
ABSTRACT
Bone sialoprotein-binding protein (Bbp), a MSCRAMMs (Microbial Surface Components Recognizing Adhesive Matrix Molecules) family protein expressed on the surface of Staphylococcus aureus (S. aureus), mediates adherence to fibrinogen α (Fg α), a component in the extracellular matrix of the host cell and is important for infection and pathogenesis. In this study, we solved the crystal structures of apo-Bbp273−598 and Bbp273−598-Fg α561−575 complex at a resolution of 2.03 Å and 1.45 Å, respectively. Apo-Bbp273−598 contained the ligand binding region N2 and N3 domains, both of which followed a DE variant IgG fold characterized by an additional D1 strand in N2 domain and D1′ and D2′ strands in N3 domain. The peptide mapped to the Fg α561−575 bond to Bbp273−598 on the open groove between the N2 and N3 domains. Strikingly, the disordered C-terminus in the apo-form reorganized into a highly-ordered loop and a β-strand G′′ covering the ligand upon ligand binding. BbpAla298–Gly301 in the N2 domain of the Bbp273−598-Fg α561−575 complex, which is a loop in the apo-form, formed a short α-helix to interact tightly with the peptide. In addition, BbpSer547–Gln561 in the N3 domain moved toward the binding groove to make contact directly with the peptide, while BbpAsp338–Gly355 and BbpThr365–Tyr387 in N2 domain shifted their configurations to stabilize the reorganized C-terminus mainly through strong hydrogen bonds. Altogether, our results revealed the molecular basis for Bbp-ligand interaction and advanced our understanding of S. aureus infection process.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Staphylococcus aureus (S. aureus) has been one of the leading causes of bacterial infections worldwide. Each year, some 500,000 patients in United States’ hospitals contract staphylococcal infections. S. aureus resides as part of the normal flora in the healthy human body until there is damage to skin surface or mucosal barrier, when it can gain access to tissues or the bloodstream, ultimately resulting in a wide range of infections and diseases, including impetigo, bacteremia, endocarditis, sepsis and arthritis (Lowy, 1998). Several antibiotics have been introduced to successfully treat S. aureus infections in patients over the past few decades. However, the infections became a growing concern lately due to the emergence of highly virulent and antibiotic-resistant strains, leading to increased morbidity and mortality (Zetola et al., 2005). Effective vaccines against S. aureus at early stages of infection are highly desirable, although all efforts to develop these vaccines have failed to date (Deresinski and Herrera, 2010; Vazquez et al., 2011).
S. aureus has evolved multiple strategies to promote colonization and evade the immune system. The initial adhesion of the pathogen to the extracellular matrix (ECM) components of the host is believed to be a critical step for successful infection. This is mediated by S. aureus surface adhesins called Microbial Surface Components Recognizing Adhesive Matrix Molecules (MSCRAMMs) (Gillaspy et al., 1998; Patti et al., 1994). Several structurally related proteins characterized by serine-aspartate dipeptide repeats (SD repeats) make up a family of MSCRAMMs (McCrea et al., 2000). The serine-aspartate repeat (Sdr) family include SdrF and SdrG in S. epidermidis and clumping factor A (ClfA), ClfB, SdrC, SdrD, SdrE and Bbp in S. aureus (Josefsson et al., 1998; McDevitt et al., 1994; Ni Eidhin et al., 1998; Tung et al., 2000). S. aureus bone sialoprotein-binding protein (Bbp) is an allelic variant of SdrE (Peacock et al., 2002). The members of Sdr family are predicted to adopt a similar structural pattern (Trad et al., 2004). A secretary signal sequence locates at the N-terminus followed by a ligand-binding A region and a characterized R region composed of SD repeats. The C-terminus features a cell wall-anchoring motif including the conserved LPXTG sequence (W), a hydrophobic membrane-spanning domain (M) and a short positively charged cytoplasmic tail (C) (Downer, 2002). In addition, SdrC, SdrD, SdrE and Bbp have different numbers of B repeats inserted between region A and R with the presence of a well-defined 12 residues cation-binding EF-hand loop (Josefsson et al., 1998). Our recent work showed that B1 domain interacted with N2 domain and opened the ligand binding cleft between N2 and N3 domains in SdrD (Wang et al., 2013).
