
Abstract
Being attracted with their cardiotonic, antidiabetic, cough relieving activity, treatment of fever, absorbent, anti-asthmatic, etc. activities reported in ancient Ayurvedic literature, phytochemicals of Onosma bracteata wall should be evaluated for their activity against SARS-CoV-2 virus. The main objective of this study is to identify a hit molecule for the inhibition of entry, replication, and protein synthesis of SARS CoV-2 virus into the host. To achieve given objective, computational virtual screening of phytochemicals of Onosma bracteata wall has been performed against three main viral targets: spike, RdRp, and Mpro. Further, the analysis of Lipinski’s Ro5 and their estimation of ADMET profiles were performed using computational tools. The MD simulations studies of top hits against each viral target have also been performed for 20 ns to ensure their stability. The analysis of results revealed that Pulmonarioside C (9) and other plant compounds showed better binding affinity towards targets than existing antiviral compounds, making them probable lead compounds against SARS-CoV-2. Structural modifications and studies through in silico analysis provided the founding stone for the establishment of SARS CoV-2 inhibitory potential of phytoconstitutents of Onosma bracteata wall.
Similar content being viewed by others
Introduction
Corona Virus Disease-2019 (COVID-19), caused by infectious coronaviruses or severe acute respiratory syndrome coronavirus-2 (SARS CoV-2), was first reported in Wuhan of the Hubei region of China in December 2019 and was declared a pandemic on 13 March 2020. As of 26 April 2023, it has caused the infection to 764,474,387 people across the globe, along with 6,915,286 confirmed deaths (WHO Coronavirus Disease (COVID-19) Dashboard 2023). The disease conditions of COVID-19 vary from asymptomatic, pre-symptomatic, mild pneumonia, severe pneumonia, acute respiratory distress syndrome, and critical stages, which may lead to fatalities (Adhikari et al. 2020). The control of the pandemic spreading is challenging because there is no specific drug to fight against it, as usually happens in virus-originated diseases. The unprecedented rapid spread of COVID-19 and its unclear mortality rates have created urgency for a treatment to be developed worldwide. In addition, due to the higher mutagenicity of the virus, several variants such as ‘gamma’, ‘delta’, and ‘omicron’ have increased the state of urgency (Update on Omicron 2022; Lopez Bernal et al. 2021).
The identification of the crucial proteins and enzymes involved in the virus’ entry and replication processes have been regarded as the basic steps to identify the viral drug targets for SARS-CoV-2 (Sorouri et al. 2021). The genomic sequence of the virus has been reported similar to that of SARS CoV-1 by 79.5% and that of bats infecting strain of coronaviruses by 96% (Wang et al. 2020). The SARS CoV-2 spike protein binds to the angiotensin-converting enzyme-2 (ACE2) receptor through the receptor-binding domain (RBD) for inoculation into the host cell, followed by the membrane fusion with host cells allowing the entry of the viral genome. The alternative entry of the virus may be via direct proteolytic cleavage of transmembrane protease 2 (TMPRSS2) to the ACE2 receptor. The high binding preference of the RBD, pre-activation of the spike, and covered RBD in the spike possibly help efficient viral entry as well as evading the immunity surveillance (Shang et al. 2020). The viral genome translation has followed this into polyproteins pp1a and pp1b host cell machinery. These polyproteins, along with proteases like the 3 Chymotrypsin-Like Protease (3CLpro, also called main protease Mpro), are transformed into replicase-transcriptase complex, which generates viral genome for viral replication mediated through RNA-dependent RNA polymerase (RdRp). Thus, ACE2 of the host, Mpro, and RdRp of the virus are vital enzymes for developing therapeutics against COVID-19 (Świderek and Moliner 2020; Abuo-Rahma et al. 2020; Zhang and Kutateladze 2020; Gajjar et al. 2021; Nagar et al. 2021).
