group 2 innate lymphoid cells in the inflamed lung. Mucosal Immunol 8:1313–1323
Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, Littman DR, O’Shea JJ (2009) Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J Exp Med 206:35–41
Taniguchi A, Miyahara N, Waseda K, Kurimoto E, Fujii U, Tanimoto Y, Kataoka M, Yamamoto Y, Gelfand EW, Yamamoto H et al (2015) Contrasting roles for the receptor for advanced glycation end-products on structural cells in allergic airway inflammation vs. airway hyperresponsiveness. Am J Physiol Lung Cell Mol Physiol 309:L789–L800
Taube C, Tertilt C, Gyulveszi G, Dehzad N, Kreymborg K, Schneeweiss K, Michel E, Reuter S, Renauld JC, Arnold-Schild D et al (2011) IL-22 is produced by innate lymphoid cells and limits inflammation in allergic airway disease. PLoS ONE 6:e21799
Thawer S, Auret J, Schnoeller C, Chetty A, Smith K, Darby M, Roberts L, Mackay RM, Whitwell HJ, Timms JF et al (2016) Surfactant protein-D is essential for immunity to helminth infection. PLoS Pathog 12:e1005461
Turner JE, Morrison PJ, Wilhelm C, Wilson M, Ahlfors H, Renauld JC, Panzer U, Helmby H, Stockinger B (2013) IL-9-mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J Exp Med 210:2951–2965
Van Maele L, Carnoy C, Cayet D, Songhet P, Dumoutier L, Ferrero I, Janot L, Erard F, Bertout J, Leger H et al (2010) TLR5 signaling stimulates the innate production of IL-17 and IL-22 by CD3(neg)CD127 + immune cells in spleen and mucosa. J Immunol 185:1177–1185
Van Maele L, Carnoy C, Cayet D, Ivanov S, Porte R, Deruy E, Chabalgoity JA, Renauld JC, Eberl G, Benecke AG et al (2014) Activation of Type 3 innate lymphoid cells and interleukin 22 secretion in the lungs during Streptococcus pneumoniae infection. J Infect Dis 210:493–503
Vonarbourg C, Mortha A, Bui VL, Hernandez PP, Kiss EA, Hoyler T, Flach M, Bengsch B, Thimme R, Holscher C et al (2010a) Progressive loss of RORγt expression confers distinct functional fates to natural killer cell receptor-expressing RORγt + innate lymphocytes. Immunity 33:736–751
Vonarbourg C, Mortha A, Bui VL, Hernandez PP, Kiss EA, Hoyler T, Flach M, Bengsch B, Thimme R, Holscher C et al (2010b) Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity 33:736–751
Vosshenrich CA, Ranson T, Samson SI, Corcuff E, Colucci F, Rosmaraki EE, Di SJ (2005) Roles for common cytokine receptor gamma-chain-dependent cytokines in the generation, differentiation, and maturation of NK cell precursors and peripheral NK cells in vivo. J Immunol 174:1213–1221
Walker JA, Oliphant CJ, Englezakis A, Yu Y, Clare S, Rodewald HR, Belz G, Liu P, Fallon PG, McKenzie AN (2015) Bcl11b is essential for group 2 innate lymphoid cell development. J Exp Med 212:875–882
Waugh DJ, Wilson C (2008) The interleukin-8 pathway in cancer. Clin Cancer Res 14:6735–6741
Wilhelm C, Hirota K, Stieglitz B, Van Snick J, Tolaini M, Lahl K, Sparwasser T, Helmby H, Stockinger B (2011) An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nat Immunol 12:1071–1077
Wohlfahrt T, Usherenko S, Englbrecht M, Dees C, Weber S, Beyer C, Gelse K, Distler O, Schett G, Distler JH et al (2016) Type 2 innate lymphoid cell counts are increased in patients with systemic sclerosis and correlate with the extent of fibrosis. Ann Rheum Dis 75:623–626
Wynn TA (2011) Integrating mechanisms of pulmonary fibrosis. J Exp Med 208:1339–1350
Xiong H, Keith JW, Samilo DW, Carter RA, Leiner IM, Pamer EG (2016) Innate lymphocyte/Ly6C(hi) Monocyte crosstalk promotes Klebsiella pneumoniae clearance. Cell 165:679–689
Xu X, Weiss ID, Zhang HH, Singh SP, Wynn TA, Wilson MS, Farber JM (2014) Conventional NK cells can produce IL-22 and promote host defense in Klebsiella pneumoniae pneumonia. J Immunol 192:1778–1786