The gene identified from chromosomal DNA isolated from S. aureus subsp. aureus TCH60 encodes bone sialoprotein-binding protein (Bbp) with 1149 amino acids, containing SD-repeats of 154 residues and the ligand-binding A region from 53 to 601 residues further divided into N1, N2 and N3 domains. S. aureus isolated from patients suffering from septic arthritis and osteomyelitis specifically interacts with bone sialoprotein (BSP), a noncollagenous protein of bone and dentine extracellular matrix, mediated by Bbp (Ganss et al., 1999; Ryden et al., 1987; Tung et al., 2000). BSP is proposed to induce hydroxyapatite crystal formation and distributes predominantly in the newly formed bone, which is more likely to be infected (Hultenby, 1994; Hunter and Goldberg, 1993).
Fibrinogen (Fg), a hexameric glycoprotein consisting of three different chains α2, β2 and γ2, plays critical roles in blood clotting and thrombosis (Gailit et al., 1997; Kollman et al., 2009; Mosesson et al., 2001). ClfB binds to fibrinogen α (Fg α) chain (Ganesh et al., 2011; Xiang et al., 2012). ClfA and the fibronectin-binding proteins FnbpA and FnbpB all bind to the C-terminal residues of fibrinogen γ (Fg γ) chain (Rivera et al., 2007; Wann et al., 2000). SdrG is reported to attack the thrombin cleavage site of fibrinogen β (Fg β) chain (Davis et al., 2001). A “dock, lock and latch” (DLL) model is identified in SdrG-Fg β complex to elucidate the ligand binding mechanism, where the ligand docks in the opened groove between N2 and N3 domains and the C-terminus across the groove stretches into N2 domain (Ponnuraj et al., 2003).
As a bifunctional MSCRAMM, Bbp also recognizes the human Fg α chain and inhibits thrombin induced blood coagulation (Vazquez et al., 2011). The molecular basis for Bbp-ligand interaction remains unknown. In our study, we solved the crystal structures of apo-Bbp273−598 and Bbp273−598 complexed with the peptide of Fg α561−575. We described the N2-N3 domains of Bbp similar to Dev-IgG fold (Deivanayagam et al., 2002). The Bbp273−598-Fg α561−575 complex revealed the ligand-binding basis through the rearrangement of the C-terminus and the significant changes in four regions. These results advance our understanding of the ligand binding mechanism of Bbp during S. aureus infection. Moreover, our study should shed light on the further identification of the substrate or ligand of other closely related Sdr proteins, and provide novel targets for the development of potent antagonists, vaccines or antibiotics.
RESULTS
Structure of apo-Bbp273−598
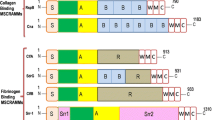
It was previously reported that on other MSCRAMMs of the Sdr family, the ligand-binding region was mapped to the N2 and N3 domains of the N-terminal A region. Based on sequence alignment of Bbp, SdrG and ClfA (Davis et al., 2001; Josefsson et al., 1998), we engineered a plasmid that would generate a recombinant fusion protein covering residues Asn273–Pro598 of Bbp from S. aureus, a segment containing both N2 and N3 domains (Fig. 1A), with N-terminal GST-tag for purification purposes in Escherichia coli. We obtained apo-Bbp273−598 protein and solved the structure at 2.03 Å resolution (Table 1), consisting of residues Asn273–Leu584 and additional residues Gly and Ser at N-terminus, two of the remaining five amino acid residues (GPLGS) from digested GST-tag (Fig. 1C). No electron density was observed for the 14 residues at C-terminus in the apo-Bbp273−598 structure.

Overall structure of the apo-Bbp 273−598. (A) Domain organization of the Bbp molecule. S (amino acid 1–52), signal sequence; N1–N3 (amino acid 53–601), Fg binding region; B (amino acid 602–935), B-repeats region; R (amino acid 936–1089), serine-aspartate repeat region; W, wall-spanning domain; M, membrane anchor; C, cytoplasmic positively charged tail. The region of N2 and N3 domains for crystallization (below). (B) Schematic representation of the topology of Bbp273−598 fold. The N2 and N3 domains are shown in cyan and violet, respectively. The N- and C-terminus are marked with red characters. (C) Cartoon representation of apo-Bbp273−598 structure with its N- and C-terminus indicated. The N2 and N3 domains are shown in cyan and violet, respectively
The apo-Bbp273−598 folds into two distinct domains N2 and N3, both of which have two layers of β-sheets and are structurally similar to the Dev-IgG fold (Fig. 1B), a variant of IgG fold (Deivanayagam et al., 2002). The two β-sheets of the N2 domain are composed of A, B and E strands on one side and C, D, D1, F and G strands on the opposite side. In N3 domain, C′, D1′, D2′, F′ and G′ strands form one principal sheet and A′, B′, D′ and E′ strands contribute to the facing sheet. The additional D1′ and D2′ strands present the featured Dev-IgG fold. One difference occurs with regard to the D strand in N2 domain, which parallels with E strand, although exhibiting an antiparallel orientation with the corresponding strand in the description of SdrG, ClfB and ClfA (Ganesh et al., 2008; Ponnuraj et al., 2003; Xiang et al., 2012).