There has been a continuous need for novel approaches and identifying novel molecules or hits to counter the disease. In ancient texts, ayurvedic therapies mention epidemic diseases and their management descriptively under the term Janapadodhwamsa (Goyal 2019; Rastogi et al. 2022). In addition, the Ministry of Ayurveda, Yoga, and Naturopathy, Unani, Siddha, and Homeopathy (AYUSH), Government of India has recommended immunity-boosting remedies for self-care during the COVID-19 crisis. It mentions various Ayurveda interventions, including general measures, immunity-promoting therapies, simple ayurvedic procedures, and dry cough/sore throat measures (Ayurveda’s immunity boosting measures for self care during COVID 19 crisis 2022). These remedies have been recommended by eminent Ayurveda professionals across India, and scientific literature supports immunity-boosting claims (Vegad and Pandya 2021). Additionally, ancient Ayurvedic literatures have indicated various plants useful in respiratory disease conditions.
O. bracteata Wall., a perennial herb explicitly found in the north-western Himalayas at 3500–4500 m, has been acknowledged in ancient Ayurvedic literature Bhavprakasha Nighantu as Gojihva, which falls in Saka Varga (vegetable class) as per Charak and Sushruta. The Gojihva has been reported to have cardiotonic, antidiabetic, anti-tussive, anti-pyretic, absorbent, anti-asthmatic, antitoxic, anti-inflammatory, anti-leprotic, and diuretic activities (Vegad and Pandya 2022a). Preliminary pharmacological studies have revealed it as hepatoprotective (Kumar et al. 2020), diuretic (Vegad and Pandya 2022b), anticancer (Albaqami et al. 2018; Kumar et al. 2021), anxiolytic (Asif et al. 2019), smooth muscle relaxant, and anti-aging substances (Farooq et al. 2019). Allomicrophyllone (1) and ehretiquinone (2), along with three novel benzoquinones ehretiquinones B-D (3–5) with anti-aging activity, have been isolated from O. bracteata (Farooq et al. 2019). Hernacorizine (6) and two naphthoquinones derivatives (7–8) have been separated from methanol extract of the plant in an unpublished thesis work (Khan 2017). Three new compounds, Pulmonarioside C (9), 9′-methoxyl salvianolic acid R (10), and (−)-4-O-(E)-p-coumaroyl-l-threonic acid methyl ester (11), have been reported for the first time from the plant (Sun et al. 2021). Now-a-days, computational drug discovery is very emerging and widely used approach for identification of hit molecules through protein–ligand interactions (Singh et al. 2022; Singh and Purohit 2023a, b) In continuation of our quest for effective SARS CoV-2 inhibitors (Nagar et al. 2021; Sureja et al. 2022), these novel compounds attracted us for in silico screening against ACE2, Mpro and RdRp viral targets through measurement of binding energies, ADMET analysis and MD simulations.
Methods
Collection of data
3-D structures of the target proteins such as RdRp, spike glycoprotein, and main protease were obtained from RCSB protein data bank by following their PDB IDs (7BV2, 6M0J, 6LU7) (Protein Data Bank 2022; Jin et al. 2020; Lan et al. 2020; Yin et al. 2020). For further molecular modelling, proteins were prepared using AutoDock Tools. (Trott and Olson 2010). Following the deletion of water molecules, addition of polar hydrogens and Kollman charges, the polarity and charges of protein structures were corrected. Receptor grid box of the dimensions of 90.768 Å (x) × 99.896 Å (y) × 99.788 Å (z) with the center coordinates of 70 Å (x) × 70 Å (y) × 70 Å (z) were generated around co-crystallized ligand remdesivir with RdRp (Nagar et al. 2021). The receptor grid box of the size of 50 Å (x) × 42 Å (y) × 50 Å (z) and the center coordinates of − 36.258 Å × 40.262 Å × 13.154 Å was generated in spike glycoprotein (Surti et al. 2020). Similarly, the receptor grid box of 24 Å (x) × 20 Å (y) × 22 Å (z) and the center coordinates of − 12.855 Å × 16.974 Å × 66.782 Å (x, y, and z) were generated around co-crystallized ligand N3 in Mpro after validating the pose of the co-crystallized ligand with the docked pose of N3. Structures of eleven Onosma bracteata phytochemicals were downloaded from PubChem in SDF format (PubChem 2021). After optimization and their conversion into PDBQT files using OpenBabel, the ligands were subjected to molecular docking (O’Boyle et al. 2011).