Yasuda K, Muto T, Kawagoe T, Matsumoto M, Sasaki Y, Matsushita K, Taki Y, Futatsugi-Yumikura S, Tsutsui H, Ishii KJ et al (2012) Contribution of IL-33-activated type II innate lymphoid cells to pulmonary eosinophilia in intestinal nematode-infected mice. Proc Natl Acad Sci U S A 109:3451–3456
Ying X, Su Z, Bie Q, Zhang P, Yang H, Wu Y, Xu Y, Wu J, Zhang M, Wang S et al (2016) Synergistically increased ILC2 and Th9 cells in lung tissue jointly promote the pathological process of asthma in mice. Mol Med Rep 13:5230–5240
Yoshida H, Honda K, Shinkura R, Adachi S, Nishikawa S, Maki K, Ikuta K, Nishikawa SI (1999) IL-7 receptor alpha + CD3(-) cells in the embryonic intestine induces the organizing center of Peyer’s patches. Int Immunol 11:643–655
Yu X, Pappu R, Ramirez-Carrozzi V, Ota N, Caplazi P, Zhang J, Yan D, Xu M, Lee WP, Grogan JL (2014) TNF superfamily member TL1A elicits type 2 innate lymphoid cells at mucosal barriers. Mucosal Immunol 7:730–740
Yu Y, Wang C, Clare S, Wang J, Lee SC, Brandt C, Burke S, Lu L, He D, Jenkins NA et al (2015) The transcription factor Bcl11b is specifically expressed in group 2 innate lymphoid cells and is essential for their development. J Exp Med 212:865–874
Zhou W, Toki S, Zhang J, Goleniewksa K, Newcomb DC, Cephus JY, Dulek DE, Bloodworth MH, Stier MT, Polosuhkin V et al (2016) Prostaglandin I2 Signaling and Inhibition of Group 2 Innate Lymphoid Cell Responses. Am J Respir Crit Care Med 193:31–42
Zook EC, Ramirez K, Guo X, van der Voort G, Sigvardsson M, Svensson EC, Fu YX, Kee BL (2016) The ETS1 transcription factor is required for the development and cytokine-induced expansion of ILC2. J Exp Med 213:687–696
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (Grant No. 81571534), and Youth Development Fund of First Hospital of Jilin University (No. JDYY72016041). We are grateful to Ying Sun for critical reading of this manuscript. We sincerely apologize to the authors whose work was not cited in this review due to the space limitations.
ABBREVIATIONS
AHR, airway hyper-responsiveness; APC, antigen-presenting cell; Bcl11b, B cell leukemia/lymphoma 11b; BLF, bronchoalveolar lavage fluid; CCR6, C-C motif chemokine receptor 6; COPD, chronic obstructive pulmonary disease; CysLT1, cysteinyl leukotriene receptor 1; DC, dendritic cell; EPE, eosinophilic pleural effusion; G9a, lysine methyltransferase G9a; GATA3, GATA binding protein 3; GFP, green fluorescent protein; GM-CSF, granulocyte-macrophage colony-stimulating factor; HDM, house dust mite; HES, Heligmosomoides polygyrus excretory/secretory products; ICAM-1, intercellular adhesion molecule 1; IFN-γ, interferon-γ; ILCs, innate lymphoid cells; IL-2Rγ, common γ chain of the interleukin-2 receptor; IRF4, interferon regulatory factor 4; Lin, lineage; LTi, lymphoid tissue inducer; NCR, natural cytotoxicity triggering receptor; NK cell, nature killer cell; NSCLC, non-small cell lung cancer; OVA, ovalbumin; PSP, primary spontaneous pneumothorax; RAG, recombination-activating gene; ROR, retinoic acid receptor-related orphan receptor; TGF-β, transforming growth factor-β; Th cells, T helper cells; TLR5, Toll-like receptor 5; TNF, tumor necrosis factor; TSLP, thymic stromal lymphopoietin; VCAM-1, vascular cell adhesion molecule-1
COMPLIANCE WITH ETHICS GUIDELINES
Hang Cheng, Chengyan Jin, Jing Wu, Shan Zhu, Yong-Jun Liu, and Jingtao Chen declare that they have no competing interest. This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding authors
Additional information
An erratum to this article is available at https://doi.org/10.1007/s13238-017-0399-1.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Cheng, H., Jin, C., Wu, J. et al. Guards at the gate: physiological and pathological roles of tissue-resident innate lymphoid cells in the lung. Protein Cell 8, 878–895 (2017). https://doi.org/10.1007/s13238-017-0379-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-017-0379-5