In the apo-Bbp273−598 structure, the C-terminus with poor electron density extends into the solvent region, thus leading to an open groove. Presumably, a ligand-binding site could exist in the groove. (All structural figures in this paper were generated by PyMOL).
Structure of the Bbp273−598-Fibrinogen α (Fg α)561−575 complex
The ITC result showed that Fg α561−575, a synthesized polypeptide Fg α-chain containing residues 561–575 (Vazquez et al., 2011), has binding affinity to Bbp273−598 with a K D of 0.290 μmol/L (Fig. 2A). To study the molecular mechanism underlying Bbp-ligand recognition, Bbp273−598 was crystallized in complex with the ligand Fg α561−575. We solved the crystal structure of Bbp273−598-Fg α561−575 complex at 1.45 Å (Fig. 2B and Table 1).

Overall structure of the Bbp 273−598 -Fg α 561−575 complex. (A) ITC curves of Fg α561−575 titrated into Bbp273−598 protein. The first peak in the thermogram has not been used for analysis. (B) Cartoon representation of Bbp273−598-Fg α561−575 complex structure with its N- and C-terminus indicated. The N2 and N3 domains are colored the same as in Fig. 1C. The peptide is shown as sticks in yellow. (C) 2Fo-Fc map of Fg α561−575 peptide. The map is contoured at the level of 1.0 δ. The peptide residues are marked with red characters
Each crystallographic asymmetric unit contains two independent Bbp273−598-Fg α561−575 molecules. The bound peptide Fg α561−575 threads into the groove between the N2 and N3 domains in a snug conformation, with a well-defined 2Fo-Fc electron density map observed for the residues Lys562–Ser569 of the N-terminus of the peptide (Fig. 2C). The six residues of the C-terminus of Fg α561−575 have few interactions with the ligand binding groove and are not traceable in the density map. Due to the poor side-chain density, the N-terminal residue Fg αSer561 was replaced with an Alanine during structure refinement.
Structure comparison of Bbp273−598 between the apo-protein and in complex with the Fg α561−575 shows that the RMSD for the Cα atoms is 0.679 Å. Even though the overall topology of Bbp273−598 is similar in the two structures, significant conformational changes were observed around the peptide-binding groove, including the C-terminus of Bbp273−598 and four additional regions containing BbpAsp338–Gly355, BbpThr365–Tyr387, BbpAla298–Gly301 in N2 domain and BbpSer547–Gln561 in N3 domain, respectively (Fig. 3).