Molecular docking
The multi-ligand molecular docking was carried out using AutoDock Vina to identify ligand interactions with the selected proteins. Selected ligands were docked on the sites on which the receptor grid boxes were generated for each protein. Ten poses per ligand were obtained, and the binding energy of each ligand’s top poses was compared with the known antiviral drugs like remdesivir, lopinavir, ritonavir, and favipiravir. Biovia Discovery studio 2020 was used to generate the 3-dimensional docking poses by visualizing the docked complexes and their interactions 3-dimentionally (Dassault Systѐmes BIOVIA 2021). For this purpose, interacting aminoacids are visualised in surface model and ligand structure is presented in ball and sticks model to get better understanding of interactions and interacting residues.
Analysis of physicochemical properties and ADMET parameters
Physicochemical properties of the selected molecules were analyzed using virtual tools like Swiss ADME (Daina et al. 2017) and pkCSM (pKCSM 2021; Pires et al. 2015) and compared with the selected known inhibitors. Required ADMET parameters were also studied using pkCSM to evaluate the pharmacokinetic properties of the selected compounds.
Molecular dynamics (MD) simulations
The compound 9 with the highest binding energies against all proteins during molecular docking was subjected to MD simulations using GROMACS 2020.1 software (Abraham et al. 2015, 2020). CHARMM36 as an all-atom force field (Huang et al. 2016) and CGenFF server was used to generate the topology of proteins and ligands, respectively (Vanommeslaeghe et al. 2010; Yu et al. 2012). After generating topologies for ligand and protein, the complex was solvated using the TIP3P water model, neutralized using Na+ and Cl− ions, equilibrated using a canonical [number of particles (N), system volume (V) and temperature (T), NVT] and isobaric-isothermic [number of particles (N), system pressure (P) and temperature (T), NPT] ensemble for 100 picoseconds. The final MD run of protein–ligand complexes for 20 ns was subjected in NPT group at 300 K temperature and 1 bar pressure as reported previously (Gajjar et al. 2021; Nagar et al. 2021; Bhakhar et al. 2021; Dhameliya et al. 2022). After completion of MD run, calculation of an interaction energies is carried out by the extraction of average short-range Coulombic interaction energy (Coul-SR) and the short-range Lennard–Jones energy (LJ-SR) through energy module to evaluate energies of electrostatic and hydrophobic interactions, respectively. Along with this, calculation of root mean square deviation (RMSD) for protein and ligand, Radius of gyration (RoG) of ligand in the complex, surface area of the solvent atoms assessed by the complex and number of hydrogen bonds formed during the MD simulations have also been calculated to understand the proper dynamic changes of the protein–ligand complex at molecular level.
Results and discussion
Preparation of proteins
The protein deposited in protein data banks for RdRp from SARS CoV-2 (PDB ID: 7BV2, resolution of 2.8 Å) (Protein Data Bank 2022; Yin et al. 2020) and ACE2 (PDB ID: 6M0J, resolution of 2.45 Å) (Lan et al. 2020) and Mpro (PDB ID: 6LU7, resolution of 2.16 Å) (Jin et al. 2020) were used for the present works. The proteins have been downloaded from RCSB protein data banks and have been prepared using AutoDock Tools for further molecular modeling (Morris et al. 2009).
Molecular docking
The prepared proteins using AutoDock Vina have been subjected to identify the binding energies of the selected eleven ligands and their interactions at the active site and the 2D-structures of natural products were downloaded from the PubChem database (PubChem 2021) were converted into PDBQT formats to use in molecular modeling. The energies of the bound ligands were analyzed in comparison with N3, and other antiviral drugs like remdesivir, lopinavir, ritonavir, and favipiravir. From this molecular modeling, compound 9 was found with the highest binding energy against all the selected targets (Table 1). The binding energy of compound 9 was found superior to selected known drugs against every protein except lopinavir (6M0J).