Conformational changes occur to C-terminus and additional four regions. (A) Surface charge representation of Bbp273−598 binding with the Fg α561−575 peptide. The surface is colored depending on negative charge, electrically neutral area and positive charge that are visualized in red, white and blue, respectively. Close view of surface and cartoon representation of C-terminus of Bbp273−598 and the Fg α561−575 peptide shown as ribbon representation colored in cyan. The connecting loop is shown as cartoon representation, composed of the residues Thr586–Ser589 running across the open groove are marked. (B) Structure alignment of apo-Bbp273−598 and Bbp273−598-Fg α561−575 shows four changed regions. Apo-Bbp273−598 and Bbp273−598-Fg α561−575 are colored in cyan and magenta, respectively. The peptide is shown as sticks colored in yellow. The regions BbpAsp338–Gly355, BbpThr365–Tyr387, BbpAla298–Gly301 and BbpSer547–Gln561 of the Bbp273−598-Fg α561−575 complex are show in red, lemon, orange and blue, respectively
The structural basis for peptide binding
The disordered C-terminus encompassing residues Ser585–Pro598 in apo-Bbp273−598 rearranges in the Bbp273−598-Fg α561−575 structure. The connecting loop between the G′ and G′′ strands spanning residues Thr586-Gly589 runs across the central region of the groove and the following sequence forms a short β-strand G′′ which inserts into N2 domain (Fig. 3A), making regions BbpAsp338–Gly355 and BbpThr365–Tyr387 deviate significantly from the apo-form (Fig. 3B). It shows a deviation of 5.6 Å for BbpPro347 in the region BbpAsp338–Gly355 between the C and D strands in N2 domain. BbpGly590 and Gly592 of β-strand G′′ of N3 domain form two hydrogen bonds with BbpThr345 in this region. The region BbpThr365–Tyr387 in N2 domain contains β-strand E and the TYKFTDYVD sequence, a TYTFTDYVD-like motif conserved in Sdr protein family (McCrea et al., 2000). The β-strand E moves toward β-strand G′′ to stabilize the C-terminus of N3 through several hydrogen bonds. BbpTyr387 in this region interacts with BbpSer585 at the end of the G′ strand, which play an important role in redirecting the C-terminus. BbpAsp373 and Arg374 in this region form two hydrogen bonds with BbpLys597 to stabilize the tail of the reordered C-terminus (Fig. 4A).

Detailed interactions in the four changed regions. (A) Closer view of the interactions between the C-terminus of N3 and the regions BbpAsp338–Gly355 and BbpThr365–Tyr387 of N2 in Bbp273−598-Fg α561−575; the residues involved are shown as sticks and marked with red and black characters, respectively. The hydrogen bonds are indicated by magenta dashed lines. The peptide is shown as sticks in yellow. (B) Detailed interactions between the ligand and Bbp273−598 in the Bbp273−598-Fg α561−575 complex. Bbp273−598 and Fg α561−575 are shown as sticks, colored in magenta and yellow, respectively. The residues of Bbp273−598 and Fg α561−575 are marked with black and red characters, respectively. The hydrogen bonds are indicated by green dashed lines
In addition, the binding of the peptide also induces a reorganization of the region BbpAla298–Gly301 between the A and B strands in N2 domain and a large movement of the region BbpSer547–Gln561 between the E′ and F′ strands in N3 domain toward the peptide binding groove. Two newly formed α-helices are observed in both of the two regions (Fig. 3B). The residues BbpHis299 and Gly301 in the first α-helix interact with Fg αThr568 and Ser566 through two main-chain hydrogen bonds and the BbpAsp300 forms the third hydrogen bond with the side-chain of Fg αSer567. In the second α-helix, BbpAsp555 contributes a hydrogen bond with Fg αLys562 and the side chain of BbpLeu557 contacts with the aromatic ring of Fg αPhe564 mediated by a hydrophobic interaction (Fig. 4B).
The structural rearrangements and the direct protein-ligand interactions formed upon peptide binding result in an effectively stabilized Bbp-Fg α complex compared to its apo-form.
Structural insights into Bbp273−598 and Fg α561−575 interactions
Apart from the interactions between the Fg α561−575 peptide and the residues from the two newly formed α-helices we have described above, there are several contacts with distances of less than 4 Å marked (Fig. 4B). Among them, there are three pairs of antiparallel main-chain hydrogen bonds between residues Fg αSer567, Thr565 and Gln563 and BbpThr582, Leu584 and Thr586 in the G′ strand and the connecting loop region. The carbonyl group of Fg αSer567 interacts with the side-chain polar group of BbpAsn581 forming another hydrogen bond. The residue Fg αSer569 from the C-terminus of the peptide forms a hydrogen bond with BbpThr580 in the G′ strand. The side chain hydroxyl group of Fg αSer566 interacts with the backbone atom of BbpPhe433 in N3 domain and Fg αSer566 forms the second hydrogen bond with the side-chain carbonyl group of BbpAsp334 in N2 domain. Fg αPhe564 contributes two hydrogen bonds with BbpAsp334 and Ile335 in the loop region between the C and D strands in N2 domain. The backbone atom of BbpAsn581 interacts with the carbonyl group of Fg αSer569, which plays a significant role in anchoring the C-terminus of the Fg α561−575 peptide. Two hydrogen bonds formed between the polar group of Fg αGln563 and BbpAsp588 from the loop region are involved in locking the peptide N-terminus.
Analysis of the interactions between Bbp273−598 mutants and Fg α561−575
Mutagenesis studies were conducted to further verify the binding of Fg α561−575 to Bbp273−598. We mutated the residues Thr582 and Leu584 to Ala respectively which form two pairs of hydrogen bonds with the peptide. The mutant proteins were purified to homogeneity and tested for their interaction with the Fg α561−575 peptide by surface plasmon resonance (SPR) (Fig. 5). The results indicate that the mutated protein BbpT582A or BbpL584A exhibits higher binding affinities with the peptide than wild type (WT) Bbp273−598 protein. This is probably because the side chain of Ala occupies less space compared to Thr or Leu, which brings an alteration of steric hindrance. Thus, the alteration presumably makes the peptide more easily dock into the groove and contacts more tightly with Bbp273−598. Even though BbpThr582 and Leu584 could interact with the peptide by two pairs of main chain-main chain hydrogen bonds (Fig. 3C), the replacement to alanine might serve a similar role instead of breaking the interaction with the peptide according to the results here. Perhaps, we can speculate that the residues BbpThr582 and Leu584 are only involved in the binding with the peptide, but not showing specificities on ligand recognition.

SPR analyses of the interactions between Bbp 273−598 WT and mutants (T582A and L584A) and the Fg α 561−575 (A–C), respectively. The proteins were immobilized on a BIA-core sensor chip. The concentration of Fg α561−575 peptide ranges from 62.5 μmol/L to 0.976 μmol/L: Night blue, 62.5 μmol/L; Magenta, 31.25 μmol/L; Peach, 15.625 μmol/L; Bright blue, 7.812 μmol/L; Red, 3.9 μmol/L; Lime, 1.953 μmol/L; Black, 0.976 μmol/L. KD values of individual binding assays are indicated below the sensorgrams
DISCUSSION
In this study, we have solved the crystal structures of apo-Bbp273−598 and the Bbp273−598-Fg α561−575 complex. Based on the structural information, we analyzed the structural basis for ligand binding.
In our study, tight interactions between the protein and the ligand result in a stable binding state. Due to the “Dock” of the ligand, the rearrangements occur to C-terminus and additional four regions of Bbp273−598. The connecting loop covers the open groove, resulting in “Lock” of the ligand. And then the G′′ strand forms compact interactions with the E strand in the region BbpThr365–Tyr387 of N2 domain, which “Latch” the ligand binding site and thus stabilize the overall structure. Our structure further supports the DLL model described for the SdrG-Fg β complex (Fig. 6A) (Ponnuraj et al., 2003).

Structure comparison of Bbp, ClfB and SdrG. (A) Superimposition of Bbp-Fg α and SdrG-Fg β (PDB entry: 1R17), colored in cyan and wheat, respectively. The peptides in both structures are shown as cartoon. The C-termini of Bbp and SdrG are indicated. (B) Superimposition of Bbp-Fg α and ClfB-Fg α (PDB entry: 3AT0), colored in cyan and blue, respectively. The peptides in both structures are shown as cartoon. The C-termini of Bbp and ClfB are indicated
In the work of V. Ganesh et al. on the ClfB-ligand complex, the ligand binding mechanism was described as the “DL” model due to the absence of the “Latch” process (Ganesh et al., 2011). In their structure, peptide Fg α336−347 in ClfB adopts a reverse orientation compared to the peptide Fg α561−575 in our structure. The C-terminus of ClfB does not stretch into N2 domain to interact with the E strand but exhibits a different direction. Upon ligand binding, no rearrangements are observed in the region between the D and D′ strands in N2 and the region between the E and F strands in N3 (Fig. 6B). However, large movements occur to the corresponding regions BbpAsp338–Gly355 in N2 and BbpSer547–Gln561 in N3 of Bbp273−598 in our studies. The diversity of the ligands binding pattern of MSCRAMMs adds to the necessity for structural analysis of individual members of this family.
Altogether, our findings have promoted the understanding of the ligand binding mechanism of Bbp in Sdr family, a critical step in the S. aureus infection process. In addition, potential new target sites based on these pathogen-host interactions could be explored for development of potent antibiotics and new therapeutic methods.