The 3D-poses of the top four ligands having the highest binding energies against each protein were presented using Biovia Discovery Studio 2020 (Dassault Systѐmes BIOVIA 2021). Against RdRp, compounds 9, 10, 1 and 3 were found with binding energies of − 12.5, − 9.3, − 8.9, and − 8.8 kCal/mol, respectively. Compound 9 was found to form hydrogen bonds (HBs) with Asn496 and Lys577 (chain A) and Ade13, Cyt15, and Urd17 (chain P), Urd12, Ade13, Ade14, Cyt15, Urd16 and Urd17 (chain T, Fig. 1a). Next, compound 10 was found to form HBs with the residues Asn496 and Asn497, nucleotides, Urd13 and Ade15 (chain P), and Ade14 of chain T. Additionally, it formed a π-alkyl interactions with Lys500 and van der Waals interactions with Ade11, Urd12, and Ade13 (chain T, Fig. 1b). Compound 1 formed HBs by means of amino acids, Asn497 and Lys500 and Gua16 and Urd18 and Ade13. The π-π interaction was also observed with the Ade15 (Fig. 1c). Compound 3 interacted with Ade19 and Urd20 and amino acid Lys45 through HBs. Alkyl groups present in the ligand structures formed π-alkyl interactions with Urd17, Urd18 of chain P and amino acids, Ala547, Ile548 (Fig. 1d). These interactions hypothesized us to predict the strong binding of these compounds with RdRp into its active site.

The 3D-interactions of compounds Pulmonarioside C (9) (a), 9′-Methoxyl salvianolic acid R (10) (b), Allomicrophyllone (1) (c), and Ehretiquinone –B (3) (d) complexed with RdRp. Ligand is shown in ball and stick model which interacts with the amino acids of RdRp present in surface model
Next, we analyzed the binding modes of the best four ligands against the spike-ACE2 complex (PDB ID: 6M0J, Fig. 2). Ligands 9, 5, 2, and 3 had the binding energies of − 7.2, − 6.5, − 6.5, and − 6.2 kcal/mol, respectively (Table 1). Compound 9 interacted via HBs with amino acids, Lys26, Asn33, Asn90, Gln96, Gln388, Arg559, and Ser563 (Fig. 2a). Due to nominal structural differences, compounds 5, 2, and 3 were found to form HBs with Asn33, and two π-alkyl interactions with Pro389 (Fig. 2b–d).

3-D interactions of compounds Pulmonarioside C 9 (a), Ehretiquinone-D (5) (b), Ehretiquinone (2) (c), and Ehretiquinone-B (3) (d) with Spike glycoprotein-ACE2 (PDB ID: 6M0J). Ligand is shown in ball and stick model which interacts with the amino acids of Spike glycoprotein-ACE2 present in surface model
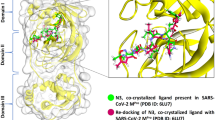
With a view to a better understanding of the binding mode of identified hits against Mpro (PDB ID: 6LU7), we studied the binding interactions (Fig. 3) of the selected compounds 9, 3, 4, and 5 with the binding energies of − 7.9, − 6.7, − 6.7 and − 6.6 kcal/mol, correspondingly. Compound 9 was found to interact via HBs with amino acids, Leu4, Thr24, Thr26, Asn142, Ser144, and Cys145, along with the van der Waals interactions with Thr45 (Fig. 3a). Compound 3 formed HBs with His41 and π-alkyl bonds with Met49, Cys143, and His163 (Fig. 3b). Next, compound 4 formed HBs with His41 and π-alkyl bonds with Val3 (chain C), Cys145 (chain A), and His163 (chain A) (Fig. 3c). The almost similar interactions were observed in the case of compound 5 due to their close structural similarity; only amino acid, Gly143 was found to interact in place of His163 (Fig. 3d).

3-D interactive poses of compounds Pulmonarioside C (9) (a), Ehretiquinone-B (3) (b), Ehretiquinone-C (4) (c), and Ehretiquinone-D (5) (d) with Mpro (PDB ID: 6LU7). Ligand is shown in ball and stick model which interacts with the amino acids of Mpro present in surface model
ADMET
For a drug molecule, the oral bioavailability of the drug candidates has been considered the critical parameters that can be devised following the Rule of Five (Ro5) given by Christopher A. Lipinski (Lipinski et al. 2001; Lipinski 2004). In this regard, we analyzed the molecules for their physicochemical properties (Ro5, Table 2) like formula weight, hydrogen bond donors/acceptors, rotational bonds and permeability using virtual tools such as SwissADME (Daina et al. 2017) and pkCSM (Pkcsm 2021; Pires et al. 2015) along with the assessment of the number of violated parameters. As per our analysis, all the compounds (except compounds 9 and 10) have been found to follow Ro5 with zero violations. Compounds 9 and 10 were found to violate three properties as per Ro5 being larger in molecular size. However, three of the known inhibitors also found to violate two parameters. Thus, most of the natural ligands may possess enough candidature to be a better drug.
The absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties have been reported to yield a better understanding and vision of the pharmacokinetics of identified drug candidates to assess the risk factors for the human body (Cheng et al. 2013) (Table 3). All the molecules (except compounds 9 and 10) were satisfied with the criteria for the ADMET parameters. In majority, the compounds possess better absorption in guts due to their good oral bioavailability. Compounds 9, 5, 2, and 3 inhibited the P-gp I substrate in the body, requiring the dose adjustment for better therapeutic effectivity.
Further, additional parameters for predicting the metabolism, excretion, and toxicity were also evaluated for these ligands (Table 4). Most of the molecules not inhibited CYP2D6 and CYP3A4 (except compounds 1 and 6 inhibited the CYP3A4). Compounds 9 and 10 were found with very low renal clearance limiting their effects to cause renal toxicity. None of the molecules was observed as the substrates for renal uptake transporter and all the compounds except 11 (producing AMES toxicity) were found non-cytotoxic, non-mutagenic, non-toxic to the dermis with safer window for chronic toxicity. In fine, the ligands have the potential to be selected as the promising candidates if explored against COVID-19.
MD simulation
Molecular dynamics (MD) simulation gives a better understanding of ligand stability in the active site of the receptor (Durrant and McCammon 2011; Hospital et al. 2015) being widely used in drug discovery for COVID-19 (Padhi et al. 2021). To assess the stability of selected ligands using GROMACS 2020.1 (Abraham et al. 2015, 2020), the complexes of most promising compound 9 with selected three different targets were subjected for MD simulations.
The root mean square deviation (RMSD) with an average of 0.847 nm (Fig. 4a) for the ligand 9 and 0.263 nm (Fig. 4b) for the RdRp, indicated that enough stability of the ligand. However, some alterations have been observed as evident from the RMSD of ligand after 18 ns, which may be due to better accommodation of compound 9 into the binding site of RdRp. Herein, the RMSD values for protein–ligand complex are almost similar to RMSD values of ligand fitted in backbone. The range of 5.56 to 5.71 nm with an average value of 5.636 nm (Fig. 4c) for radius of gyration (RoG) suggested the tight compactness of ligand into the complex. The solvent assessable surface area (SASA) was observed ranging from of 31–48 nm2 with the mid-point of 98.81 nm2 (Fig. 4d). A maximum of seven HBs were observed between the ligand 9 and RdRp protein in plotting the number of HB vs. time (Fig. 4e).

The representative charts of RMSD for ligand (a), RMSD for protein (b), radius of gyrations (c), surface area (d), and hydrogen bonds (e) for the complex of 9 with RdRp. Plots are showing considerable deviation of ligand from the initial position. Sufficient number of hydrogen bonds were observed between protein and ligand
Next, we analyzed the time-dependent stability of the complex of 9 with Spike-ACE2 protein (Fig. 5) using the above statistical parameters. The satisfactory stability of the ligand was observed from the RMSD ranging from 0.3 to 0.7 nm (Fig. 5a) and 0.07–0.25 nm (Fig. 5b) for the ligand and receptor, respectively. The radius of gyration (2.49–2.57 nm) was found with an average value of 2.524 nm (Fig. 5c). SASA has been noted in the range of 11–27 nm2 with an average of 20.94 nm2 (Fig. 5d). A maximum of seven hydrogen bonding interactions were observed between 9 and Spike-ACE2 during the MD run (Fig. 5e). These results supported our hypothesis of sufficient stability of compound 9 with Spike-ACE2 protein.