MATERIALS AND METHODS
Cloning, expression and purification of the recombinant proteins
The fragment of the Bbp gene (corresponding to 273–598 aa) was amplified using S. aureus ATCC 25923 genomic DNA by PCR. The gene fragments of mutated proteins BbpT582A and BbpL584A followed the same protocol. After digestion with BamHI and XhoI (NEB), the amplified fragments were cloned into the prokaryotic expression vector pGEX-6p-1 (GE Healthcare Life Sciences) to produce the GST-Bbp fusion protein and were confirmed by DNA sequencing. The recombinant protein was expressed in Escherichia coli strain BL21 with a high yield.
The bacteria cells were harvested and resuspended in lysis buffer containing 1× PBS, 2 mmol/L DTT and 1 mmol/L PMSF. The cells were homogenized by sonification and cell debris was removed completely by centrifuging at 13,000 rpm for 50 min at 4°C.
The recombinant protein was firstly purified by GST-affinity column and digested with PreScission protease overnight. The eluant was collected and concentrated for further purification using gel filtration chromatography (Superdex-200 column, GE Healthcare) in buffer containing 20 mmol/L HEPES pH 7.5, 200 mmol/L NaCl, 2 mmol/L DTT on the FPLC system (GE Healthcare Life Sciences). The proteins in every step of purification were analyzed by SDS-PAGE.
Crystallization and structure determination
The apo-Bbp273−598 and its complex were concentrated to 30 mg/mL in 20 mmol/L HEPES pH 7.5, 200 mmol/L NaCl, 2 mmol/L DTT. Crystals were screened by the sitting-drop vapor diffusion method (Jancarik et al., 1991) using sparse-matrix screen kits Crystal Screen I and II (Hampton Research), followed by optimizing the crystallization conditions through the variation of protein concentrations and pH. Crystals were grown at 18°C using the hanging-drop vapor diffusion method by mixing 1.0 μL protein solution with 1.0 μL reservoir solution and equilibrating against 200 μL reservoir solution. The apo-Bbp273−598 crystals were grown in 0.2 mol/L calcium acetate hydrate, 0.1 mol/L sodium cacodylate trihydrate pH 6.5, 18% PEG8000. The synthesized Fg α561−575 peptide was added into the concentrated protein samples at 10:1 ratio and the protein-peptide complex crystals are grown in 0.2 mol/L lithium sulfate, 0.1 mol/L Tris-HCl pH 8.2, 30% PEG4000. The apo-Bbp273−598 and Bbp273−598-peptide complex crystals diffracted to 2.03 Å and 1.45 Å respectively. The data were collected at the Shanghai Synchrotron Radiation Facility (SSRF) BL17U using a MAR225 (MAR Research, Hamburg) CCD detector at 100 K and processed with the HKL2000 package (Otwinowski and Minor, 1997). Further processing was carried out using programs from the CCP4 suite (Collaborative Computational Project, 1994). The model building of the Bbp273−598 molecules was conducted in COOT and the structure with peptide was determined by molecular replacement methods in CCP4 (Emsley and Cowtan, 2004). All the structures were refined with the PHENIX packages (Adams et al., 2002). Data collection and structure statistic are summarized in Table 1.
Synthesis of Fg α561−575 chain peptide
The peptide corresponding to the fibrinogen α561−575 was synthesized as previously described (Vazquez et al., 2011).
Isothermal titration calorimetry
ITC experiments were carried out at 25°C using a Microcal iTC200 instrument (GE Healthcare). The cell contained 50 μmol/L Bbp273−598 and the syringe contained 500 μmol/L peptide in the buffer containing 200 mmol/L NaCl and 20 mmol/L HEPES pH 7.5. Injecting peptide into buffer was performed as the blank titration. The data were fitted and analyzed using the Origin 7 software package (Microcal).
Surface plasmon resonance spectroscopy
The interaction affinities between Fg α561−575 and Bbp273−598 protein were conducted by surface plasmon resonance (SPR) using BIAcore T200 instrument (GE Healthcare). The wild type Bbp273−598 protein and two mutants were immobilized, respectively, on a CM5 sensor chip in 10 mmol/L sodium acetate, pH 4.5. The immobilization level was 3375RU. The synthetic peptide in 200 mmol/L NaCl and 20 mmol/L HEPES (pH 7.5) was injected on the protein-coated surface at various concentrations at 30 μL/min. The binding and dissociation was 60 s and 120 s, respectively.