The representative charts of RMSD for ligand (a), RMSD for protein (b), radius of gyrations (c), surface area (d), and hydrogen bonds (e) for the complex of 9 with Spike-ACE2. Plots are showing considerable deviation of ligand from the initial position. Sufficient number of hydrogen bonds were observed between protein and ligand
Next, the RMSD value for the compound 9 complexed with Mpro was found to have the mid-points of 1.501 nm for ligand (Fig. 6a) and 0.259 nm for protein (Fig. 6b). The Deviations were observed in the RMSD of ligand after 12 ns, which can be associated with the bit of instability of compound 9 in the protein’s active site. The RoG was found with the mid-point value of 2.238 nm (Fig. 6c). However, the minute fluctuation was observed between 5–6 ns. The solvent access surface area has been found within the range from 12–25 nm2 (average: 18.377 nm2, Fig. 6d). A maximum of nine HB were observed between the ligand and protein complex during simulation (Fig. 6e). The analysis of the energies of all three complexes in terms of electrostatic and hydrophobic interactions reflected the significance of electrostatic interactions rather than hydrophobic interactions (Table 5) to stabilize compound 9 with Mpro.

The representative charts of RMSD for ligand (a), RMSD for protein (b), radius of gyrations (c), surface area (d), and hydrogen bonds (e) for the complex of 9 with Mpro. Plots are showing considerable deviation of ligand from the initial position. Sufficient number of hydrogen bonds were observed between protein and ligand
Conclusions
The in silico screening of eleven compounds of Onosma bracteata Wall. against spike, RdRp and Mpro led to identifying hits such as Compound 9 and other ligands with significantly higher binding potential as compared to antiviral SARS CoV-2 inhibitors. Various interactions between ligand and protein provided insightful knowledge regarding their binding modes in the active sites of proteins. Though compounds 9 and 10 violated Lipinski’s Ro5 with three criteria, the drug-likeliness studies and analysis of ADMET of these eleven ligands identified their suitability for drug candidates. MD simulation studies of compound 9 against each protein indicated the stability and compactness of the complex’s active site through hydrogen bond and electrostatic interactions. In the future, the structural modifications of hits and their studies through in vitro assays may open up the further probability as future SARS CoV-2 inhibitors.
Data availability
The protein and ligand required for molecular modeling have been retrieved from RCSB PDB and PubChem, respectively. The preparation of protein, ligands, and prediction of ADMET properties has been performed using AutoDock Vina, OpenBabel, and SwissADME/pKCSM, respectively. The 3D images of docked compounds with the ligands have been presented using Biovia Discovery Studio. To study the stability of ligands in the ligand–protein complexes, GROMACS 2020.1 has been used from www.zenodo.org.
Code availability
The present work does not involve any deposition of newly reported crystal structures.
References
Abraham MJ, Murtola T, Schulz R et al (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2:19–25
Abraham MJ, Berk Hess, Lindahl E, Spoel D van der (2020) GROMACS 2020.1 (Manual Version 2020.1) Zenodo. Accessed 10 Sep 2020. https://doi.org/10.5281/zenodo.4054996
Abuo-Rahma GE-DA, Mohamed MFA, Ibrahim TS et al (2020) Potential repurposed SARS-CoV-2 (COVID-19) infection drugs. RSC Adv 10:26895–26916
Adhikari SP, Meng S, Wu YJ et al (2020) Epidemiology, causes, clinical manifestation and diagnosis, prevention and control of coronavirus disease (COVID-19) during the early outbreak period: a scoping review. Infect Dis Poverty 9:29
Albaqami J, Myles EL, Tiriveedhi V et al (2018) The Effect of Onosma bracteatum in cancer cells. MOJ Bioequival Bioavailab 5:321–325
Asif HM, Hayee A, Aslam MR et al (2019) Dose-dependent, antidepressant, and anxiolytic effects of a traditional medicinal plant for the management of behavioral dysfunctions in animal models. Dose-Response 17:1559325819891262