Abbreviations
- Bbp:
-
bone sialoprotein-binding protein
- BSP:
-
bone sialoprotein
- Clf:
-
clumping factor
- ECM:
-
extracellular matrix
- Fg:
-
fibrinogen
- MSCRAMM:
-
Microbial Surface Components Recognizing Adhesive Matrix Molecules
- RMSD:
-
root mean square deviation
- S. aureus :
-
Staphylococcus aureus
- Sdr:
-
serine-aspartate repeat
- SPR:
-
surface plasmon resonance
- WT:
-
wild type
REFERENCES
Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58:1948–1954
Davis SL, Gurusiddappa S, McCrea KW, Perkins S, Hook M (2001) SdrG, a fibrinogen-binding bacterial adhesin of the microbial surface components recognizing adhesive matrix molecules subfamily from Staphylococcus epidermidis, targets the thrombin cleavage site in the Bbeta chain. J Biol Chem 276:27799–27805
Deivanayagam C, Wann ER, Chen W, Carson M, Rajashankar KR, Höök M, Narayana SV (2002) A novel variant of the immunoglobulin fold in surface adhesins of Staphylococcus aureus: crystal structure of the fibrinogen-binding MSCRAMM, clumping factor A. EMBO J 21:6660–6672
Deresinski S, Herrera V (2010) Immunotherapies for Staphylococcus aureus: current challenges and future prospects. Infect Control Hosp Epidemiol 31(Suppl 1):S45–47
Downer R, Roche F, Park PW, Mecham RP, Foster TJ (2002) The elastin-binding protein of Staphylococcus aureus (EbpS) is expressed at the cell surface as an integral membrane protein and not as a cell wall-associated protein. J Biol Chem 277:243–250
Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132
Gailit J, Clarke C, Newman D, Tonnesen MG, Mosesson MW, Clark RA (1997) Human fibroblasts bind directly to fibrinogen at RGD sites through integrin αvβ3. Exp Cell Res 232:118–126
Ganesh VK, Rivera JJ, Smeds E, Ko YP, Bowden MG, Wann ER, Gurusiddappa S, Fitzgerald JR, Hook M (2008) A structural model of the Staphylococcus aureus ClfA-fibrinogen interaction opens new avenues for the design of anti-staphylococcal therapeutics. PLoS Pathog 4:e1000226
Ganesh VK, Barbu EM, Deivanayagam CC, Le B, Anderson AS, Matsuka YV, Lin SL, Foster TJ, Narayana SV, Hook M (2011) Structural and biochemical characterization of Staphylococcus aureus clumping factor B/ligand interactions. J Biol Chem 286:25963–25972
Ganss B, Kim RH, Sodek J (1999) Bone sialoprotein. Crit Rev Oral Biol Med 10:79–98
Gillaspy AF, Lee CY, Sau S, Cheung AL, Smeltzer MS (1998) Factors affecting the collagen binding capacity of Staphylococcus aureus. Infect Immun 66:3170–3178
Hultenby K, Reinholt FP, Norgard M, Oldberg A, Wendel M, Heinegard D (1994) Dephosphorylation of osteopontin and bone sialoprotein by osteoclastic tartrate-resistant acid phosphatase. Eur J Cell Biol 63:230–239
Hunter GK, Goldberg HA (1993) Nucleation of hydroxyapatite by bone sialoprotein. Proc Natl Acad Sci USA 90:8562–8565
Jancarik J, Scott WG, Milligan DL, Koshland DE, Kim S-H (1991) Crystallization and preliminary X-ray diffraction study of the ligand-binding domain of the bacterial chemotaxis-mediating aspartate receptor of Salmonella typhimurium. J Mol Biol 221:31–34
Josefsson E, McCrea KW, Ni Eidhin D, O’Connell D, Cox J, Hook M, Foster TJ (1998) Three new members of the serine-aspartate repeat protein multigene family of Staphylococcus aureus. Microbiology 144(Pt 12):3387–3395
Kluytmans J, van Belkum A, Verbrugh H (1997) Nasal carriage of Staphylococcus aureus: epidemiology, underlying mechanisms, and associated risks. Clin Microbiol Rev 10:505–520
Kollman JM, Pandi L, Sawaya MR, Riley M, Doolittle RF (2009) Crystal structure of human fibrinogen. Biochemistry 48:3877–3886
Lowy FD (1998) Staphylococcus aureus infections. N Engl J Med 339:520–532
McCrea KW, Hartford O, Davis S, Eidhin DN, Lina G, Speziale P, Foster TJ, Hook M (2000) The serine-aspartate repeat (Sdr) protein family in Staphylococcus epidermidis. Microbiology 146(Pt 7):1535–1546