Ayurveda’s immunity boosting measures for self care during COVID 19 crisis. https://www.mohfw.gov.in/pdf/ImmunityBoostingAYUSHAdvisory.pdf. Accessed 7 Feb 2022
Bhakhar KA, Gajjar ND, Bodiwala KB et al (2021) Identification of anti-mycobacterial agents against mmpL3: virtual screening, ADMET analysis and MD simulations. J Mol Struct 1244:130941
Cheng F, Li W, Liu G, Tang Y (2013) In silico ADMET prediction: recent advances, current challenges and future trends. Curr Top Med Chem 13:1273–1289
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7:42717
Dassault Systѐmes BIOVIA, BIOVIA Workbook, Release 2021; BIOVIA DS Visualizer, Release 2021, San Diego: Dassault Systѐmes, 2021
Dhameliya TM, Nagar PR, Gajjar ND (2022) Systematic virtual screening in search of SARS CoV-2 inhibitors against spike glycoprotein: pharmacophore screening, molecular docking, ADMET analysis and MD simulations. Mol Divers 26:2775–2792
Durrant JD, McCammon JA (2011) Molecular dynamics simulations and drug discovery. BMC Biol 9:71
Farooq U, Pan Y, Disasa D, Qi J (2019) Novel anti-aging benzoquinone derivatives from Onosma bracteatum Wall. Molecules 24:1428
Gajjar ND, Dhameliya TM, Shah GB (2021) In search of RdRp and Mpro inhibitors against SARS CoV-2: molecular docking, molecular dynamic simulations and ADMET analysis. J Mol Struct 1239:130488
Goyal M (2019) Threats and challenges of emerging viral diseases and scope of Ayurveda in its prevention. Ayu 40:67–68
Hospital A, Goñi JR, Orozco M, Gelpí JL (2015) Molecular dynamics simulations: advances and applications. Adv Appl Bioinforma Chem 8:37–47
Huang J, Rauscher S, Nawrocki G et al (2016) CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods 14:71–73
Jin Z, Du X, Xu Y et al (2020) Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582:289–293
Khan S (2017) Investigation of bioactive leads for the prevention of bronchial asthmatic syndrome. Shoolini University of Biotechnology and Management Sciences, Solan
Kumar A, Kaur V, Pandit K et al (2020) Antioxidant Phytoconstituents From Onosma bracteata Wall. (Boraginaceae) ameliorate the CCl4 induced hepatic damage: in vivo study in male wistar rats. Front Pharmacol 11:1301
Kumar A, Kaur S, Pandit K et al (2021) Onosma bracteata Wall. induces G0/G1 arrest and apoptosis in MG-63 human osteosarcoma cells via ROS generation and AKT/GSK3β/cyclin E pathway. Environ Sci Pollut Res 28:14983–15004
Lan J, Ge J, Yu J et al (2020) Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581:215–220
Lipinski CA (2004) Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol 1:337–341
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and devlopment settings. Adv Drug Deliv Rev 46:3–26
Lopez Bernal J, Andrews N, Gower C et al (2021) Effectiveness of COVID-19 vaccines against the B.1.617.2 (Delta) variant. N Engl J Med 385:585–594
Morris GM, Huey R, Lindstrom W et al (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791
Nagar PR, Gajjar ND, Dhameliya TM (2021) In search of SARS CoV-2 replication inhibitors: virtual screening, molecular dynamics simulations and ADMET analysis. J Mol Struct 1246:131190
O’Boyle NM, Banck M, James CA et al (2011) Open Babel: an open chemical toolbox. J Cheminform 3:33
Padhi AK, Rath SL, Tripathi T (2021) Accelerating COVID-19 research using molecular dynamics simulation. J Phys Chem B 125:9078–9091
Pires DEV, Blundell TL, Ascher DB (2015) pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem 58:4066–4072
pkCSM: Pharmacokinetic properties. http://biosig.unimelb.edu.au/pkcsm/prediction. Accessed 19 Sep 2021
Protein Data Bank. https://www.rcsb.org/. Accessed 1 Mar 2022
PubChem. https://pubchem.ncbi.nlm.nih.gov/. Accessed 31 Aug 2021
Rastogi S, Pandey DN, Singh RH (2022) COVID-19 pandemic: a pragmatic plan for ayurveda intervention. J Ayurveda Integr Med 13:100312
Shang J, Wan Y, Luo C et al (2020) Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci USA 117:11727–11734
Singh R, Purohit R (2023a) Computational analysis of protein-ligand interaction by targeting a cell cycle restrainer. Comput Methods Programs Biomed 231:107367. https://doi.org/10.1016/j.cmpb.2023.107367