McDevitt D, Francois P, Vaudaux P, Foster TJ (1994) Molecular characterization of the clumping factor (fibrinogen receptor) of Staphylococcus aureus. Mol Microbiol 11:237–248
Mosesson MW, Siebenlist KR, Meh DA (2001) The structure and biological features of fibrinogen and fibrin. Ann N Y Acad Sci 936:11–30
Ni Eidhin D, Perkins S, Francois P, Vaudaux P, Hook M, Foster TJ (1998) Clumping factor B (ClfB), a new surface-located fibrinogen-binding adhesin of Staphylococcus aureus. Mol Microbiol 30:245–257
Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol 276:307–326
Patti JM, Allen BL, McGavin MJ, Hook M (1994) MSCRAMM-mediated adherence of microorganisms to host tissues. Annu Rev Microbiol 48:585–617
Peacock SJ, Moore CE, Justice A, Kantzanou M, Story L, Mackie K, O’Neill G, Day NP (2002) Virulent combinations of adhesin and toxin genes in natural populations of Staphylococcus aureus. Infect Immun 70:4987–4996
Ponnuraj K, Bowden MG, Davis S, Gurusiddappa S, Moore D, Choe D, Xu Y, Hook M, Narayana SV (2003) A “dock, lock, and latch” structural model for a staphylococcal adhesin binding to fibrinogen. Cell 115:217–228
Rivera J, Vannakambadi G, Hook M, Speziale P (2007) Fibrinogen-binding proteins of Gram-positive bacteria. Thromb Haemost 98:503–511
Ryden C, Maxe I, Franzen A, Ljungh A, Heinegard D, Rubin K (1987) Selective binding of bone matrix sialoprotein to Staphylococcus aureus in osteomyelitis. Lancet 2:515
Tung H, Guss B, Hellman U, Persson L, Rubin K, Ryden C (2000) A bone sialoprotein-binding protein from Staphylococcus aureus: a member of the staphylococcal Sdr family. Biochem J 345(Pt 3):611–619
Vazquez V, Liang X, Horndahl JK, Ganesh VK, Smeds E, Foster TJ, Hook M (2011) Fibrinogen is a ligand for the Staphylococcus aureus microbial surface components recognizing adhesive matrix molecules (MSCRAMM) bone sialoprotein-binding protein (Bbp). J Biol Chem 286:29797–29805
Wang X, Ge J, Liu B, Hu Y, Yang M (2013) Structures of SdrD from Staphylococcus aureus reveal the molecular mechanism of how the cell surface receptors recognize their ligands. Protein Cell 4:277–285
Wann ER, Gurusiddappa S, Hook M (2000) The fibronectin-binding MSCRAMM FnbpA of Staphylococcus aureus is a bifunctional protein that also binds to fibrinogen. J Biol Chem 275:13863–13871
Xiang H, Feng Y, Wang J, Liu B, Chen Y, Liu L, Deng X, Yang M (2012) Crystal structures reveal the multi-ligand binding mechanism of Staphylococcus aureus ClfB. PLoS pathog 8:e1002751
Zetola N, Francis JS, Nuermberger EL, Bishai WR (2005) Community-acquired meticillin-resistant Staphylococcus aureus: an emerging threat. Lancet Infect Dis 5:275–286
ACKNOWLEDGMENTS
We would like to thank the staff at the SSRF BL17U beam-line for assistance in data collection. We also thank the National Center for Protein Sciences (Beijing, China) for technical support. This work was supported by the National Basic Research Program (973 Program) (Nos. 2011CB910502 and 2012CB911101) and National Natural Science Foundation of China (Grant Nos. 31030020 and 31170679).
COMPLIANCE WITH ETHICS GUIDELINES
Xinyue Zhang, Meng Wu, Wei Zhuo, Jinke Gu, Sensen Zhang, Jingpeng Ge and Maojun Yang declare that they have no conflict of interest.
This article does not contain any studies with human or animal subjects performed by the any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
Xinyue Zhang, Meng Wu and Wei Zhuo have contributed equally to this work.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhang, X., Wu, M., Zhuo, W. et al. Crystal structures of Bbp from Staphylococcus aureus reveal the ligand binding mechanism with Fibrinogen α. Protein Cell 6, 757–766 (2015). https://doi.org/10.1007/s13238-015-0205-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-015-0205-x