Singh R, Purohit R (2023b) Multi-target approach against SARS-CoV-2 by stone apple molecules: a master key to drug design. Phyther Res. https://doi.org/10.1002/ptr.7772
Singh R, Bhardwaj VK, Purohit R (2022) Inhibition of nonstructural protein 15 of SARS-CoV-2 by golden spice: a computational insight. Cell Biochem Funct 40:926–934. https://doi.org/10.1002/cbf.3753
Sorouri F, Emamgholipour Z, Keykhaee M et al (2021) The situation of small molecules targeting key proteins in combatting SARS-CoV-2: synthesis, metabolic pathway, mechanism of action, and potential therapeutic applications. Mini Rev Med Chem 22:273–311
Sun B, Jiang H, Wang Z-N et al (2021) Phytochemical constituents of Onosma bracteatum Wall. Phytochem Lett 45:1–5
Sureja DK, Shah AP, Gajjar ND, et al (2022) In-silico computational investigations of antiviral lignan derivatives as potent inhibitors of SARS CoV-2. ChemistrySelect e202202069
Surti M, Patel M, Adnan M et al (2020) Ilimaquinone (marine sponge metabolite) as a novel inhibitor of SARS-CoV-2 key target proteins in comparison with suggested COVID-19 drugs: Designing, docking and molecular dynamics simulation study. RSC Adv 10:37707–37720
Świderek K, Moliner V (2020) Revealing the molecular mechanisms of proteolysis of SARS-CoV-2 Mproby QM/MM computational methods. Chem Sci 11:10626–10630
Trott O, Olson AJ (2010) AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comb Chem 31:455–461
Update on Omicron. https://www.who.int/news/item/28-11-2021-update-on-omicron. Accessed 7 Feb 2022
Vanommeslaeghe K, Hatcher E, Acharya C et al (2010) CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 31:671–690
Vegad UG, Pandya DJ (2021) Tackling COVID-19 through Ayurveda: a review on the herbs of recommended Indian remedies. Curr Tradit Med 7:e100221191285
Vegad UG, Pandya DJ (2022a) A comprehensive review on Onosma bracteata. Wall: a controversial medicinal herb in Ayurveda. Curr Tradit Med. https://doi.org/10.2174/2215083808666220816105019
Vegad UG, Pandya DJ (2022b) evaluation of diuretic and laxative potential of Onosma bracteata Wall.: a Species of the Controversial Drug ‘Gojihva.’ Int J Pharm Investig 12:358–362. https://doi.org/10.5530/ijpi.2022.3.60
Wang L, Wang Y, Ye D, Liu Q (2020) Review of the 2019 novel coronavirus (SARS-CoV-2) based on current evidence. Int J Antimicrob Agents 55:105948
WHO Coronavirus Disease (COVID-19) Dashboard. https://covid19.who.int/. Accessed 27 Apr 2023
Yin W, Mao C, Luan X et al (2020) Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 368:1499–1504
Yu W, He X, Vanommeslaeghe K, MacKerell AD (2012) Extension of the CHARMM general force field to sulfonyl-containing compounds and its utility in biomolecular simulations. J Comput Chem 33:2451–2468
Zhang Y, Kutateladze TG (2020) Molecular structure analyses suggest strategies to therapeutically target SARS-CoV-2. Nat Commun 11:2920
Funding
The authors did not receive any funding for the work.
Author information
Authors and Affiliations
Contributions
UGV and NDG contributed equally to the present work. UGV: conceptualization, writing- original draft and manuscript preparation; NDG: methodology, data generation, docking, MD simulations, ADMET assay, writing- original draft and manuscript preparation; PRN: docking; SPC: reviewing and editing; DJP: reviewing and editing; TMD: supervision, writing-original draft and manuscript preparation, reviewing and editing.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing financial interest.
Research involving human participants and/or animals
Not Applicable.
Informed consent
Not Applicable.
Accession numbers
Not Applicable.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Vegad, U.G., Gajjar, N.D., Nagar, P.R. et al. In silico screening, ADMET analysis and MD simulations of phytochemicals of Onosma bracteata Wall. as SARS CoV-2 inhibitors. 3 Biotech 13, 221 (2023). https://doi.org/10.1007/s13205-023-03635-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-023-03635-7